
ATM/ATR Phosphorylation of CtIP on Its Conserved Sae2-like Domain Is Required for Genotoxin-Induced DNA Resection but Dispensable for Animal Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of CtipT855A Mice
2.2. Generation of Cell Lines
2.3. Clonogenic Survival Assays
2.4. Alkaline Comet Assays
2.5. T-FISH Assays
2.6. Western Blotting
2.7. Immunofluorescence Microscopy
2.8. DR-GFP and Single-Molecule Analysis of Resection Tracks (SMART) Assays
2.9. Statistical Analyses
3. Results
3.1. Mice Homozygous for the CtipT855A Allele Develop Normally, but Are Hypersensitive to Ionizing Radiation
3.2. Homozygous CtipT855A/T855A Cells Are Hypersensitive to a Subset of Genotoxic Agents
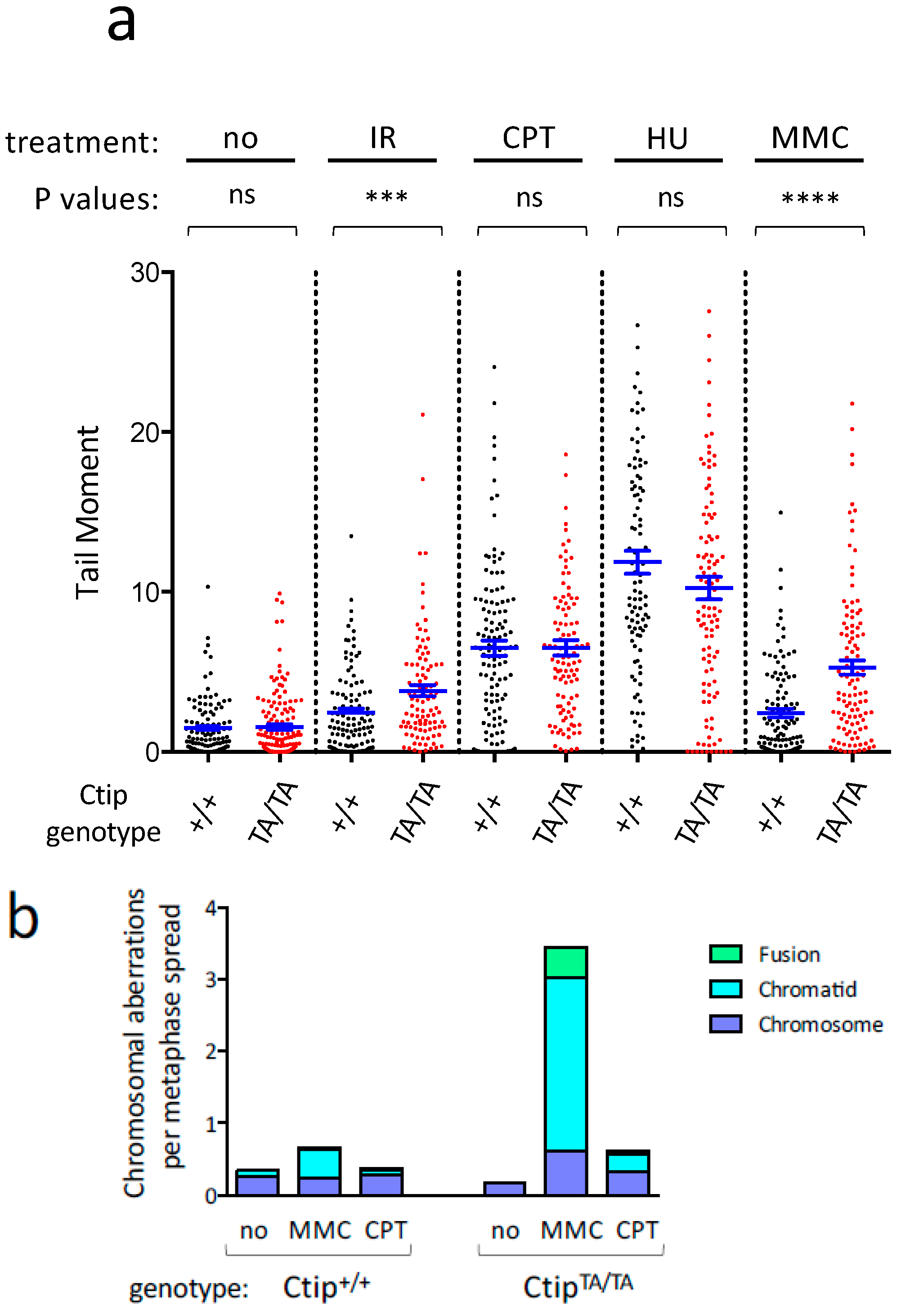
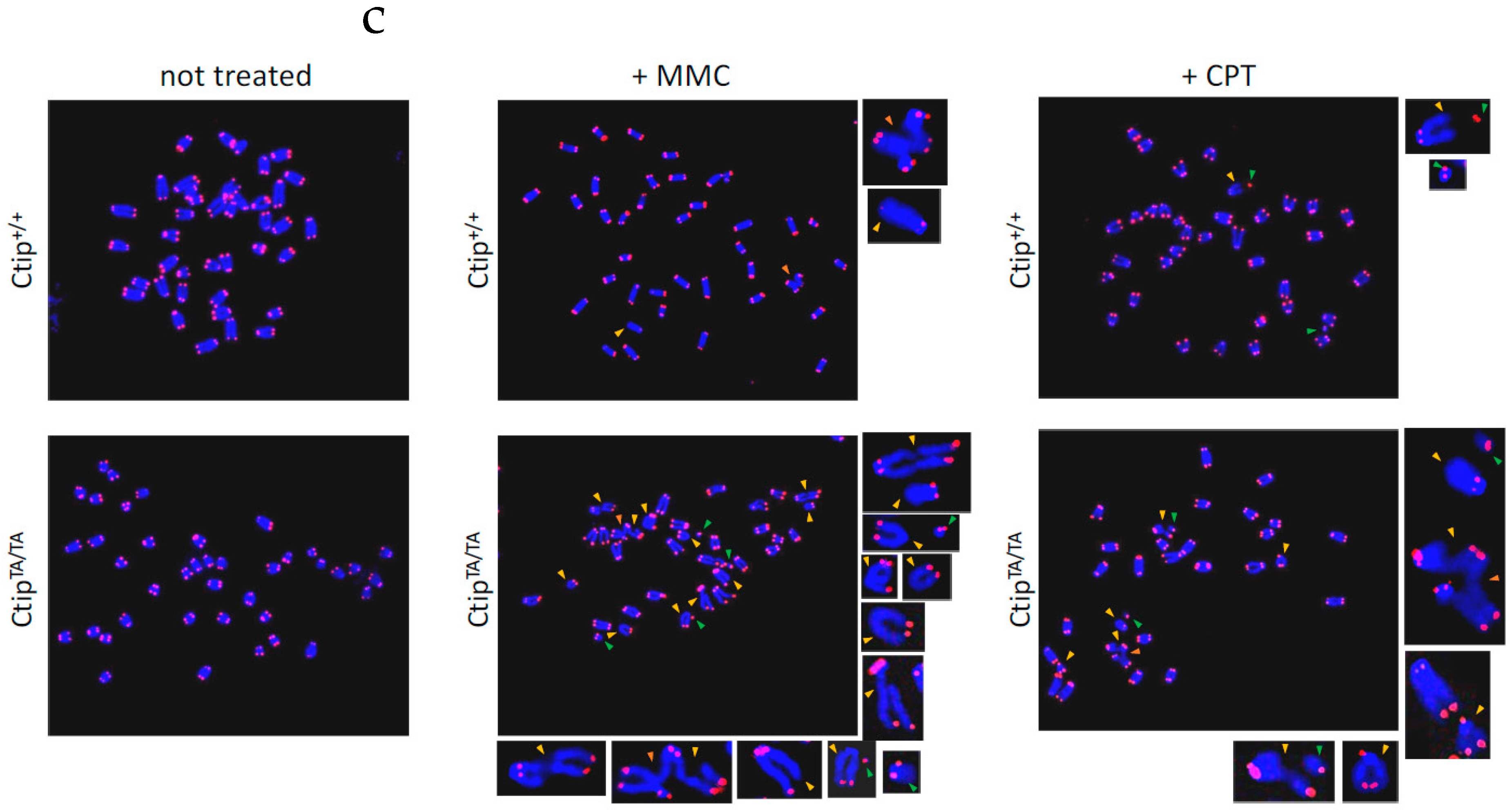
3.3. Homozygous CtipT855A/T855A Cells Display Elevated DNA Damage in Response to Certain Genotoxic Agents
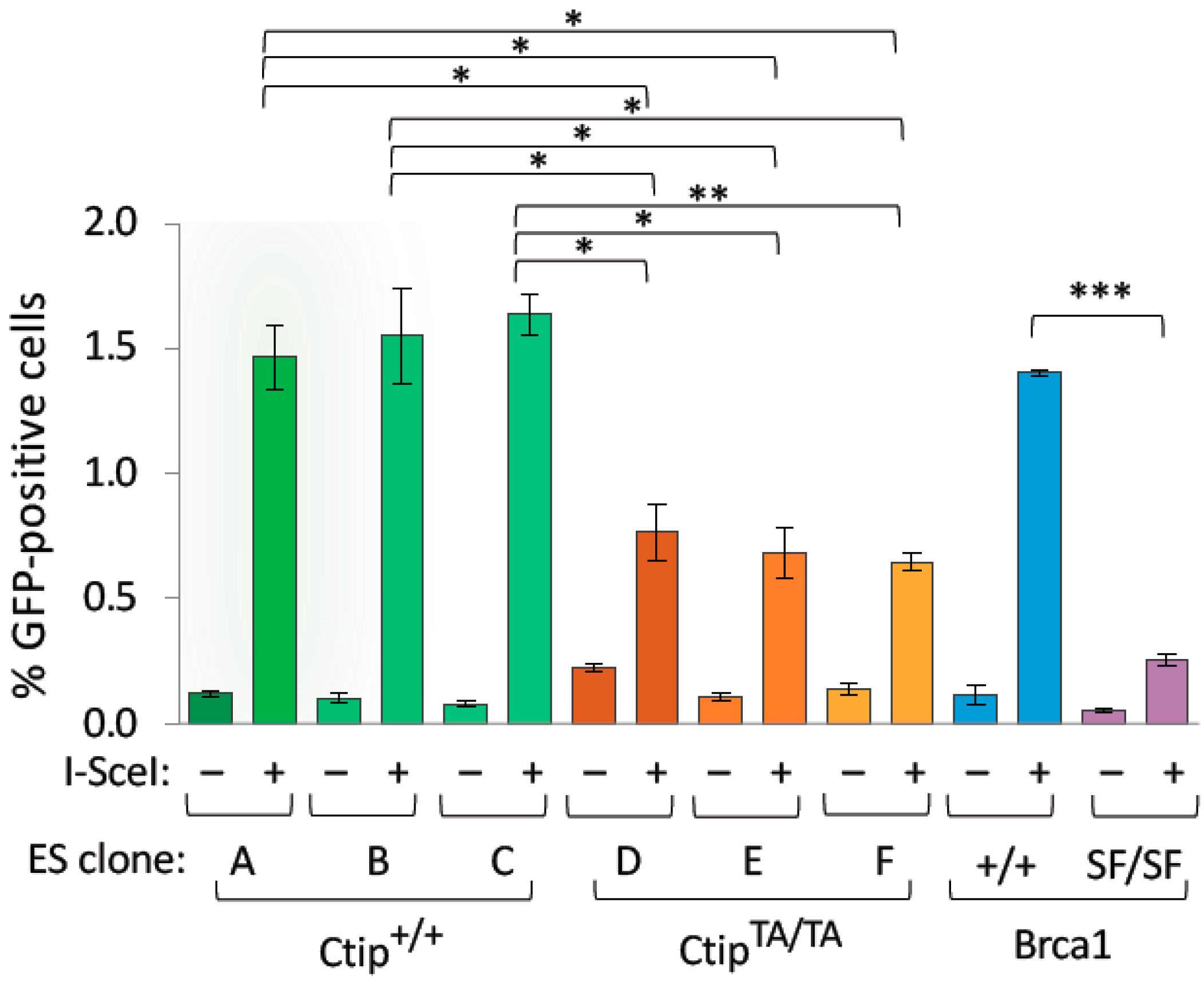
3.4. Homozygous CtipT855A/T855A Cells Are Impaired for Homology-Directed Repair of DNA Double-Strand Breaks
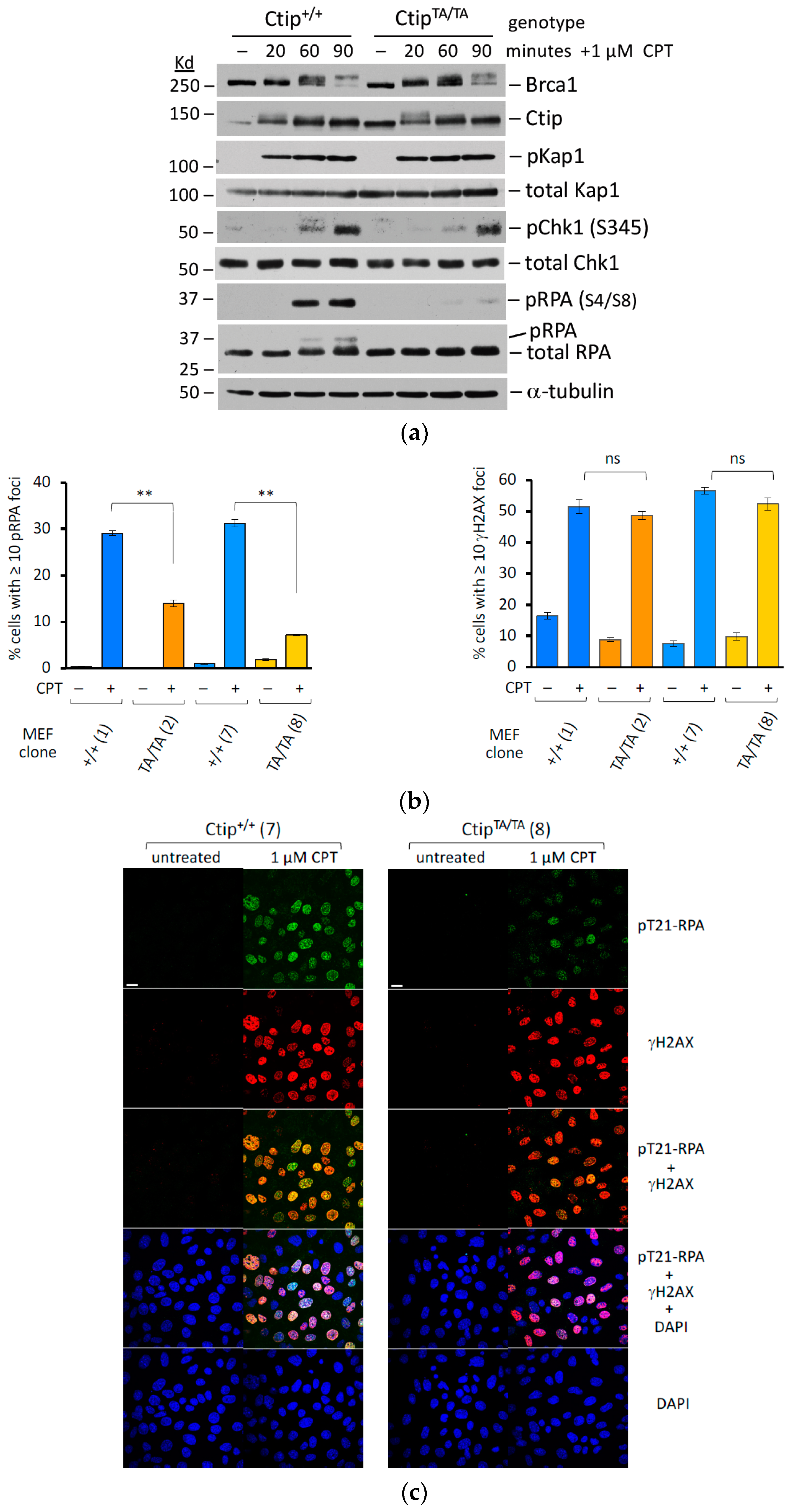
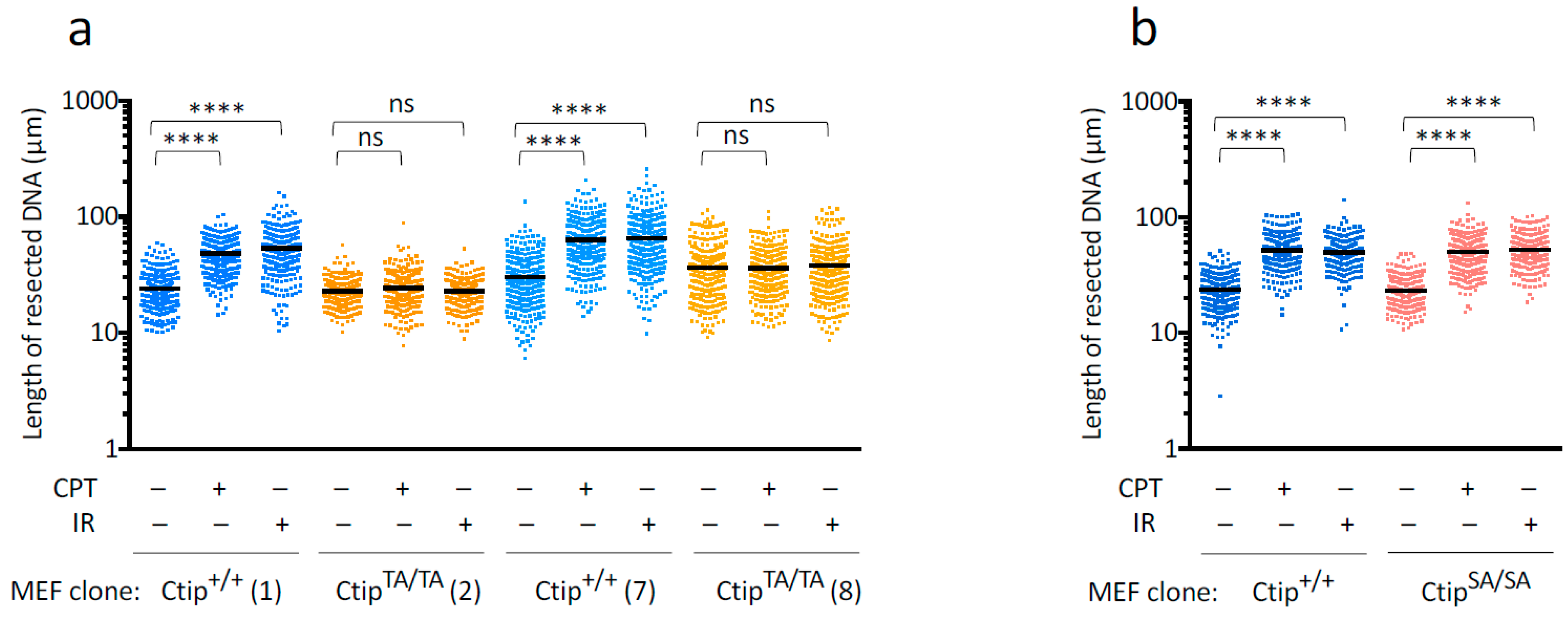
3.5. Genotoxin-Induced DNA Resection Is Compromised in Homozygous CtipT855A/T855A Cells
3.6. Genotoxin-Induced DNA Resection Is Dependent on Phosphorylation of Ctip-T855, but Not the Phospho-Dependent Interaction between Ctip and Brca1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schaeper, U.; Subramanian, T.; Lim, L.; Boyd, J.M.; Chinnadurai, G. Interaction between a cellular protein that binds to the C-terminal region of Adenovirus E1A (CtIP) and a novel cellular protein is disrupted by E1A through a conserved PLDLS motif. J. Biol. Chem. 1998, 273, 8549–8552. