
PTPN1 Deficiency Modulates BMPR2 Signaling and Induces Endothelial Dysfunction in Pulmonary Arterial Hypertension

 ,
, 
 , , , , and
, , , , and 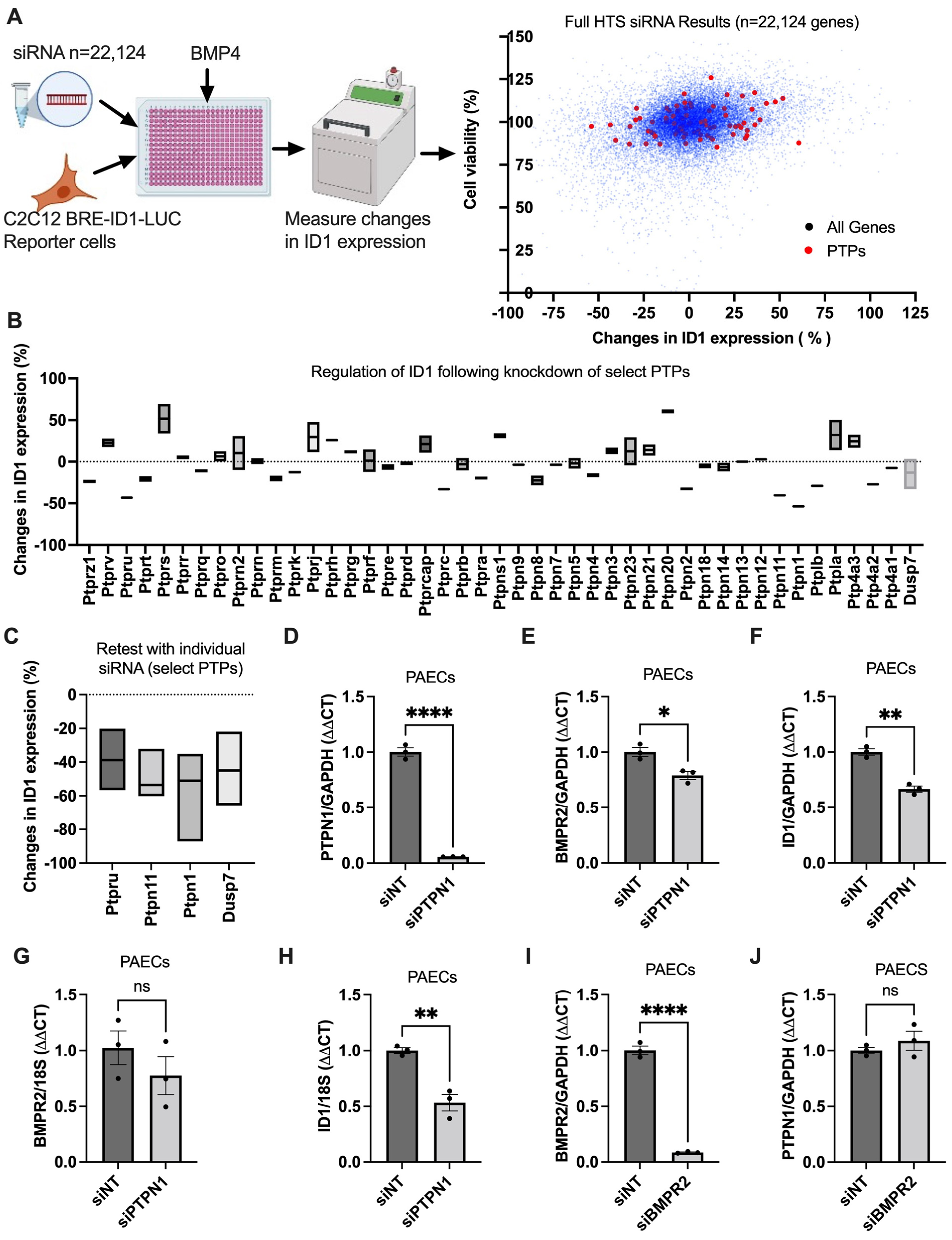{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. High-Throughput siRNA Screen (HTS)
2.2. Cell Culture
2.3. RNAi
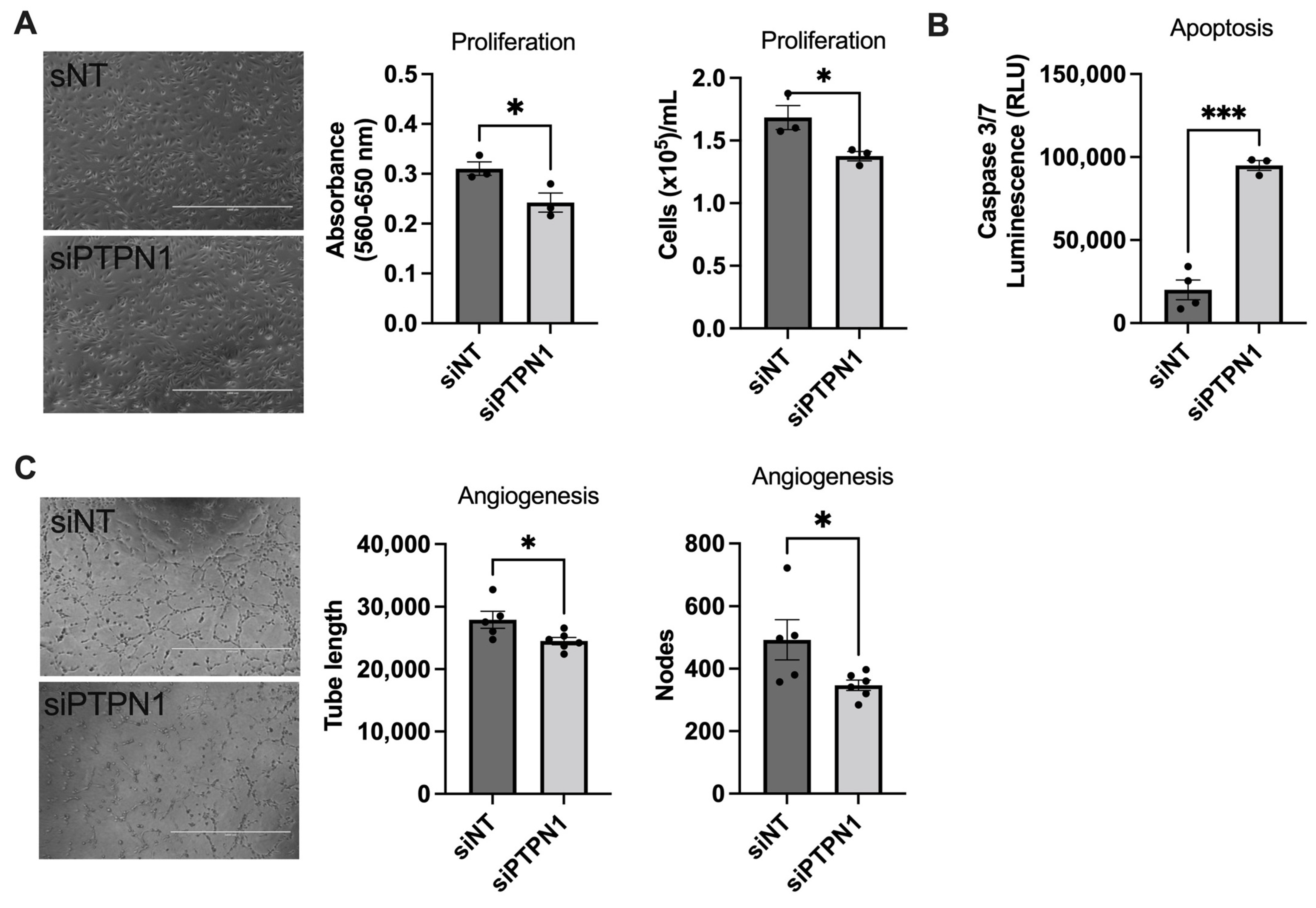
2.4. Cell Proliferation, Apoptosis, and Angiogenesis
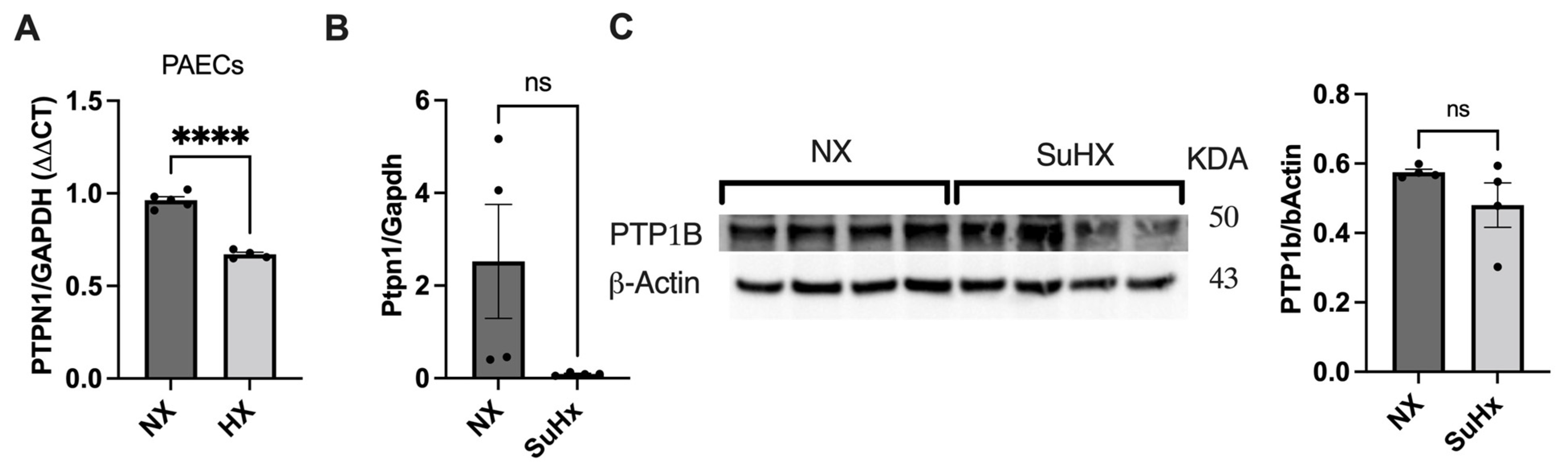
2.5. Hypoxia Induction
2.6. Sugen5416/Hypoxia-Induced PH Rat Models
2.7. Gene Expression Quantification
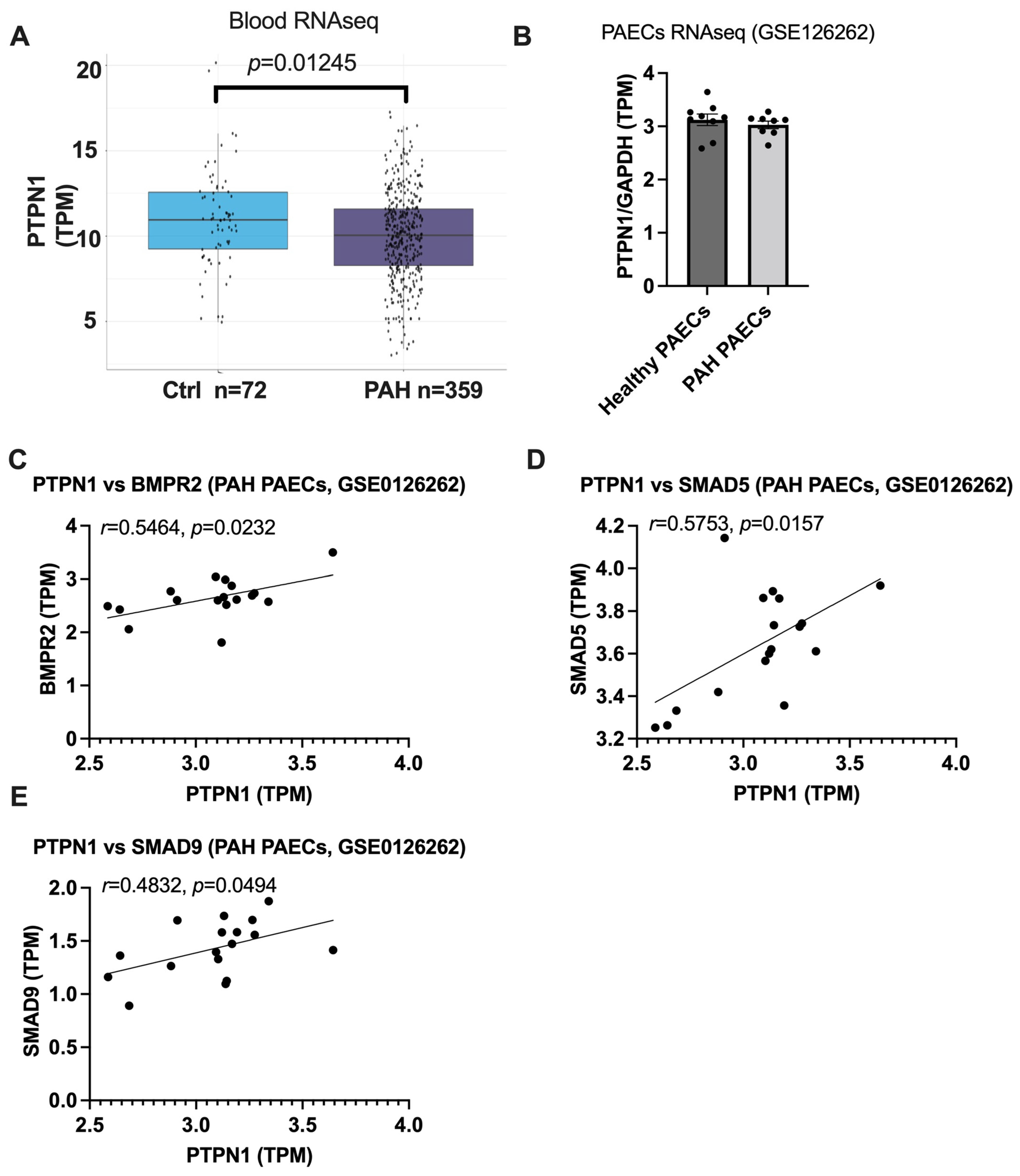
2.8. RNA-Seq Analysis of Whole Blood and PAECs from PAH Patients and Healthy Controls
2.9. Western Blotting
2.10. Statistical Analysis
3. Results
3.1. PTPN1, a Novel BMPR2 Signaling Modifier
3.2. PTPN1 Inhibition Induces Endothelial Dysfunction in hPAECs In Vitro
3.3. PTPN1 Is Downregulated in Hypoxic hPAECs and in the Lung of the Sugen5416/Hypoxia/Normoxia Rat Model
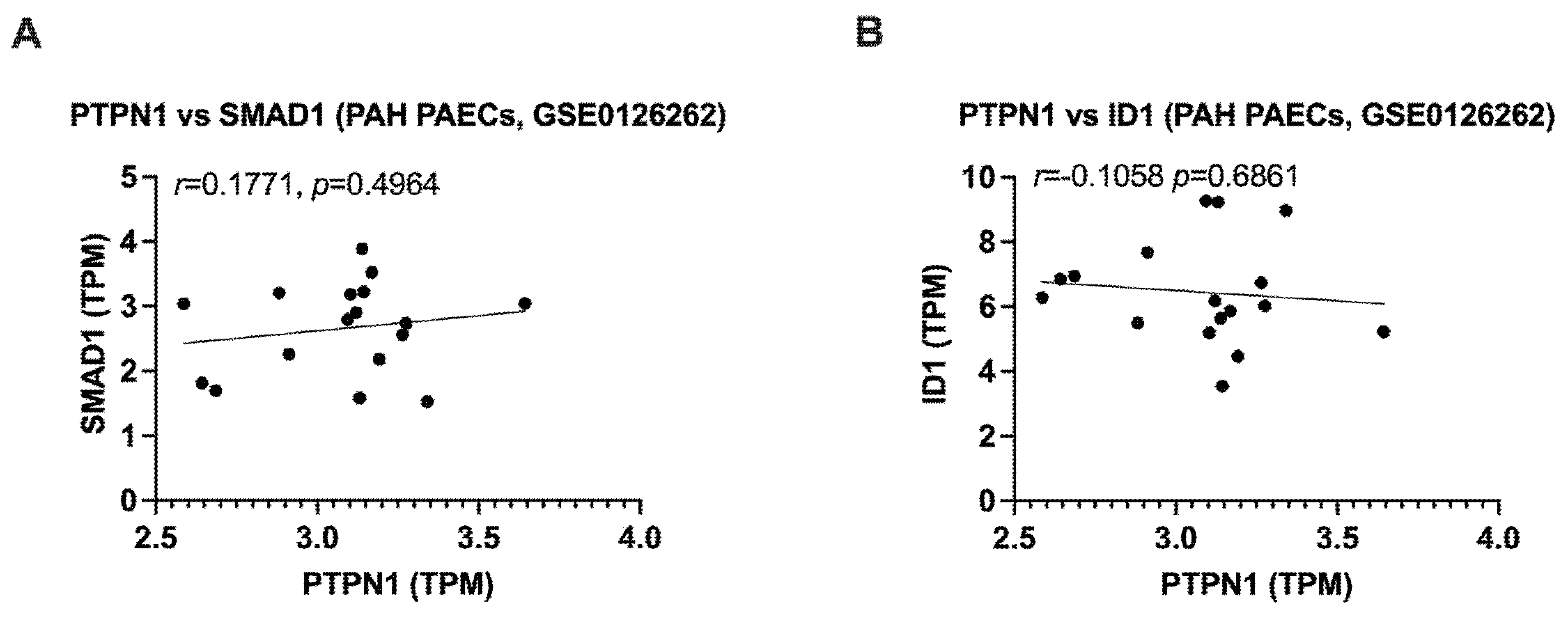
3.4. PTPN1 Is Downregulated in the Blood of PAH Patients and the Expression of PTPN1 Is Correlated with BMPR2 Signaling in PAECs of Healthy and PAH Patients
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ali, M.K.; Ichimura, K.; Spiekerkoetter, E. Promising therapeutic approaches in pulmonary arterial hypertension. Curr. Opin. Pharmacol. 2021, 59, 127–139. [Google Scholar] [CrossRef]
- Dannewitz Prosseda, S.; Ali, M.K.; Spiekerkoetter, E. Novel Advances in Modifying BMPR2 Signaling in PAH. Genes 2020, 12, 8. [Google Scholar] [CrossRef]
