
CRISPR-Cas9 KO Cell Line Generation and Development of a Cell-Based Potency Assay for rAAV-FKRP Gene Therapy

 and
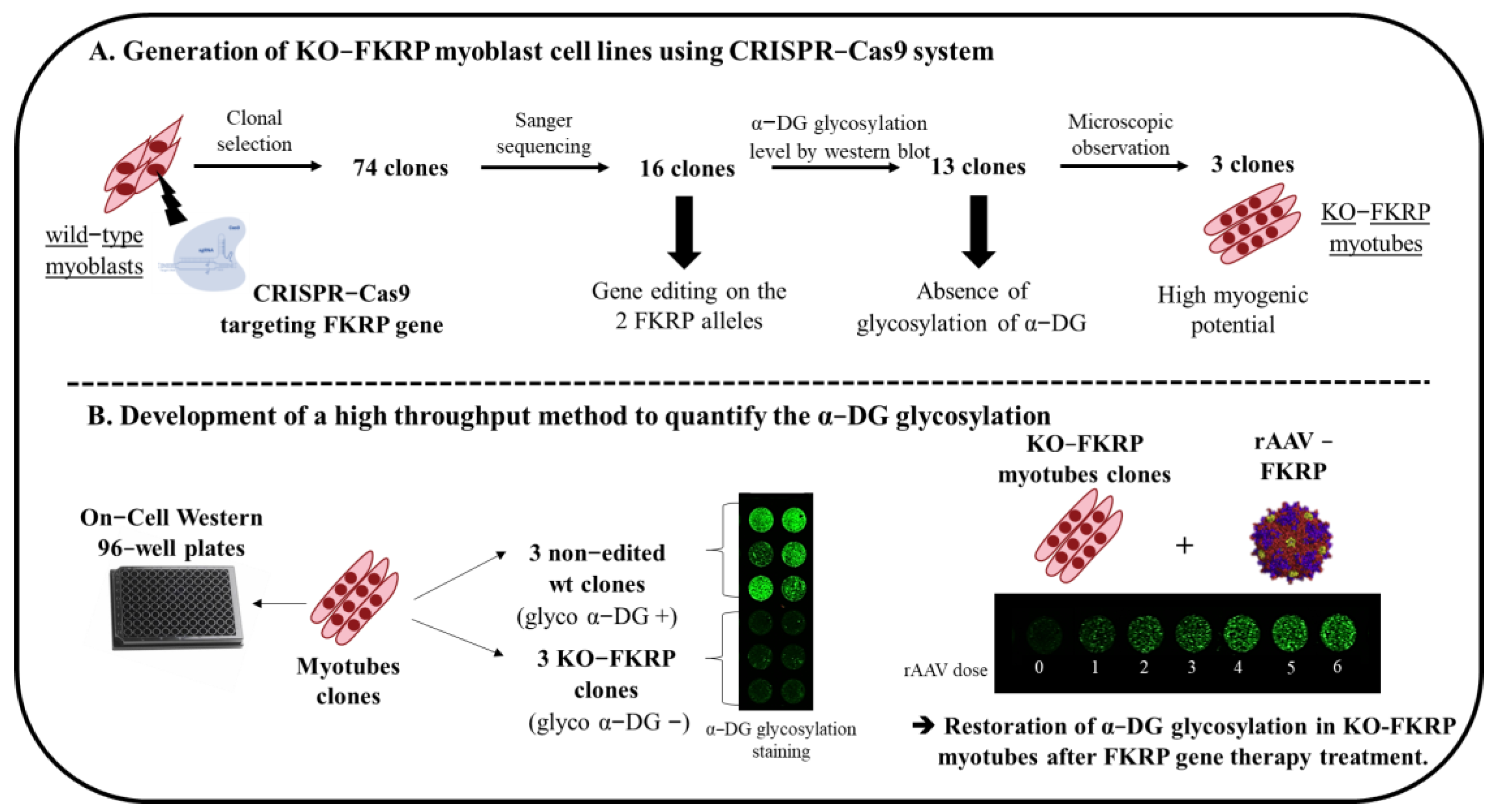
and 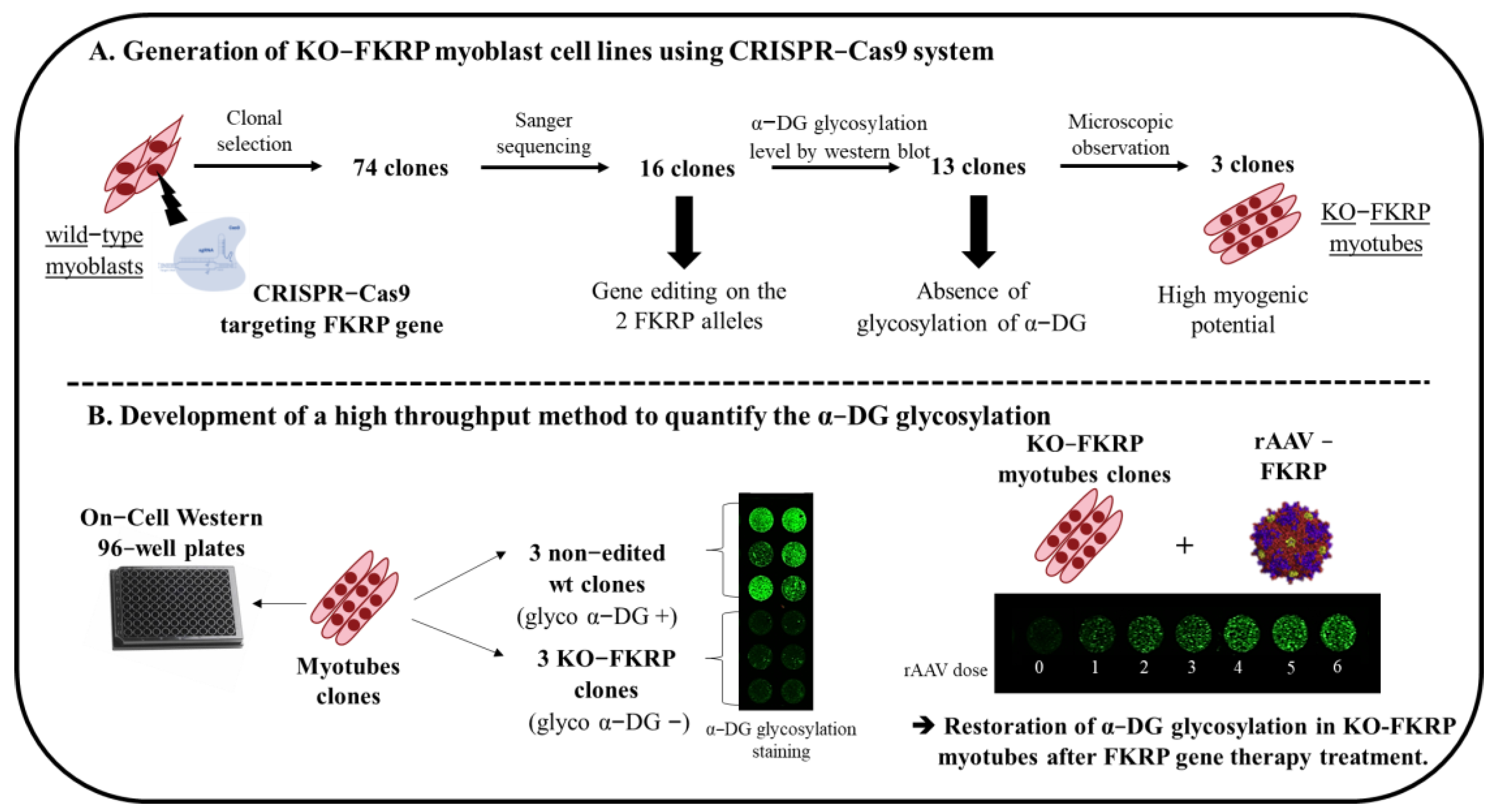{kind=link}
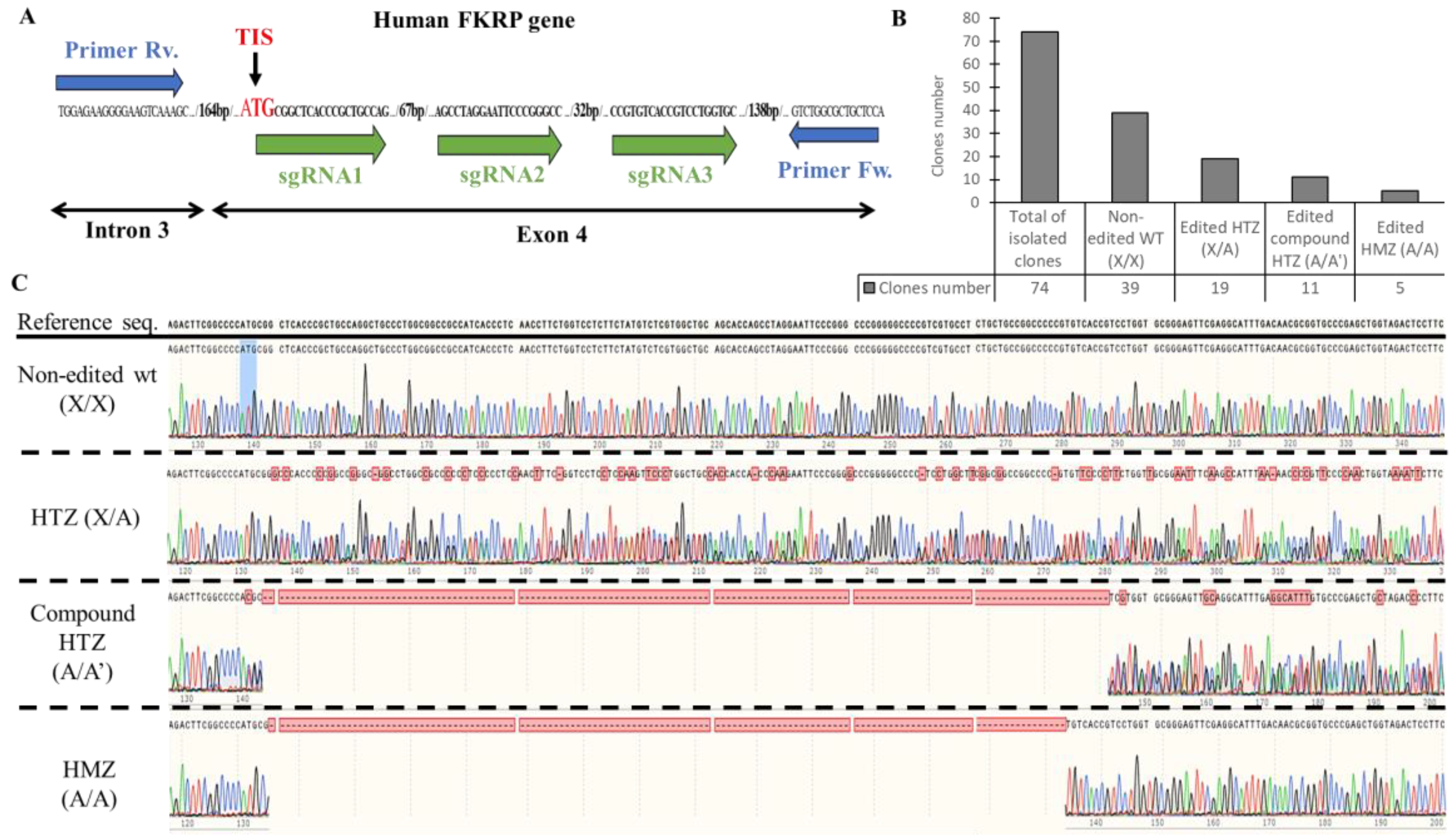{kind=link}
{kind=link}
{kind=link}
{kind=link}
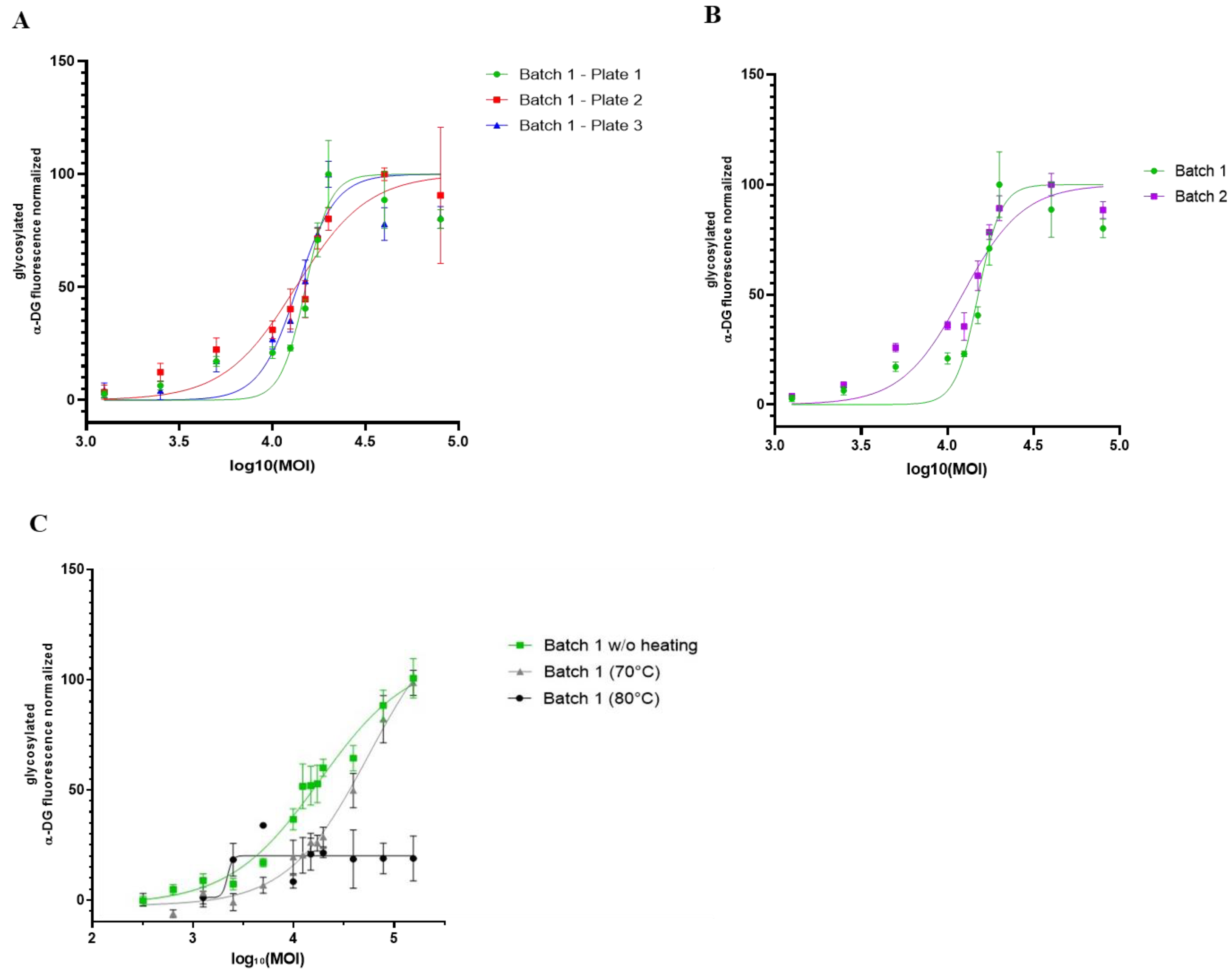{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Differentiation
2.2. Generation of KO-FKRP Cell Line
2.3. DNA Analysis by Sanger Sequencing
2.4. Immunofluorescence Staining
2.5. AAV Vectors and Transduction
2.6. Western Blot
2.7. JESS Simple Western™
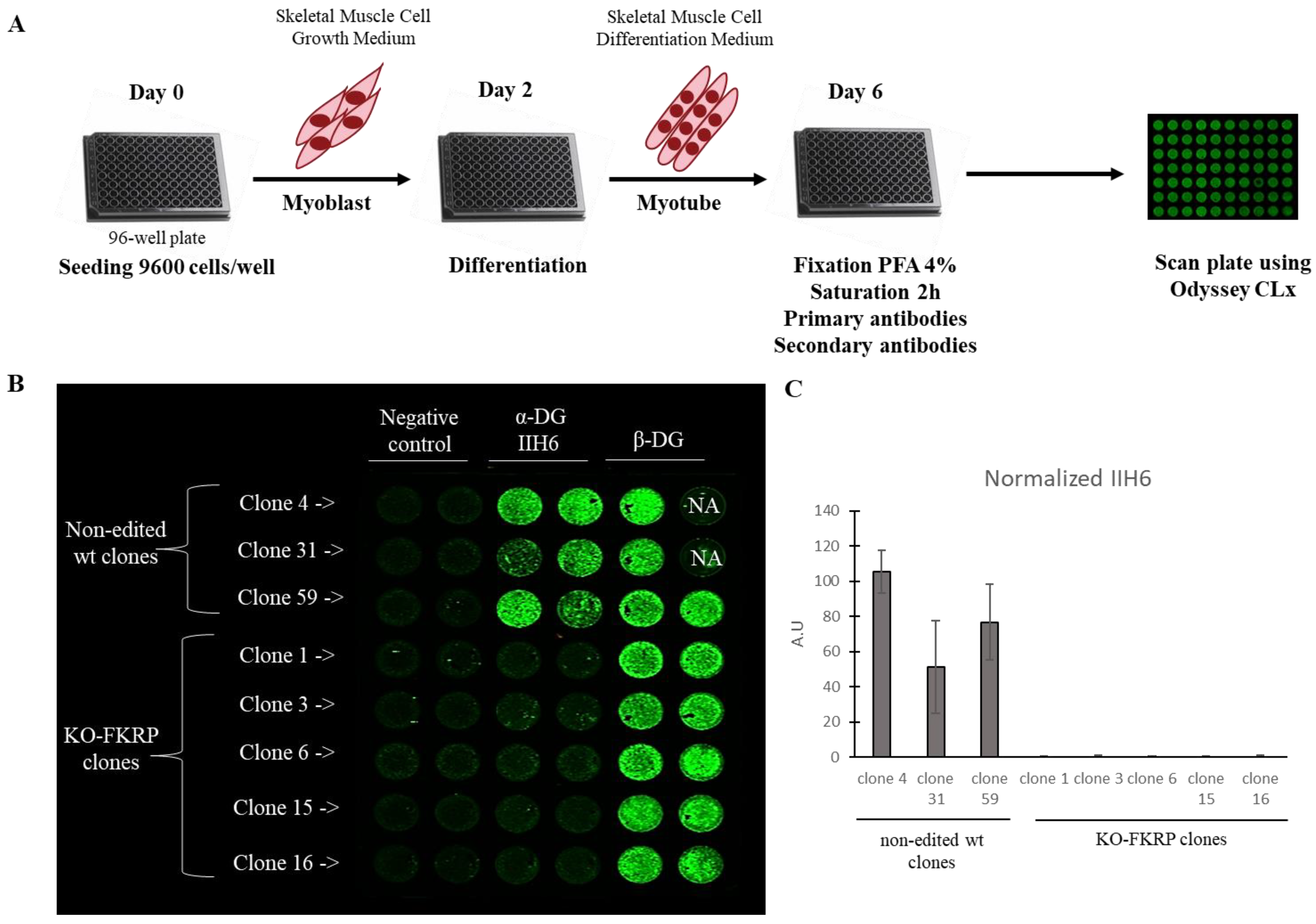
2.8. On-Cell Western
2.9. Statistics
3. Results
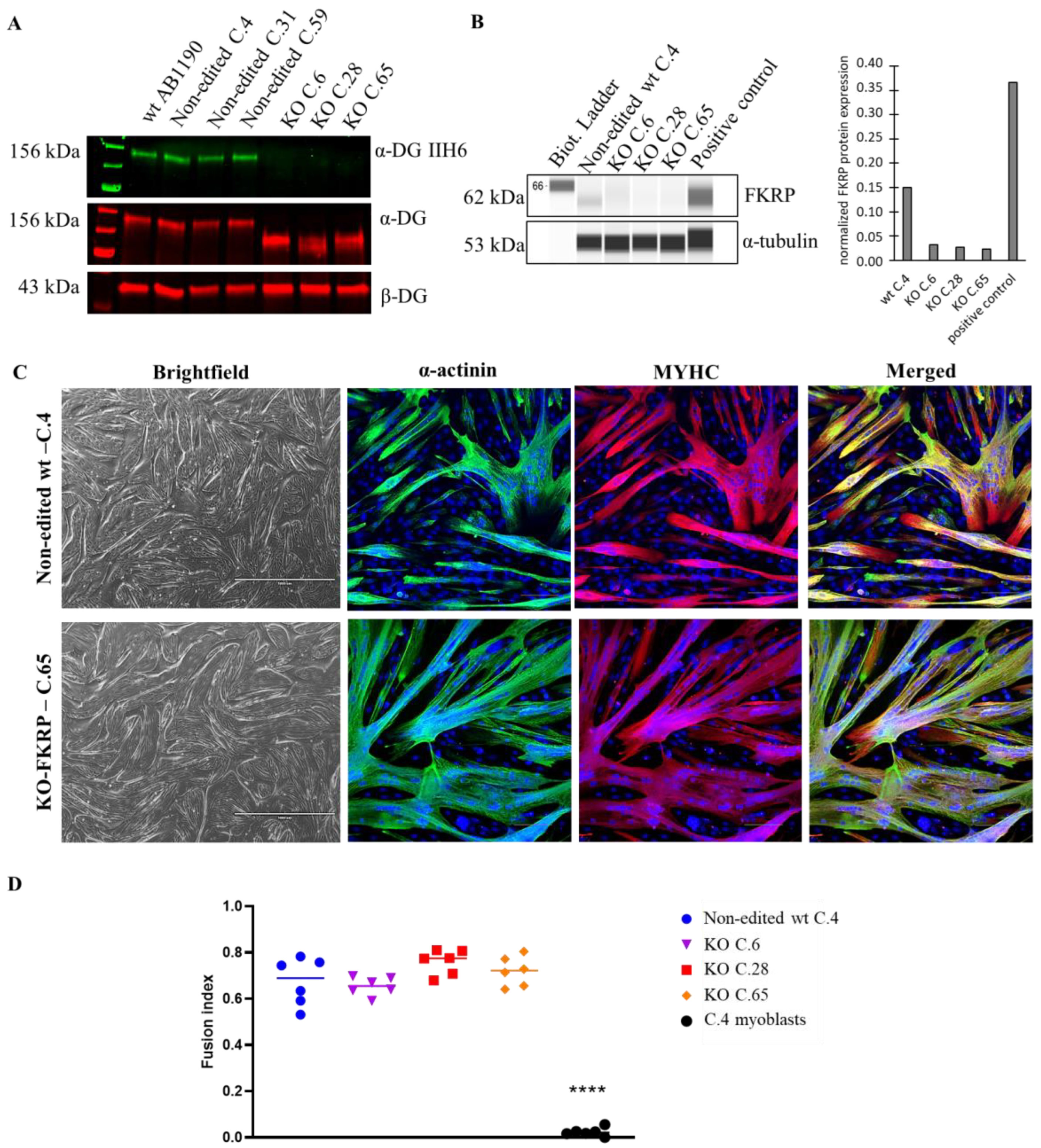
3.1. Generation of KO-FKRP Myoblasts by CRISPR-Cas9 System
3.2. Characterization of α-DG Glycosylation in Selected KO-FKRP Clones
3.3. Development of On-Cell Western Method to Detect the Glycosylated α-DG
3.4. Restoration of Biological Activity in KO-FKRP Clones after Transduction by rAAV-FKRP Vector
3.5. Proof of Concept of rAAV Batch-to-Batch Comparison
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kanagawa, M. Dystroglycanopathy: From Elucidation of Molecular and Pathological Mechanisms to Development of Treatment Methods. Int. J. Mol. Sci. 2021, 22, 13162. [Google Scholar] [CrossRef]
- Talts, J.F.; Andac, Z.; Göhring, W.; Brancaccio, A.; Timpl, R. Binding of the G domains of laminin α1 and α2 chains and perlecan to heparin, sulfatides, α-dystroglycan and several extracellular matrix proteins. EMBO J. 1999, 18, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, E.; Tisi, D.; Talts, J.F.; Timpl, R. The crystal structure of a laminin G–like module reveals the molecular basis of α-dystroglycan binding to laminins, perlecan, and agrin. Mol. Cell 1999, 4, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Ervasti, J.M.; Campbell, K.P. A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 1993, 122, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Endo, T. Glycobiology of α-dystroglycan and muscular dystrophy. J. Biochem. 2015, 157, 1–12. [Google Scholar] [CrossRef]