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wu, L.C.; Bowcock, A.M.; Aronheim, A.; Baer, R. The carboxy-terminal (BRCT) motifs of BRCA interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J. Biol. Chem. 1998, 273, 25388–25392. [Google Scholar] [CrossRef]
- Wong, A.K.; Ormonde, P.A.; Pero, R.; Chen, Y.; Lian, L.; Salada, G.; Berry, S.; Lawrence, Q.; Dayananth, P.; Ha, P.; et al. Characterization of a carboxy-terminal BRCA1 interacting protein. Oncogene 1998, 17, 2279–2285. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Fu, S.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Keeney, S.; Kleckner, N. Covalent protein-DNA complexes at the 5’ strand termini of meiosis-specific double-strand breaks in yeast. Proc. Natl. Acad. Sci. USA 1995, 92, 11274–11278. [Google Scholar] [CrossRef]
- Cejka, P.; Symington, L.S. DNA End Resection: Mechanism and Control. Annu. Rev. Genet. 2021, 55, 285–307. [Google Scholar] [CrossRef]
- Mozaffari, N.L.; Pagliarulo, F.; Sartori, A.A. Human CtIP: A ‘double agent’ in DNA repair and tumorigenesis. Semin. Cell Dev. Biol. 2021, 113, 47–56. [Google Scholar] [CrossRef]
- Chen, L.; Nievera, C.; Lee, A.Y.; Wu, X. Cell cycle-dependent complex formation of BRCA1/CtIP/MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef]
- Wang, H.; Shao, Z.; Shi, L.Z.; Hwang, P.Y.; Truong, L.N.; Berns, M.W.; Chen, D.J.; Wu, X. CtIP protein dimerization is critical for its recruitment to chromosomal DNA double-stranded breaks. J. Biol. Chem. 2012, 287, 21471–21480. [Google Scholar] [CrossRef]
