

Opposing Roles for the α Isoform of the Catalytic Subunit of Protein Phosphatase 1 in Inside–Out and Outside–In Integrin Signaling in Murine Platelets
 , and
, and
Abstract
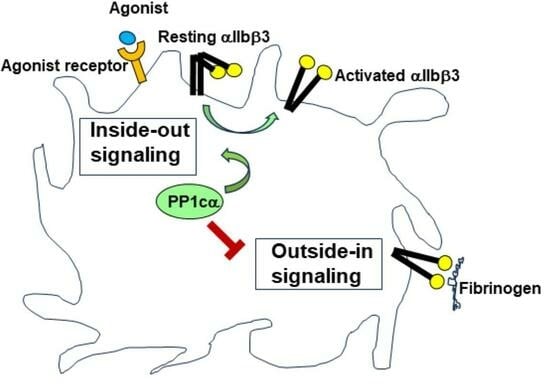
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Mice
2.3. Mice Platelet Preparation
2.4. Flow Cytometry Studies for Integrin Activation, Binding of Soluble Fibrinogen, Granule Secretion, and Platelet Aggregation
2.5. p38 MAPK Immunoblotting Studies
2.6. Platelet Adhesion, Clot Retraction and In Vitro Thrombus Formation
2.7. Intra Vital Microscopy and Vivo Platelet Thrombus Formation
2.8. Hemostasis Studies
2.9. Statistics
3. Results
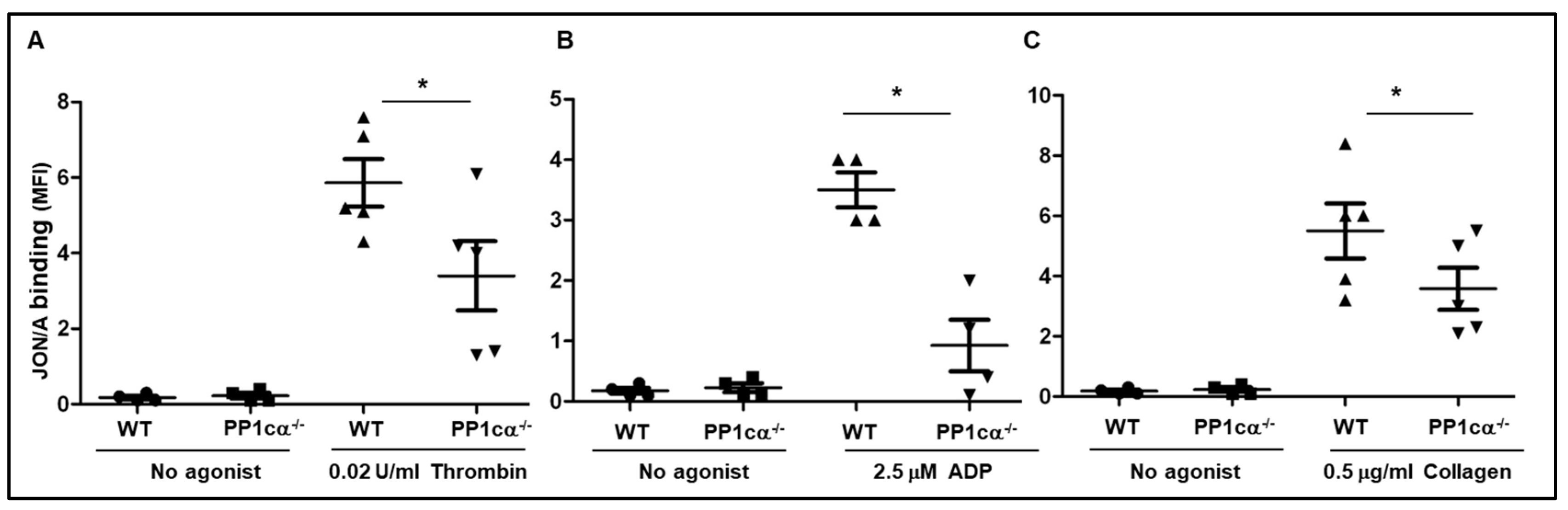
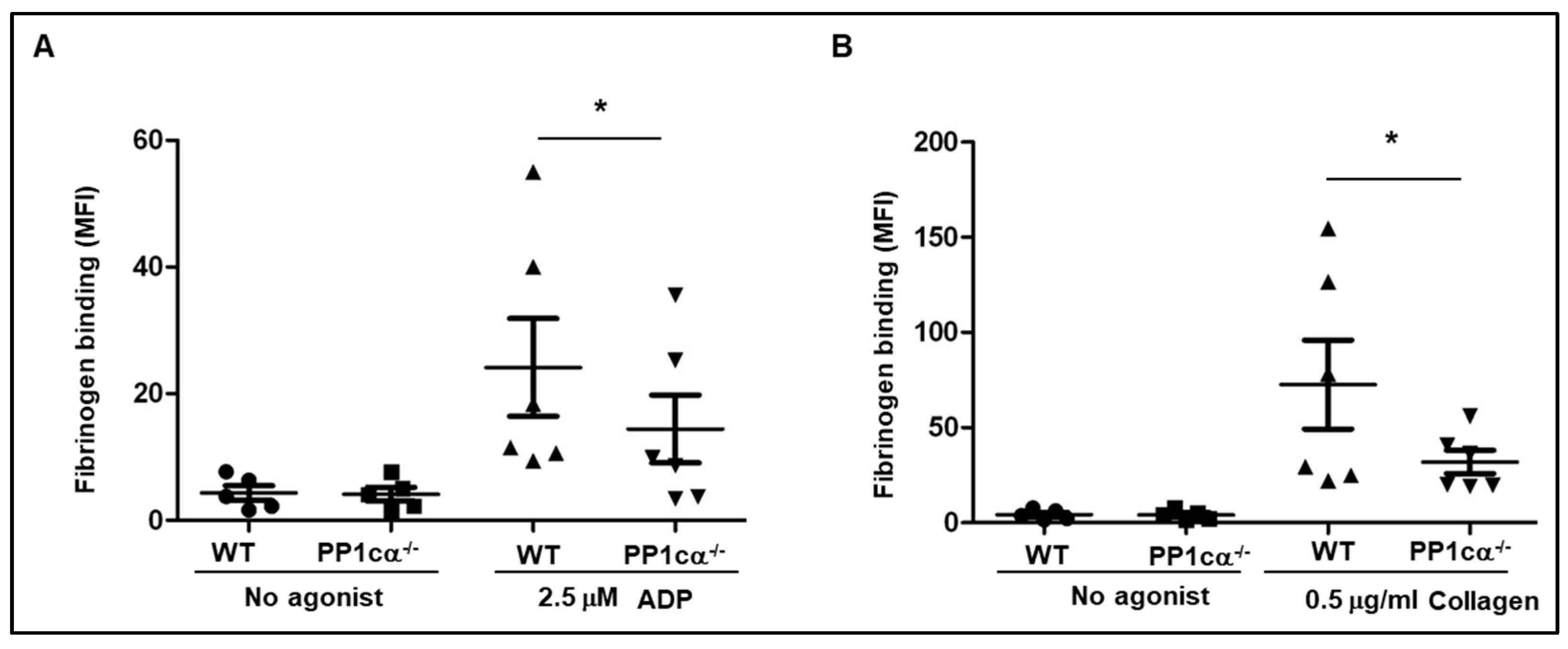
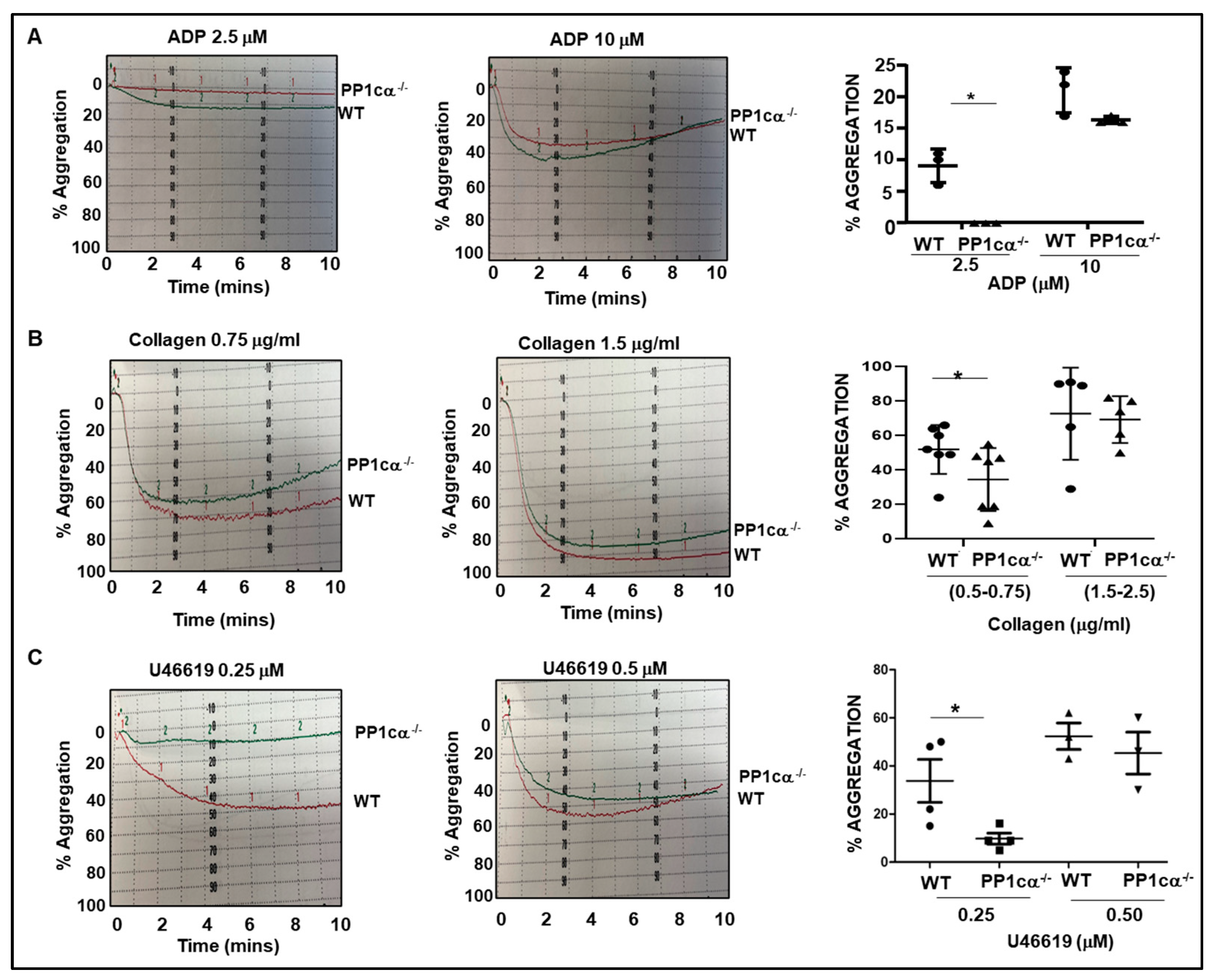
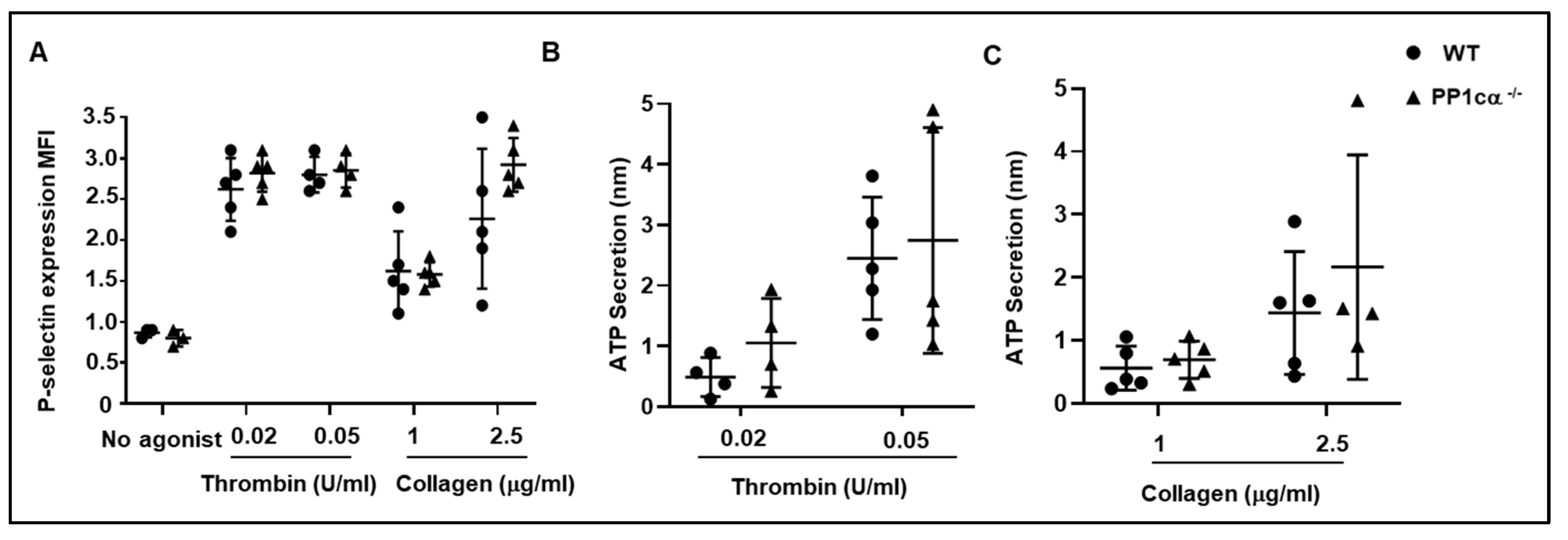
3.1. Loss of PP1cα Moderately Reduced Low Dose Agonist-Induced Integrin Signaling
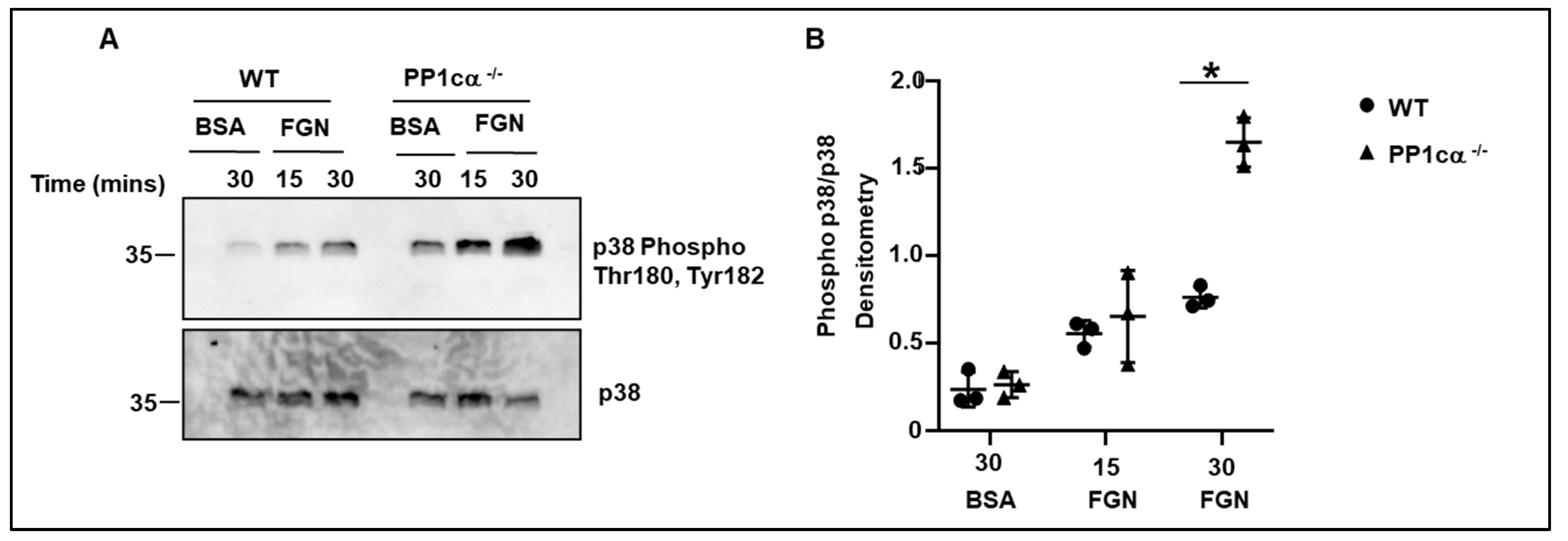
3.2. Loss of PP1cα Enhanced Activation of p38 MAPK and Integrin αIIbβ3 Outside–In Signaling
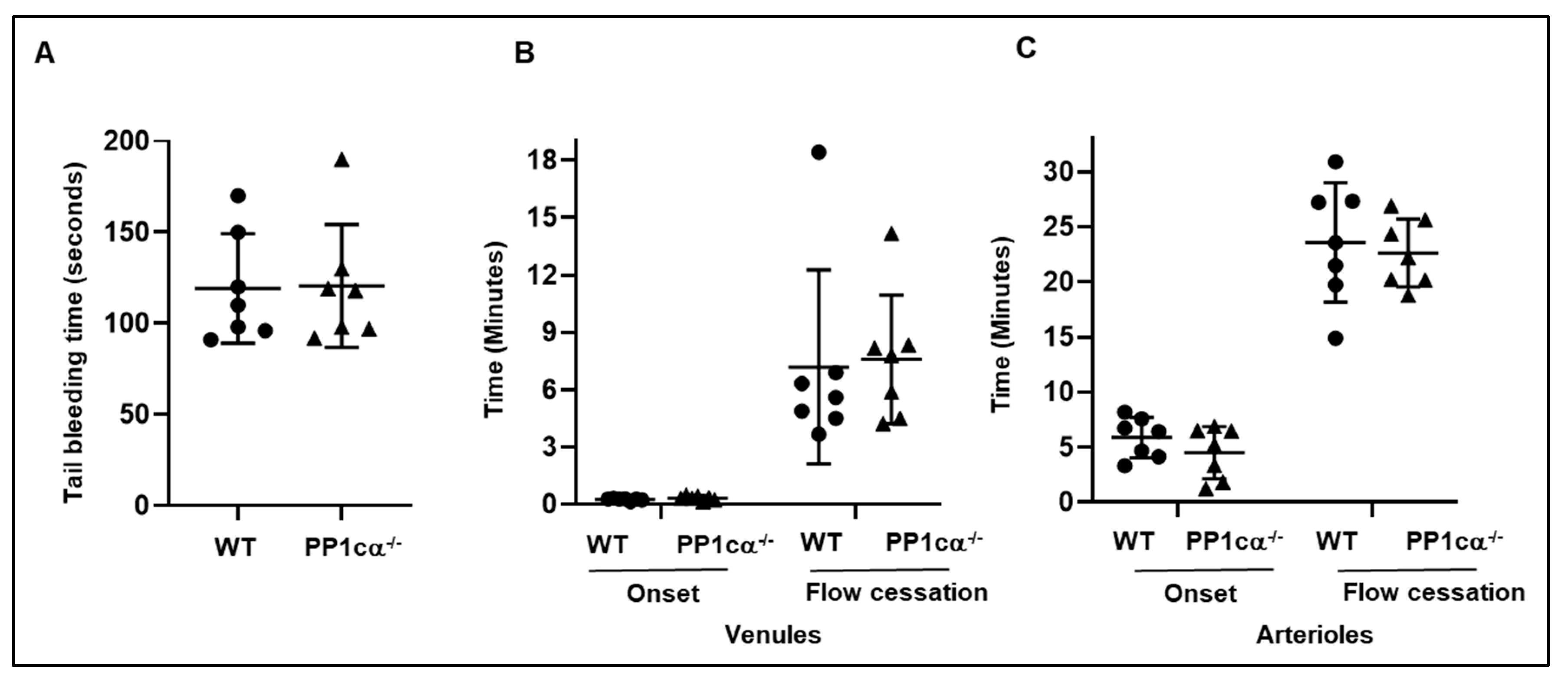
3.3. Loss of PP1cα Does Not Impact Secretion and In Vivo Hemostasis and Thrombosis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Estevez, B.; Du, X. New Concepts and Mechanisms of Platelet Activation Signaling. Physiology 2017, 32, 162–177. [Google Scholar] [CrossRef] [PubMed]