- Hong, K.H.; Lee, Y.J.; Lee, E.; Park, S.O.; Han, C.; Beppu, H.; Li, E.; Raizada, M.K.; Bloch, K.D.; Oh, S.P. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation 2008, 118, 722–730. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Fagan, K.; Steudel, W.; Fouty, B.; Lane, K.; Harral, J.; Hoedt-Miller, M.; Tada, Y.; Ozimek, J.; Tuder, R.; et al. Pulmonary hypertension in transgenic mice expressing a dominant-negative BMPRII gene in smooth muscle. Circ. Res. 2004, 94, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- West, J.; Harral, J.; Lane, K.; Deng, Y.; Ickes, B.; Crona, D.; Albu, S.; Stewart, D.; Fagan, K. Mice expressing BMPR2R899X transgene in smooth muscle develop pulmonary vascular lesions. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L744–L755. [Google Scholar] [CrossRef]
- Hautefort, A.; Mendes-Ferreira, P.; Sabourin, J.; Manaud, G.; Bertero, T.; Rucker-Martin, C.; Riou, M.; Adao, R.; Manoury, B.; Lambert, M.; et al. Bmpr2 Mutant Rats Develop Pulmonary and Cardiac Characteristics of Pulmonary Arterial Hypertension. Circulation 2019, 139, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Tian, X.; Cai, J.; Hopper, R.K.; Sudheendra, D.; Li, C.G.; El-Bizri, N.; Sawada, H.; Haghighat, R.; Chan, R.; et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Investig. 2013, 123, 3600–3613. [Google Scholar] [CrossRef]
- Dannewitz Prosseda, S.; Tian, X.; Kuramoto, K.; Boehm, M.; Sudheendra, D.; Miyagawa, K.; Zhang, F.; Solow-Cordero, D.; Saldivar, J.C.; Austin, E.D.; et al. FHIT, a Novel Modifier Gene in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2019, 199, 83–98. [Google Scholar] [CrossRef]