- Wang, X.S.; Zhao, J.; Wu-Baer, F.; Shao, Z.; Lee, B.J.; Cupo, O.M.; Rabadan, R.; Gautier, J.; Baer, R.; Zha, S. CtIP-mediated DNA resection is dispensable for IgH class switch recombination by alternative end-joining. Proc. Natl. Acad. Sci. USA 2020, 117, 25700–25711. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, R.A.; Lee, J.-H.; Arora, S.; Paull, T.T. Nbs1 Converts the Human Mre11/Rad50 Nuclease Complex into an Endo/Exonuclease Machine Specific for Protein-DNA Adducts. Mol. Cell 2016, 64, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 2016, 64, 940–950. [Google Scholar] [CrossRef]
- Cannavo, E.; Cejka, P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 2014, 514, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 2008, 455, 770–774. [Google Scholar] [CrossRef]
- Zhu, Z.; Chung, W.H.; Shim, E.Y.; Lee, S.E.; Ira, G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008, 134, 981–994. [Google Scholar] [CrossRef]
- Wang, H.; Shi, L.Z.; Wong, C.C.L.; Han, X.; Hwang, P.Y.-H.; Truong, L.N.; Zhu, Q.; Shao, Z.; Chen, D.J.; Berns, M.W.; et al. The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genet. 2013, 9, e1003277. [Google Scholar] [CrossRef]
- Ceppi, I.; Howard, S.M.; Kasaciunaite, K.; Pinto, C.; Anand, R.; Seidel, R.; Cejka, P. CtIP promotes the motor activity of DNA2 to accelerate long-range DNA end resection. Proc. Natl. Acad. Sci. USA 2020, 117, 8859–8869. [Google Scholar] [CrossRef]
- Daley, J.M.; Jimenez-Sainz, J.; Wang, W.; Miller, A.S.; Xue, X.; Nguyen, K.A.; Jensen, R.B.; Sung, P. Enhancement of BLM-DNA2-Mediated Long-Range DNA End Resection by CtIP. Cell Rep. 2017, 21, 324–332. [Google Scholar] [CrossRef]
- Shiotani, B.; Zou, L. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol. Cell 2009, 33, 547–558. [Google Scholar] [CrossRef]