- Shattil, S.J.; Kashiwagi, H.; Pampori, N. Integrin signaling: The platelet paradigm. Blood 1998, 91, 2645–2657. [Google Scholar] [CrossRef]
- Shattil, S.J. Signaling through platelet integrin aIIbb3: Inside-out, outside-in, and sideways. Thromb. Haemost. 1999, 82, 318–325. [Google Scholar] [PubMed]
- Shiravand, Y.; Walter, U.; Jurk, K. Fine-Tuning of Platelet Responses by Serine/Threonine Protein Kinases and Phosphatases-Just the Beginning. Hamostaseologie 2021, 41, 206–216. [Google Scholar] [CrossRef]
- Beck, F.; Geiger, J.; Gambaryan, S.; Solari, F.A.; Dell’Aica, M.; Loroch, S.; Mattheij, N.J.; Mindukshev, I.; Pötz, O.; Jurk, K.; et al. Temporal quantitative phosphoproteomics of ADP stimulation reveals novel central nodes in platelet activation and inhibition. Blood 2017, 129, e1–e12. [Google Scholar] [CrossRef]
- Qureshi, A.H.; Chaoji, V.; Maiguel, D.; Faridi, M.H.; Barth, C.J.; Salem, S.M.; Singhal, M.; Stoub, D.; Krastins, B.; Ogihara, M.; et al. Proteomic and phospho-proteomic profile of human platelets in basal, resting state: Insights into integrin signaling. PLoS ONE 2009, 4, e7627. [Google Scholar] [CrossRef]