- Andruska, A.M.; Ali, M.K.; Tian, X.; Spiekerkoetter, E. Selective Src-Family B Kinase Inhibition Promotes Pulmonary Artery Endothelial Cell Dysfunction. bioRxiv 2021. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Wang, Y.; Pandey, R.N.; York, A.J.; Mallela, J.; Nichols, W.C.; Hu, Y.C.; Molkentin, J.D.; Wikenheiser-Brokamp, K.A.; Hegde, R.S. The EYA3 tyrosine phosphatase activity promotes pulmonary vascular remodeling in pulmonary arterial hypertension. Nat. Commun. 2019, 10, 4143. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Jiang, W.; Du, H.; Shao, J.; Lu, B.; Wang, J.; Zou, D. Protein tyrosine phosphatase 1B gene polymorphisms and essential hypertension: A case-control study in Chinese population. J. Endocrinol. Investig. 2010, 33, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Pu, Q.; Chang, Y.; Zhang, C.; Cai, Y.; Hassid, A. Chronic insulin treatment suppresses PTP1B function, induces increased PDGF signaling, and amplifies neointima formation in the balloon-injured rat artery. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H132–H139. [Google Scholar] [CrossRef] [PubMed]
- Belin de Chantemele, E.J.; Muta, K.; Mintz, J.; Tremblay, M.L.; Marrero, M.B.; Fulton, D.J.; Stepp, D.W. Protein tyrosine phosphatase 1B, a major regulator of leptin-mediated control of cardiovascular function. Circulation 2009, 120, 753–763. [Google Scholar] [CrossRef]