- Praissman, J.L.; Willer, T.; Sheikh, M.O.; Toi, A.; Chitayat, D.; Lin, Y.-Y.; Lee, H.; Stalnaker, S.H.; Wang, S.; Prabhakar, P.K.; et al. The functional O-mannose glycan on α-dystroglycan contains a phospho-ribitol primed for matriglycan addition. Elife 2016, 5, e14473. [Google Scholar] [CrossRef]
- Kanagawa, M.; Kobayashi, K.; Tajiri, M.; Manya, H.; Kuga, A.; Yamaguchi, Y.; Akasaka-Manya, K.; Furukawa, J.-I.; Mizuno, M.; Kawakami, H.; et al. Identification of a Post-translational Modification with Ribitol-Phosphate and Its Defect in Muscular Dystrophy. Cell Rep. 2016, 14, 2209–2223. [Google Scholar] [CrossRef]
- Kuwabara, N.; Imae, R.; Manya, H.; Tanaka, T.; Mizuno, M.; Tsumoto, H.; Kanagawa, M.; Kobayashi, K.; Toda, T.; Senda, T.; et al. Crystal structures of fukutin-related protein (FKRP), a ribitol-phosphate transferase related to muscular dystrophy. Nat. Commun. 2020, 11, 303. [Google Scholar] [CrossRef]
- Brockington, M.; Blake, D.J.; Prandini, P.; Brown, S.C.; Torelli, S.; Benson, M.A.; Ponting, C.P.; Estournet, B.; Romero, N.B.; Mercuri, E.; et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin α2 deficiency and abnormal glycosylation of α-dystroglycan. Am. J. Hum. Genet. 2001, 69, 1198–1209. [Google Scholar] [CrossRef]
- Brockington, M.; Yuva, Y.; Prandini, P.; Brown, S.C.; Torelli, S.; Benson, M.A.; Herrmann, R.; Anderson, L.V.; Bashir, R.; Burgunder, J.-M.; et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum. Mol. Genet. 2001, 10, 2851–2859. [Google Scholar] [CrossRef]
- de Bernabe, D.B.-V.; Voit, T.; Longman, C.; Steinbrecher, A.; Straub, V.; Yuva, Y.; Herrmann, R.; Sperner, J.; Korenke, C.; Diesen, C.; et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J. Med. Genet. 2004, 41, e61. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.F.; Gicquel, E.; Marsolier, J.; Richard, I. Functional and cellular localization diversity associated with Fukutin-related protein patient genetic variants. Hum. Mutat. 2019, 40, 1874–1885. [Google Scholar] [CrossRef] [PubMed]