- Cohen, P.T. Protein phosphatase 1—Targeted in many directions. J. Cell Sci. 2002, 115, 241–256. [Google Scholar] [CrossRef]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73–e82. [Google Scholar] [CrossRef]
- Higashihara, M.; Takahata, K.; Kurokawa, K.; Ikebe, M. The inhibitory effects of okadaic acid on platelet function. FEBS Lett. 1992, 307, 206–210. [Google Scholar] [CrossRef]
- Nishikawa, M.; Toyoda, H.; Saito, M.; Morita, K.; Tawara, I.; Deguchi, K.; Kuno, T.; Shima, H.; Nagao, M.; Shirakawa, S. Calyculin A and okadiac acid inhibit human platelet aggregation by blocking protein phosphatases types 1 and 2A. Cell Signal 1994, 6, 59–71. [Google Scholar] [CrossRef]
- Hoyt, C.H.; Lerea, K.M. Aggregation-dependent signaling in human platelets is sensitive to protein serine/threonine phosphatase inhibitors. Biochemistry 1995, 34, 9565–9570. [Google Scholar] [CrossRef] [PubMed]
- Simon, Z.; Kiss, A.; Erdödi, F.; Setiadi, H.; Beke Debreceni, I.; Nagy, B., Jr.; Kappelmayer, J. Protein phosphatase inhibitor calyculin-A modulates activation markers in TRAP-stimulated human platelets. Platelets 2010, 21, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Lerea, K.M.; Cordero, K.P.; Sakariassen, K.S.; Kirk, R.I.; Fried, V.A. Phosphorylation sites in the integrin b3 cytoplasmic domain in intact platelets. J. Biol. Chem. 1999, 274, 1914–1919. [Google Scholar] [CrossRef] [PubMed]