- Jager, M.; Hubert, A.; Gogiraju, R.; Bochenek, M.L.; Munzel, T.; Schafer, K. Inducible Knockdown of Endothelial Protein Tyrosine Phosphatase-1B Promotes Neointima Formation in Obese Mice by Enhancing Endothelial Senescence. Antioxid. Redox Signal. 2019, 30, 927–944. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Ceacareanu, B.; Zhuang, D.; Zhang, C.; Pu, Q.; Ceacareanu, A.C.; Hassid, A. Counter-regulatory function of protein tyrosine phosphatase 1B in platelet-derived growth factor- or fibroblast growth factor-induced motility and proliferation of cultured smooth muscle cells and in neointima formation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 501–507. [Google Scholar] [CrossRef]
- Gogiraju, R.; Gachkar, S.; Velmeden, D.; Bochenek, M.L.; Zifkos, K.; Hubert, A.; Munzel, T.; Offermanns, S.; Schafer, K. Protein Tyrosine Phosphatase 1B Deficiency in Vascular Smooth Muscle Cells Promotes Perivascular Fibrosis following Arterial Injury. Thromb. Haemost. 2022, 122, 1814–1826. [Google Scholar] [CrossRef]
- Ten Freyhaus, H.; Dagnell, M.; Leuchs, M.; Vantler, M.; Berghausen, E.M.; Caglayan, E.; Weissmann, N.; Dahal, B.K.; Schermuly, R.T.; Ostman, A.; et al. Hypoxia enhances platelet-derived growth factor signaling in the pulmonary vasculature by down-regulation of protein tyrosine phosphatases. Am. J. Respir. Crit. Care Med. 2011, 183, 1092–1102. [Google Scholar] [CrossRef]
- Berdnikovs, S.; Pavlov, V.I.; Abdala-Valencia, H.; McCary, C.A.; Klumpp, D.J.; Tremblay, M.L.; Cook-Mills, J.M. PTP1B deficiency exacerbates inflammation and accelerates leukocyte trafficking in vivo. J. Immunol. 2012, 188, 874–884. [Google Scholar] [CrossRef]