- Esapa, C.T.; McIlhinney, R.A.J.; Blake, D.J. Fukutin-related protein mutations that cause congenital muscular dystrophy result in ER-retention of the mutant protein in cultured cells. Hum. Mol. Genet. 2005, 14, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Margeta, M.; Connolly, A.M.; Winder, T.L.; Pestronk, A.; Moore, S.A. Cardiac pathology exceeds skeletal muscle pathology in two cases of limb-girdle muscular dystrophy type 2I. Muscle Nerve 2009, 40, 883–889. [Google Scholar] [CrossRef] [PubMed]
- D'Amico, A.; Petrini, S.; Parisi, F.; Tessa, A.; Francalanci, P.; Grutter, G.; Santorelli, F.M.; Bertini, E. Heart transplantation in a child with LGMD2I presenting as isolated dilated cardiomyopathy. Neuromuscul. Disord. 2008, 18, 153–155. [Google Scholar] [CrossRef]
- Ortiz-Cordero, C.; Azzag, K.; Perlingeiro, R.C.R. Fukutin-Related Protein: From Pathology to Treatments. Trends Cell Biol. 2021, 31, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Vannoy, C.H.; Leroy, V.; Lu, Q.L. Dose-Dependent Effects of FKRP Gene-Replacement Therapy on Functional Rescue and Longevity in Dystrophic Mice. Mol. Ther. Methods Clin. Dev. 2018, 11, 106–120. [Google Scholar] [CrossRef]
- Xu, L.; Lu, P.J.; Wang, C.-H.; Keramaris, E.; Qiao, C.; Xiao, B.; Blake, D.J.; Xiao, X.; Lu, Q.L. Adeno-associated virus 9 mediated FKRP gene therapy restores functional glycosylation of α-dystroglycan and improves muscle functions. Mol. Ther. 2013, 21, 1832–1840. [Google Scholar] [CrossRef]
- Benasutti, H.; Maricelli, J.W.; Seto, J.; Hall, J.; Halbert, C.; Wicki, J.; Heusgen, L.; Purvis, N.; Regnier, M.; Lin, D.C.; et al. Efficacy and muscle safety assessment of fukutin-related protein gene therapy. Mol. Ther. Methods Clin. Dev. 2023, 30, 65–80. [Google Scholar] [CrossRef]
- Gicquel, E.; Maizonnier, N.; Foltz, S.J.; Martin, W.J.; Bourg, N.; Svinartchouk, F.; Charton, K.; Beedle, A.M.; Richard, I. AAV-mediated transfer of FKRP shows therapeutic efficacy in a murine model but requires control of gene expression. Hum. Mol. Genet. 2017, 26, 1952–1965. [Google Scholar] [CrossRef]