- Moscardó, A.; Santos, M.T.; Latorre, A.; Madrid, I.; Vallés, J. Serine/threonine phosphatases regulate platelet αIIbβ3 integrin receptor outside-in signaling mechanisms and clot retraction. Life Sci. 2013, 93, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Hudák, R.; Vincze, J.; Csernoch, L.; Beke Debreceni, I.; Oláh, T.; Erdődi, F.; Clemetson, K.J.; Kappelmayer, J. The Phosphatase Inhibitor Calyculin-A Impairs Clot Retraction, Platelet Activation, and Thrombin Generation. BioMed Res. Int. 2017, 2017, 9795271. [Google Scholar] [CrossRef]
- Pradhan, S.; Khatlani, T.; Nairn, A.C.; Vijayan, K.V. The heterotrimeric G protein Gβ1 interacts with the catalytic subunit of protein phosphatase 1 and modulates G protein-coupled receptor signaling in platelets. J. Biol. Chem. 2017, 292, 13133–13142. [Google Scholar] [CrossRef]
- Liu, R.; Correll, R.N.; Davis, J.; Vagnozzi, R.J.; York, A.J.; Sargent, M.A.; Nairn, A.C.; Molkentin, J.D. Cardiac-specific deletion of protein phosphatase 1β promotes increased myofilament protein phosphorylation and contractile alterations. J. Mol. Cell Cardiol. 2015, 87, 204–213. [Google Scholar] [CrossRef]