- Chen, H.; Donnianni, R.A.; Handa, N.; Deng, S.K.; Oh, J.; Timashev, L.A.; Kowalczykowski, S.C.; Symington, L.S. Sae2 promotes DNA damage resistance by removing the Mre11-Rad50-Xrs2 complex from DNA and attenuating Rad53 signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E1880–E1887. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.Y.; Kimble, M.T.; Symington, L.S. Sae2 antagonizes Rad9 accumulation at DNA double-strand breaks to attenuate checkpoint signaling and facilitate end resection. Proc. Natl. Acad. Sci. USA 2018, 115, E11961–E11969. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.L.; Liu, F.; Cai, S.; Lin, X.; Li, A.; Chen, Y.; Gu, B.; Lee, E.Y.; Lee, W.H. Inactivation of CtIP leads to early embryonic lethality mediated by G1 restraint and to tumorigenesis by haploid insufficiency. Mol. Cell. Biol. 2005, 25, 3535–3542. [Google Scholar] [CrossRef] [PubMed]
- Reczek, C.R.; Shakya, R.; Miteva, Y.; Szabolcs, M.; Ludwig, T.; Baer, R. The DNA resection protein CtIP promotes mammary tumorigenesis. Oncotarget 2016, 7, 32172–32183. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, X.S.; Lee, B.J.; Wu-Baer, F.K.; Lin, X.; Shao, Z.; Estes, V.M.; Gautier, J.; Baer, R.; Zha, S. CtIP is essential for early B cell proliferation and development in mice. J. Exp. Med. 2019, 216, 1648–1663. [Google Scholar] [CrossRef] [PubMed]
- Limbo, O.; Chahwan, C.; Yamada, Y.; de Bruin, R.A.; Wittenberg, C.; Russell, P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol. Cell 2007, 28, 134–146. [Google Scholar] [CrossRef]