- Ali, M.K.; Liu, Y.; Zhao, L.; Wu, J.C.; de Jesus Perez, V.; Rhodes, C.J.; Cao, A.; Wilkins, M.R.; Nicolls, M.R.; Spiekerkoetter, E.F. Long non-coding RNA RGMB-AS1 as a novel modulator of Bone Morphogenetic Protein Receptor 2 signaling in pulmonary arterial hypertension. bioRxiv 2022. [Google Scholar] [CrossRef]
- Ali, M.K.; Kim, R.Y.; Brown, A.C.; Mayall, J.R.; Karim, R.; Pinkerton, J.W.; Liu, G.; Martin, K.L.; Starkey, M.R.; Pillar, A.L.; et al. Crucial role for lung iron level and regulation in the pathogenesis and severity of asthma. Eur. Respir. J. 2020, 55, 1901340. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Otero-Nunez, P.; Wharton, J.; Swietlik, E.M.; Kariotis, S.; Harbaum, L.; Dunning, M.J.; Elinoff, J.M.; Errington, N.; Thompson, A.A.R.; et al. Whole-Blood RNA Profiles Associated with Pulmonary Arterial Hypertension and Clinical Outcome. Am. J. Respir. Crit. Care Med. 2020, 202, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Palomares, A.; Gu, M.; Grubert, F.; Berest, I.; Sa, S.; Kasowski, M.; Arnold, C.; Shuai, M.; Srivas, R.; Miao, S.; et al. Remodeling of active endothelial enhancers is associated with aberrant gene-regulatory networks in pulmonary arterial hypertension. Nat. Commun. 2020, 11, 1673. [Google Scholar] [CrossRef]
- Jiang, Z.X.; Zhang, Z.Y. Targeting PTPs with small molecule inhibitors in cancer treatment. Cancer Metastasis Rev. 2008, 27, 263–272. [Google Scholar] [CrossRef]
- Tautz, L.; Critton, D.A.; Grotegut, S. Protein tyrosine phosphatases: Structure, function, and implication in human disease. Methods Mol. Biol. 2013, 1053, 179–221. [Google Scholar] [CrossRef]