- Tiedt, R.; Schomber, T.; Hao-Shen, H.; Skoda, R.C. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood 2007, 109, 1503–1506. [Google Scholar] [CrossRef]
- Gushiken, F.C.; Hyojeong, H.; Pradhan, S.; Langlois, K.W.; Alrehani, N.; Cruz, M.A.; Rumbaut, R.E.; Vijayan, K.V. The catalytic subunit of protein phosphatase 1 gamma regulates thrombin-induced murine platelet alphaIIbbeta3 function. PLoS ONE 2009, 4, e8304. [Google Scholar] [CrossRef]
- Khatlani, T.; Pradhan, S.; Da, Q.; Gushiken, F.C.; Bergeron, A.L.; Langlois, K.W.; Molkentin, J.D.; Rumbaut, R.E.; Vijayan, K.V. The β isoform of the catalytic subunit of protein phosphatase 2B restrains platelet function by suppressing outside-in αII b β3 integrin signaling. J. Thromb. Haemost. 2014, 12, 2089–2101. [Google Scholar] [CrossRef]
- Patel, K.N.; Soubra, S.H.; Bellera, R.V.; Dong, J.F.; McMullen, C.A.; Burns, A.R.; Rumbaut, R.E. Differential role of von Willebrand factor and P-selectin on microvascular thrombosis in endotoxemia. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2225–2230. [Google Scholar] [CrossRef] [PubMed]