- Uanschou, C.; Siwiec, T.; Pedrosa-Harand, A.; Kerzendorfer, C.; Sanchez-Moran, E.; Novatchkova, M.; Akimcheva, S.; Woglar, A.; Klein, F.; Schlogelhofer, P. A novel plant gene essential for meiosis is related to the human CtIP and the yeast COM1/SAE2 gene. EMBO J. 2007, 26, 5061–5070. [Google Scholar] [CrossRef]
- Penkner, A.; Portik-Dobos, Z.; Tang, L.; Schnabel, R.; Novatchkova, M.; Jantsch, V.; Loidl, J. A conserved function for a Caenorhabditis elegans Com1/Sae2/CtIP protein homolog in meiotic recombination. EMBO J. 2007, 26, 5071–5082. [Google Scholar] [CrossRef]
- Huertas, P.; Cortes-Ledesma, F.; Sartori, A.A.; Aguilera, A.; Jackson, S.P. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 2008, 455, 689–692. [Google Scholar] [CrossRef]
- Huertas, P.; Jackson, S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef]
- Peterson, S.E.; Li, Y.; Wu-Baer, F.; Chait, B.T.; Baer, R.; Yan, H.; Gottesman, M.E.; Gautier, J. Activation of DSB processing requires phosphorylation of CtIP by ATR. Mol. Cell 2013, 49, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Jasrotia, A.; Bundschuh, D.; Howard, S.M.; Ranjha, L.; Stucki, M.; Cejka, P. NBS1 promotes the endonuclease activity of the MRE11-RAD50 complex by sensing CtIP phosphorylation. EMBO J. 2019, 38, e101005. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, E.; Johnson, D.; Andres, S.N.; Kissling, V.M.; Reinert, J.K.; Garcia, V.; Erie, D.A.; Hess, D.; Thoma, N.H.; Enchev, R.I.; et al. Regulatory control of DNA end resection by Sae2 phosphorylation. Nat. Commun. 2018, 9, 4016. [Google Scholar] [CrossRef] [PubMed]