- (FDA), U.F.a.D.A. Potency Tests for Cellular and Gene Therapy Products. Final Guidance for Industry. 2011. Available online: https://www.fda.gov/files/vaccines,%20blood%20%26%20biologics/published/Final-Guidance-for-Industry--Potency-Tests-for-Cellular-and-Gene-Therapy-Products.pdf (accessed on 10 October 2023).
- Salmikangas, P.; Carlsson, B.; Klumb, C.; Reimer, T.; Thirstrup, S. Potency testing of cell and gene therapy products. Front. Med. 2023, 10, 1190016. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.C.; Torelli, S.; Brockington, M.; Yuva, Y.; Jimenez, C.; Feng, L.; Anderson, L.; Ugo, I.; Kroger, S.; Bushby, K.; et al. Abnormalities in α-dystroglycan expression in MDC1C and LGMD2I muscular dystrophies. Am. J. Pathol. 2004, 164, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-C.; Murakami, T.; Hayashi, Y.K.; Nishino, I.; Nonaka, I.; Yuo, C.Y.; Jong, Y.J. A novel FKRP gene mutation in a Taiwanese patient with limb-girdle muscular dystrophy 2I. Brain Dev. 2007, 29, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef]
- Wang, J.Y.; Doudna, J.A. CRISPR technology: A decade of genome editing is only the beginning. Science 2023, 379, eadd8643. [Google Scholar] [CrossRef]
- Beaufils, M.; Tourel, A.; Petiot, A.; Halmai, N.B.; Segal, D.J.; Rendu, J.; Marty, I. Development of Knock-Out Muscle Cell Lines using Lentivirus-Mediated CRISPR/Cas9 Gene Editing. J. Vis. Exp. 2022, 184, e64114. [Google Scholar] [CrossRef]
- Hu, O.; Provvido, A.; Zhu, Y. Generation of IL17RB Knockout Cell Lines Using CRISPR/Cas9-Based Genome Editing. Methods Mol. Biol. 2020, 2108, 345–353. [Google Scholar] [CrossRef]
- Aguilar, H.N.; Zielnik, B.; Tracey, C.N.; Mitchell, B.F. Quantification of rapid Myosin regulatory light chain phosphorylation using high-throughput in-cell Western assays: Comparison to Western immunoblots. PLoS ONE 2010, 5, e9965. [Google Scholar] [CrossRef]
- Coevoets, R.; Arican, S.; Hoogeveen-Westerveld, M.; Simons, E.; van den Ouweland, A.; Halley, D.; Nellist, M. A reliable cell-based assay for testing unclassified TSC2 gene variants. Eur. J. Hum. Genet. 2009, 17, 301–310. [Google Scholar] [CrossRef]