- Rumbaut, R.E.; Randhawa, J.K.; Smith, C.W.; Burns, A.R. Mouse cremaster venules are predisposed to light/dye-induced thrombosis independent of wall shear rate, CD18, ICAM-1, or P-selectin. Microcirculation 2004, 11, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Alrehani, N.; Pradhan, S.; Khatlani, T.; Kailasam, L.; Vijayan, K.V. Distinct roles for the α, β and γ1 isoforms of protein phosphatase 1 in the outside-in αIIbβ3 integrin signalling-dependent functions. Thromb. Haemost. 2013, 109, 118–126. [Google Scholar] [PubMed]
- McCluskey, A.; Sim, A.T.; Sakoff, J.A. Serine-threonine protein phosphatase inhibitors: Development of potential therapeutic strategies. J. Med. Chem. 2002, 45, 1151–1175. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, K.V.; Liu, Y.; Li, T.T.; Bray, P.F. Protein phosphatase 1 associates with the integrin alphaIIb subunit and regulates signaling. J. Biol. Chem. 2004, 279, 33039–33042. [Google Scholar] [CrossRef]
- Eto, M.; Kirkbride, J.; Elliott, E.; Lo, S.H.; Brautigan, D.L. Association of the tensin N-terminal protein-tyrosine phosphatase domain with the alpha isoform of protein phosphatase-1 in focal adhesions. J. Biol. Chem. 2007, 282, 17806–17815. [Google Scholar] [CrossRef]
- Flevaris, P.; Li, Z.; Zhang, G.; Zheng, Y.; Liu, J.; Du, X. Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood 2009, 113, 893–901. [Google Scholar] [CrossRef]