- Polato, F.; Callen, E.; Wong, N.; Faryabi, R.; Bunting, S.; Chen, H.T.; Kozak, M.; Kruhlak, M.J.; Reczek, C.R.; Lee, W.H.; et al. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. J. Exp. Med. 2014, 211, 1027–1036. [Google Scholar] [CrossRef]
- Wang, X.S.; Menolfi, D.; Wu-Baer, F.; Fangazio, M.; Meyer, S.N.; Shao, Z.; Wang, Y.; Zhu, Y.; Lee, B.J.; Estes, V.M.; et al. DNA damage-induced phosphorylation of CtIP at a conserved ATM/ATR site T855 promotes lymphomagenesis in mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2105440118. [Google Scholar] [CrossRef]
- Paragas, N.; Qiu, A.; Zhang, Q.; Samstein, B.; Deng, S.X.; Schmidt-Ott, K.M.; Viltard, M.; Yu, W.; Forster, C.S.; Gong, G.; et al. The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat. Med. 2011, 17, 216–222. [Google Scholar] [CrossRef]
- Billing, D.; Horiguchi, M.; Wu-Baer, F.; Taglialatela, A.; Leuzzi, G.; Nanez, S.A.; Jiang, W.; Zha, S.; Szabolcs, M.; Lin, C.-S.; et al. The BRCT Domains of the BRCA1 and BARD1 Tumor Suppressors Differentially Regulate Homology-Directed Repair and Stalled Fork Protection. Mol. Cell 2018, 72, 127–139. [Google Scholar] [CrossRef]
- Reczek, C.R.; Szabolcs, M.; Stark, J.M.; Ludwig, T.; Baer, R. The interaction between CtIP and BRCA1 is not essential for resection-mediated DNA repair or tumor suppression. J. Cell Biol. 2013, 201, 693–707. [Google Scholar] [CrossRef]
- Pierce, A.J.; Hu, P.; Han, M.; Ellis, N.; Jasin, M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001, 15, 3237–3242. [Google Scholar] [CrossRef]
- Shakya, R.; Reid, L.J.; Reczek, C.R.; Cole, F.; Egli, D.; Lin, C.-S.; deRooij, D.G.; Hirsch, S.; Kandasamy, R.; Hicks, J.B.; et al. BRCT phosphoprotein recognition, but not E3 ligase activity, is essential for BRCA1 tumor suppression. Science 2011, 334, 525–528. [Google Scholar] [CrossRef]