- Mamchaoui, K.; Trollet, C.; Bigot, A.; Negroni, E.; Chaouch, S.; Wolff, A.; Kandalla, P.K.; Marie, S.; Di Santo, J.; St Guily, J.L.; et al. Immortalized pathological human myoblasts: Towards a universal tool for the study of neuromuscular disorders. Skelet. Muscle 2011, 1, 34. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, M.; Poupiot, J.; Goyenvalle, A.; Perez, N.; Garcia, L.; Danos, O.; Richard, I. Noninvasive monitoring of therapeutic gene transfer in animal models of muscular dystrophies. Gene Ther. 2006, 13, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Apparailly, F.; Khoury, M.; Vervoordeldonk, M.J.B.; Adriaansen, J.; Gicquel, E.; Perez, N.; Riviere, C.; Louis-Plence, P.; Noel, D.; Danos, O.; et al. Adeno-associated virus pseudotype 5 vector improves gene transfer in arthritic joints. Hum. Gene Ther. 2005, 16, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Kostrominova, T.Y.; Tanzer, M.L. Temporal and spatial appearance of α-dystroglycan in differentiated mouse myoblasts in culture. J. Cell. Biochem. 1995, 58, 527–534. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Campbell, K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991, 66, 1121–1131. [Google Scholar] [CrossRef]
- De, B.P.; Chen, A.; Salami, C.O.; Van de Graaf, B.; Rosenberg, J.B.; Pagovich, O.E.; Sondhi, D.; Crystal, R.G.; Kaminsky, S.M. In Vivo Potency Assay for Adeno-Associated Virus–Based Gene Therapy Vectors Using AAVrh.10 as an Example. Hum. Gene Ther. Methods 2018, 29, 146–155. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, X.; He, Q.; Wang, J.; Mao, Q.; Liang, Z.; Xu, M. Research progress on substitution of in vivo method(s) by in vitro method(s) for human vaccine potency assays. Expert Rev. Vaccines 2023, 22, 270–277. [Google Scholar] [CrossRef]
- Stalpers, C.A.L.; Retmana, I.A.; Pennings, J.L.A.; Vandebriel, R.J.; Hendriksen, C.F.M.; Akkermans, A.M.; Hoefnagel, M.H.N. Variability of in vivo potency tests of Diphtheria, Tetanus and acellular Pertussis (DTaP) vaccines. Vaccine 2021, 39, 2506–2516. [Google Scholar] [CrossRef]