- Cheng, Y.; Yu, M.; Xu, J.; He, M.; Wang, H.; Kong, H.; Xie, W. Inhibition of Shp2 ameliorates monocrotaline-induced pulmonary arterial hypertension in rats. BMC Pulm. Med. 2018, 18, 130. [Google Scholar] [CrossRef]
- Xu, J.; Zhong, Y.; Yin, H.; Linneman, J.; Luo, Y.; Xia, S.; Xia, Q.; Yang, L.; Huang, X.; Kang, K.; et al. Methylation-mediated silencing of PTPRD induces pulmonary hypertension by promoting pulmonary arterial smooth muscle cell migration via the PDGFRB/PLCgamma1 axis. J. Hypertens. 2022, 40, 1795–1807. [Google Scholar] [CrossRef]
- Ferguson, B.S.; Wennersten, S.A.; Demos-Davies, K.M.; Rubino, M.; Robinson, E.L.; Cavasin, M.A.; Stratton, M.S.; Kidger, A.M.; Hu, T.; Keyse, S.M.; et al. DUSP5-mediated inhibition of smooth muscle cell proliferation suppresses pulmonary hypertension and right ventricular hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H382–H389. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, H.; Nakanishi, N.; Kyotani, S.; Takashima, A.; Tomoike, H.; Morisaki, T. BMPR2 mutations found in Japanese patients with familial and sporadic primary pulmonary hypertension. Hum. Mutat. 2004, 23, 632. [Google Scholar] [CrossRef]
- Kabata, H.; Satoh, T.; Kataoka, M.; Tamura, Y.; Ono, T.; Yamamoto, M.; Huqun; Hagiwara, K.; Fukuda, K.; Betsuyaku, T.; et al. Bone morphogenetic protein receptor type 2 mutations, clinical phenotypes and outcomes of Japanese patients with sporadic or familial pulmonary hypertension. Respirology 2013, 18, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, C.; Caja, L.; Bertran, E.; Gonzalez-Rodriguez, A.; Valverde, A.M.; Fabregat, I.; Sancho, P. Protein-tyrosine phosphatase 1B (PTP1B) deficiency confers resistance to transforming growth factor-beta (TGF-beta)-induced suppressor effects in hepatocytes. J. Biol. Chem. 2012, 287, 15263–15274. [Google Scholar] [CrossRef]