- Yu, X.; Baer, R. Nuclear localization and cell cycle-specific expression of CtIP, a protein that associates with the BRCA1 tumor suppressor. J. Biol. Chem. 2000, 275, 18541–18549. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Garcia, A.; Lopez-Saavedra, A.; Huertas, P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 2014, 9, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Kousholt, A.N.; Fugger, K.; Hoffmann, S.; Larsen, B.D.; Menzel, T.; Sartori, A.A.; Sorensen, C.S. CtIP-dependent DNA resection is required for DNA damage checkpoint maintenance but not initiation. J. Cell Biol. 2012, 197, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, R.A.; Myler, L.R.; Soniat, M.M.; Makharashvili, N.; Lee, L.; Lees-Miller, S.P.; Finkelstein, I.J.; Paull, T.T. DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci. Adv. 2020, 6, eaay0922. [Google Scholar] [CrossRef] [PubMed]
- Barton, O.; Naumann, S.C.; Diemer-Biehs, R.; Kunzel, J.; Steinlage, M.; Conrad, S.; Makharashvili, N.; Wang, J.; Feng, L.; Lopez, B.S.; et al. Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. J. Cell Biol. 2014, 206, 877–894. [Google Scholar] [CrossRef] [PubMed]
- Biehs, R.; Steinlage, M.; Barton, O.; Juhasz, S.; Kunzel, J.; Spies, J.; Shibata, A.; Jeggo, P.A.; Lobrich, M. DNA Double-Strand Break Resection Occurs during Non-homologous End Joining in G1 but Is Distinct from Resection during Homologous Recombination. Mol. Cell 2017, 65, 671–684.e5. [Google Scholar] [CrossRef]