- (EMA), E.M.A. Regulatory acceptance of 3R (replacement, reduction, refinement) testing approaches—Scientific guideline 2017. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-principles-regulatory-acceptance-3rs-replacement-reduction-refinement-testing-approaches_en.pdf (accessed on 10 October 2023).
- Aronson, S.J.; Bakker, R.S.; Moenis, S.; van Dijk, R.; Bortolussi, G.; Collaud, F.; Shi, X.; Duijst, S.; Ten Bloemendaal, L.; Ronzitti, G.; et al. A Quantitative In Vitro Potency Assay for Adeno-Associated Virus Vectors Encoding for the UGT1A1 Transgene. Mol. Ther. Methods Clin. Dev. 2020, 18, 250–258. [Google Scholar] [CrossRef]
- Couto, L.; Buchlis, G.; Farjo, R.; High, K.A. Poster C0048 ARVO: Potency assay for AAV vector encoding retinal pigment epithelial 65 protein. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1. [Google Scholar]
- Banning, A.; Zakrzewicz, A.; Chen, X.; Gray, S.J.; Tikkanen, R. Knockout of the CMP–Sialic Acid Transporter SLC35A1 in Human Cell Lines Increases Transduction Efficiency of Adeno-Associated Virus 9: Implications for Gene Therapy Potency Assays. Cells 2021, 10, 1259. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Yu, C.; Wang, L.; Gao, K.; Xu, G.; Wang, W.; Cao, J.; Wang, J. Development of a robust reporter gene based assay for the bioactivity determination of IL-5-targeted therapeutic antibodies. J. Pharm. Biomed. Anal. 2018, 148, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Junjhon, J.; Panyasu, K.; Chaiyaloom, S.; Saipin, K.; Somasa, P.; Sangiambut, S.; Puttikhunt, C.; Sriburi, R.; Keelapang, P.; Ekchariyawat, P.; et al. Generation and characterization of luciferase-secreting, single-round infectious DENV-2 reporter for functional antibody assays. J. Virol. Methods 2021, 291, 114119. [Google Scholar] [CrossRef]
- Wang, L.; Xu, G.-L.; Gao, K.; Wilkinson, J.; Zhang, F.; Yu, L.; Liu, C.-Y.; Yu, C.-F.; Wang, W.