- Matulka, K.; Lin, H.H.; Hribkova, H.; Uwanogho, D.; Dvorak, P.; Sun, Y.M. PTP1B is an effector of activin signaling and regulates neural specification of embryonic stem cells. Cell Stem Cell 2013, 13, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hsiung, C.A.; Chuang, L.M.; Ho, L.T.; Ting, C.T.; Bustos, V.I.; Lee, T.M.; De Witte, A.; Chen, Y.D.; Olshen, R.; et al. Single nucleotide polymorphisms in protein tyrosine phosphatase 1beta (PTPN1) are associated with essential hypertension and obesity. Hum. Mol. Genet. 2004, 13, 1885–1892. [Google Scholar] [CrossRef] [PubMed]
- Berdnikovs, S.; Abdala-Valencia, H.; Cook-Mills, J.M. Endothelial cell PTP1B regulates leukocyte recruitment during allergic inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L240–L249. [Google Scholar] [CrossRef] [PubMed]
- Foronjy, R.F.; Ochieng, P.O.; Salathe, M.A.; Dabo, A.J.; Eden, E.; Baumlin, N.; Cummins, N.; Barik, S.; Campos, M.; Thorp, E.B.; et al. Protein tyrosine phosphatase 1B negatively regulates S100A9-mediated lung damage during respiratory syncytial virus exacerbations. Mucosal. Immunol. 2016, 9, 1317–1329. [Google Scholar] [CrossRef]
- Song, D.; Adrover, J.M.; Felice, C.; Christensen, L.N.; He, X.Y.; Merrill, J.R.; Wilkinson, J.E.; Janowitz, T.; Lyons, S.K.; Egeblad, M.; et al. PTP1B inhibitors protect against acute lung injury and regulate CXCR4 signaling in neutrophils. JCI Insight 2022, 7, e158199. [Google Scholar] [CrossRef]