- McCleverty, C.J.; Lin, D.C.; Liddington, R.C. Structure of the PTB domain of tensin1 and a model for its recruitment to fibrillar adhesions. Protein Sci. 2007, 16, 1223–1229. [Google Scholar] [CrossRef]
- Hall, E.H.; Daugherty, A.E.; Choi, C.K.; Horwitz, A.F.; Brautigan, D.L. Tensin1 requires protein phosphatase-1alpha in addition to RhoGAP DLC-1 to control cell polarization, migration, and invasion. J. Biol. Chem. 2009, 284, 34713–34722. [Google Scholar] [CrossRef] [PubMed]
- Verbinnen, I.; Ferreira, M.; Bollen, M. Biogenesis and activity regulation of protein phosphatase 1. Biochem. Soc. Trans. 2017, 45, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Taylor, M.L.; Gutschner, T.; Pradeep, S.; Cho, M.S.; Sheng, J.; Lyons, Y.M.; Nagaraja, A.S.; Dood, R.L.; Wen, Y.; et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat. Commun. 2017, 8, 310. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; Rowley, J.W.; Campbell, R.A.; Grissom, C.K.; Brown, S.M.; Beesley, S.J.; Schwertz, H.; Kosaka, Y.; Manne, B.K.; Krauel, K.; et al. Sepsis alters the transcriptional and translational landscape of human and murine platelets. Blood 2019, 134, 911–923. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khatlani, T.; Pradhan, S.; Langlois, K.; Subramanyam, D.; Rumbaut, R.E.; Vijayan, K.V. Opposing Roles for the α Isoform of the Catalytic Subunit of Protein Phosphatase 1 in Inside–Out and Outside–In Integrin Signaling in Murine Platelets. Cells 2023, 12, 2424. https://doi.org/10.3390/cells12202424
Khatlani T, Pradhan S, Langlois K, Subramanyam D, Rumbaut RE, Vijayan KV. Opposing Roles for the α Isoform of the Catalytic Subunit of Protein Phosphatase 1 in Inside–Out and Outside–In Integrin Signaling in Murine Platelets. Cells. 2023; 12(20):2424. https://doi.org/10.3390/cells12202424
Chicago/Turabian StyleKhatlani, Tanvir, Subhashree Pradhan, Kimberly Langlois, Deepika Subramanyam, Rolando E. Rumbaut, and K. Vinod Vijayan. 2023. "Opposing Roles for the α Isoform of the Catalytic Subunit of Protein Phosphatase 1 in Inside–Out and Outside–In Integrin Signaling in Murine Platelets" Cells 12, no. 20: 2424. https://doi.org/10.3390/cells12202424
APA StyleKhatlani, T., Pradhan, S., Langlois, K., Subramanyam, D., Rumbaut, R. E., & Vijayan, K. V. (2023). Opposing Roles for the α Isoform of the Catalytic Subunit of Protein Phosphatase 1 in Inside–Out and Outside–In Integrin Signaling in Murine Platelets. Cells, 12(20), 2424. https://doi.org/10.3390/cells12202424