-B.; Li, M.; et al. Development of a robust reporter-based assay for the bioactivity determination of anti-VEGF therapeutic antibodies. J. Pharm. Biomed. Anal. 2016, 125, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, Y.; Yu, L.; Li, X.; Shi, X.; Qin, X.; Rao, C.; Wang, J. A novel reporter gene assay for recombinant human erythropoietin (rHuEPO) pharmaceutical products. J. Pharm. Biomed. Anal. 2014, 100, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Alhamidi, M.; Brox, V.; Stensland, E.; Liset, M.; Lindal, S.; Nilssen, Ø. Limb girdle muscular dystrophy type 2I: No correlation between clinical severity, histopathology and glycosylated α-dystroglycan levels in patients homozygous for common FKRP mutation. Neuromuscul. Disord. 2017, 27, 619–626. [Google Scholar] [CrossRef]
- Tucker, J.D.; Lu, P.J.; Xiao, X.; Lu, Q.L. Overexpression of Mutant FKRP Restores Functional Glycosylation and Improves Dystrophic Phenotype in FKRP Mutant Mice. Mol. Ther. Nucleic Acids 2018, 11, 216–227. [Google Scholar] [CrossRef]
- Dhoke, N.R.; Kim, H.; Selvaraj, S.; Azzag, K.; Zhou, H.; Oliveira, N.A.J.; Tungtur, S.; Ortiz-Cordero, C.; Kiley, J.; Lu, Q.L.; et al. A universal gene correction approach for FKRP-associated dystroglycanopathies to enable autologous cell therapy. Cell Rep. 2021, 36, 109360. [Google Scholar] [CrossRef]
- Ghosh, R.; Gilda, J.E.; Gomes, A.V. The necessity of and strategies for improving confidence in the accuracy of western blots. Expert Rev. Proteom. 2014, 11, 549–560. [Google Scholar] [CrossRef]
- Ruiz-Del-Yerro, E.; Garcia-Jimenez, I.; Mamchaoui, K.; Arechavala-Gomeza, V. Myoblots: Dystrophin quantification by in-cell western assay for a streamlined development of Duchenne muscular dystrophy (DMD) treatments. Neuropathol. Appl. Neurobiol. 2018, 44, 463–473. [Google Scholar] [CrossRef]
- Soblechero-Martín, P.; Albiasu-Arteta, E.; Anton-Martinez, A.; de la Puente-Ovejero, L.; Garcia-Jimenez, I.; González-Iglesias, G.; Larrañaga-Aiestaran, I.; López-Martínez, A.; Poyatos-García, J.; Ruiz-Del-Yerro, E.; et al. Duchenne muscular dystrophy cell culture models created by CRISPR/Cas9 gene editing and their application in drug screening. Sci. Rep. 2021, 11, 18188. [Google Scholar] [CrossRef]
- (FDA), U.F.a.D.A. Q2(R1) Validation of Analytical Procedures: Text and Methodology Guidance for Industry. 2021. Available online: https://www.fda.gov/media/152208/download (accessed on 10 October 2023).