- Steffes, L.C.; Froistad, A.A.; Andruska, A.; Boehm, M.; McGlynn, M.; Zhang, F.; Zhang, W.; Hou, D.; Tian, X.; Miquerol, L.; et al. A Notch3-Marked Subpopulation of Vascular Smooth Muscle Cells Is the Cell of Origin for Occlusive Pulmonary Vascular Lesions. Circulation 2020, 142, 1545–1561. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, M.K.; Tian, X.; Zhao, L.; Schimmel, K.; Rhodes, C.J.; Wilkins, M.R.; Nicolls, M.R.; Spiekerkoetter, E.F. PTPN1 Deficiency Modulates BMPR2 Signaling and Induces Endothelial Dysfunction in Pulmonary Arterial Hypertension. Cells 2023, 12, 316. https://doi.org/10.3390/cells12020316
Ali MK, Tian X, Zhao L, Schimmel K, Rhodes CJ, Wilkins MR, Nicolls MR, Spiekerkoetter EF. PTPN1 Deficiency Modulates BMPR2 Signaling and Induces Endothelial Dysfunction in Pulmonary Arterial Hypertension. Cells. 2023; 12(2):316. https://doi.org/10.3390/cells12020316
Chicago/Turabian StyleAli, Md Khadem, Xuefei Tian, Lan Zhao, Katharina Schimmel, Christopher J. Rhodes, Martin R. Wilkins, Mark R. Nicolls, and Edda F. Spiekerkoetter. 2023. "PTPN1 Deficiency Modulates BMPR2 Signaling and Induces Endothelial Dysfunction in Pulmonary Arterial Hypertension" Cells 12, no. 2: 316. https://doi.org/10.3390/cells12020316
APA StyleAli, M. K., Tian, X., Zhao, L., Schimmel, K., Rhodes, C. J., Wilkins, M. R., Nicolls, M. R., & Spiekerkoetter, E. F. (2023). PTPN1 Deficiency Modulates BMPR2 Signaling and Induces Endothelial Dysfunction in Pulmonary Arterial Hypertension. Cells, 12(2), 316. https://doi.org/10.3390/cells12020316