- Averbeck, N.B.; Barent, C.; Jakob, B.; Syzonenko, T.; Durante, M.; Taucher-Scholz, G. The Ubiquitin Ligase RNF138 Cooperates with CtIP to Stimulate Resection of Complex DNA Double-Strand Breaks in Human G1-Phase Cells. Cells 2022, 11, 2561. [Google Scholar] [CrossRef]
- Li, F.; Mladenov, E.; Sun, Y.; Soni, A.; Stuschke, M.; Timmermann, B.; Iliakis, G. Low CDK Activity and Enhanced Degradation by APC/C(CDH1) Abolishes CtIP Activity and Alt-EJ in Quiescent Cells. Cells 2023, 12, 1530. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu-Baer, F.; Wong, M.; Tschoe, L.; Lin, C.-S.; Jiang, W.; Zha, S.; Baer, R. ATM/ATR Phosphorylation of CtIP on Its Conserved Sae2-like Domain Is Required for Genotoxin-Induced DNA Resection but Dispensable for Animal Development. Cells 2023, 12, 2762. https://doi.org/10.3390/cells12232762
Wu-Baer F, Wong M, Tschoe L, Lin C-S, Jiang W, Zha S, Baer R. ATM/ATR Phosphorylation of CtIP on Its Conserved Sae2-like Domain Is Required for Genotoxin-Induced DNA Resection but Dispensable for Animal Development. Cells. 2023; 12(23):2762. https://doi.org/10.3390/cells12232762
Chicago/Turabian StyleWu-Baer, Foon, Madeline Wong, Lydia Tschoe, Chyuan-Sheng Lin, Wenxia Jiang, Shan Zha, and Richard Baer. 2023. "ATM/ATR Phosphorylation of CtIP on Its Conserved Sae2-like Domain Is Required for Genotoxin-Induced DNA Resection but Dispensable for Animal Development" Cells 12, no. 23: 2762. https://doi.org/10.3390/cells12232762
APA StyleWu-Baer, F., Wong, M., Tschoe, L., Lin, C.-S., Jiang, W., Zha, S., & Baer, R. (2023). ATM/ATR Phosphorylation of CtIP on Its Conserved Sae2-like Domain Is Required for Genotoxin-Induced DNA Resection but Dispensable for Animal Development. Cells, 12(23), 2762. https://doi.org/10.3390/cells12232762