- Tabebordbar, M.; Lagerborg, K.A.; Stanton, A.; King, E.M.; Ye, S.; Tellez, L.; Krunnfusz, A.; Tavakoli, S.; Widrick, J.J.; Messemer, K.A.; et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 2021, 184, 4919–4938.e22. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, M.P.; Lu, P.; Blaeser, A.; Lu, Q.L. Ribitol restores functionally glycosylated α-dystroglycan and improves muscle function in dystrophic FKRP-mutant mice. Nat. Commun. 2018, 9, 3448. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Drains, M.; Shah, S.N.; Lu, P.J.; Leroy, V.; Killilee, J.; Rawls, R.; Tucker, J.D.; Blaeser, A.; Lu, Q.L. Ribitol dose-dependently enhances matriglycan expression and improves muscle function with prolonged life span in limb girdle muscular dystrophy 2I mouse model. PLoS ONE 2022, 17, e0278482. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geoffroy, M.; Pili, L.; Buffa, V.; Caroff, M.; Bigot, A.; Gicquel, E.; Rouby, G.; Richard, I.; Fragnoud, R. CRISPR-Cas9 KO Cell Line Generation and Development of a Cell-Based Potency Assay for rAAV-FKRP Gene Therapy. Cells 2023, 12, 2444. https://doi.org/10.3390/cells12202444
Geoffroy M, Pili L, Buffa V, Caroff M, Bigot A, Gicquel E, Rouby G, Richard I, Fragnoud R. CRISPR-Cas9 KO Cell Line Generation and Development of a Cell-Based Potency Assay for rAAV-FKRP Gene Therapy. Cells. 2023; 12(20):2444. https://doi.org/10.3390/cells12202444
Chicago/Turabian StyleGeoffroy, Marine, Louna Pili, Valentina Buffa, Maëlle Caroff, Anne Bigot, Evelyne Gicquel, Grégory Rouby, Isabelle Richard, and Romain Fragnoud. 2023. "CRISPR-Cas9 KO Cell Line Generation and Development of a Cell-Based Potency Assay for rAAV-FKRP Gene Therapy" Cells 12, no. 20: 2444. https://doi.org/10.3390/cells12202444
APA StyleGeoffroy, M., Pili, L., Buffa, V., Caroff, M., Bigot, A., Gicquel, E., Rouby, G., Richard, I., & Fragnoud, R. (2023). CRISPR-Cas9 KO Cell Line Generation and Development of a Cell-Based Potency Assay for rAAV-FKRP Gene Therapy. Cells, 12(20), 2444. https://doi.org/10.3390/cells12202444