



IRF8 Regulates Intrinsic Ferroptosis through Repressing p53 Expression to Maintain Tumor Cell Sensitivity to Cytotoxic T Lymphocytes

 , and
, and
Abstract
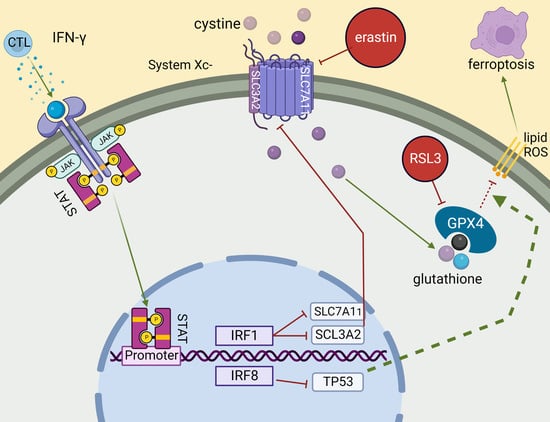
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice and Tumor Cell Line
2.2. Generation of IRF8.KO Tumor Cell Lines
2.3. Generation of p53 KO Tumor Cell Lines
2.4. Histology and Immunohistochemical Analysis
2.5. RNA Sequencing Analysis
2.6. Ferroptosis Induction and Quantification
2.7. Tumor Cell and Tumor-Specific CTL Co-Culture
2.8. Flow Cytometry
2.9. Lipid ROS Analysis
2.10. GSH Measurement
2.11. Cationic Lipid Nanoparticle
2.12. mIRF8-LNP Therapy
2.13. Western Blotting Analysis
2.14. Single-Cell RNA Sequencing Dataset Analysis
2.15. Statistical Analysis
3. Results
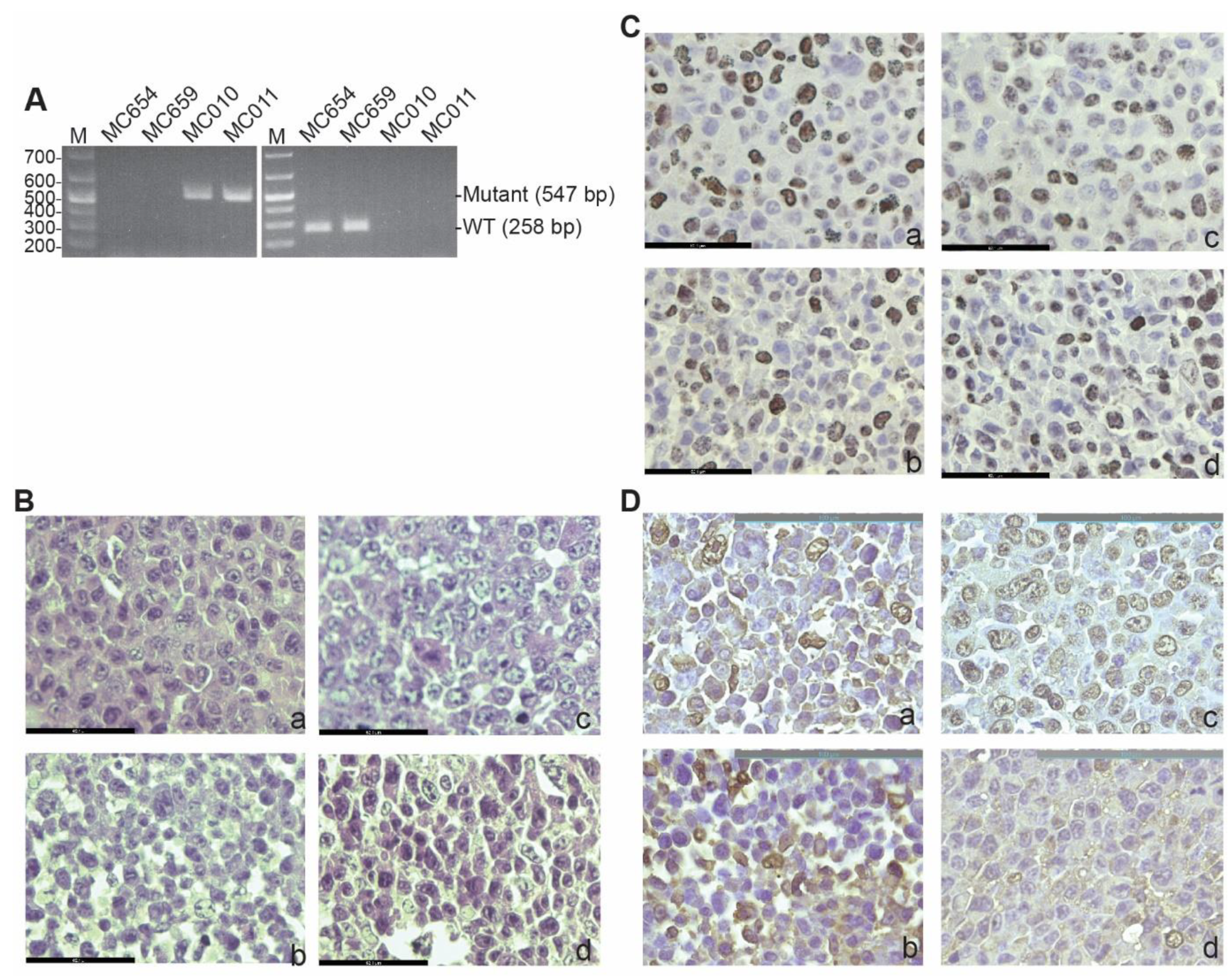
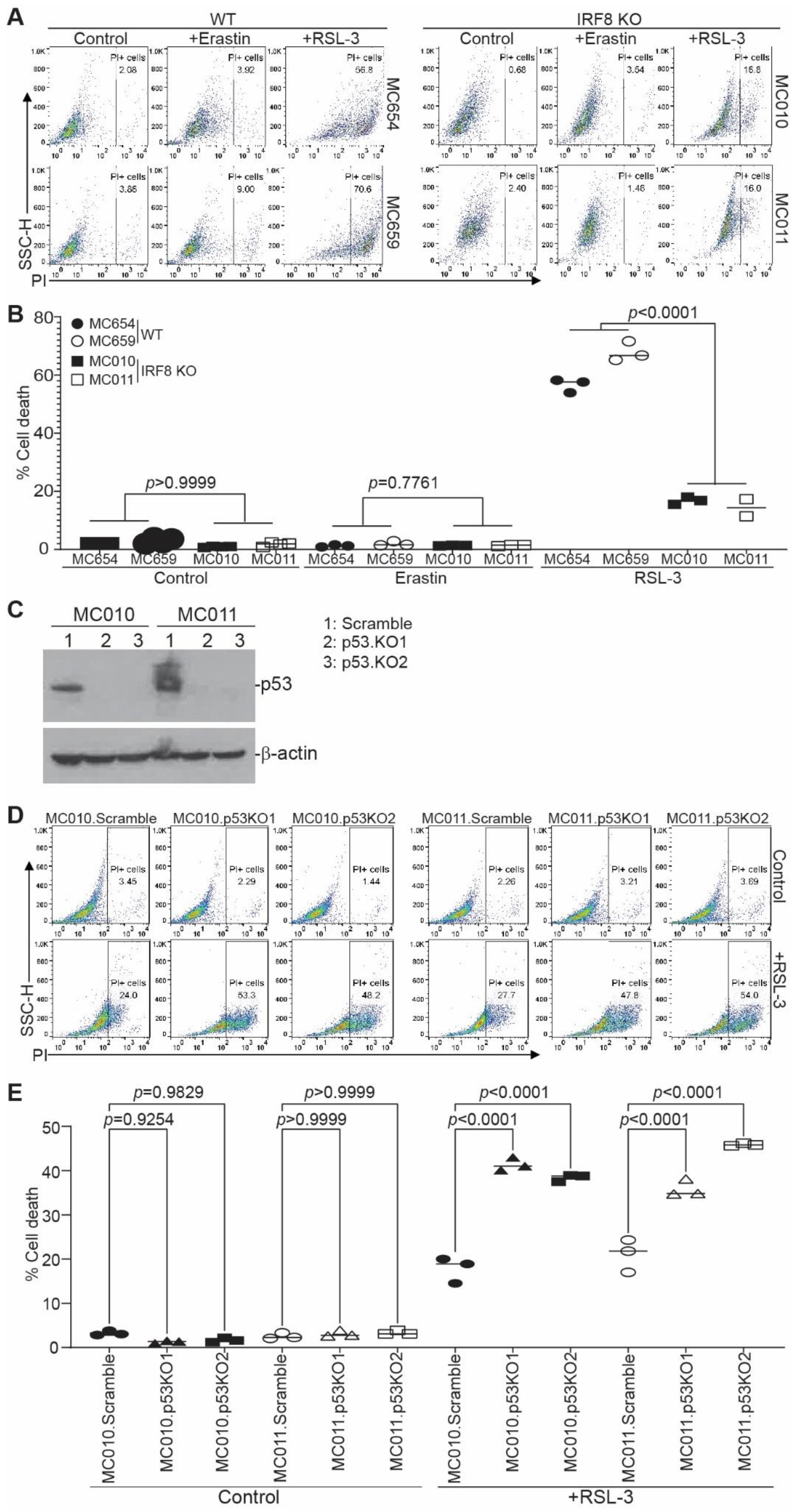
3.1. Generation and Characterization of IRF8 KO Tumor Cell Lines
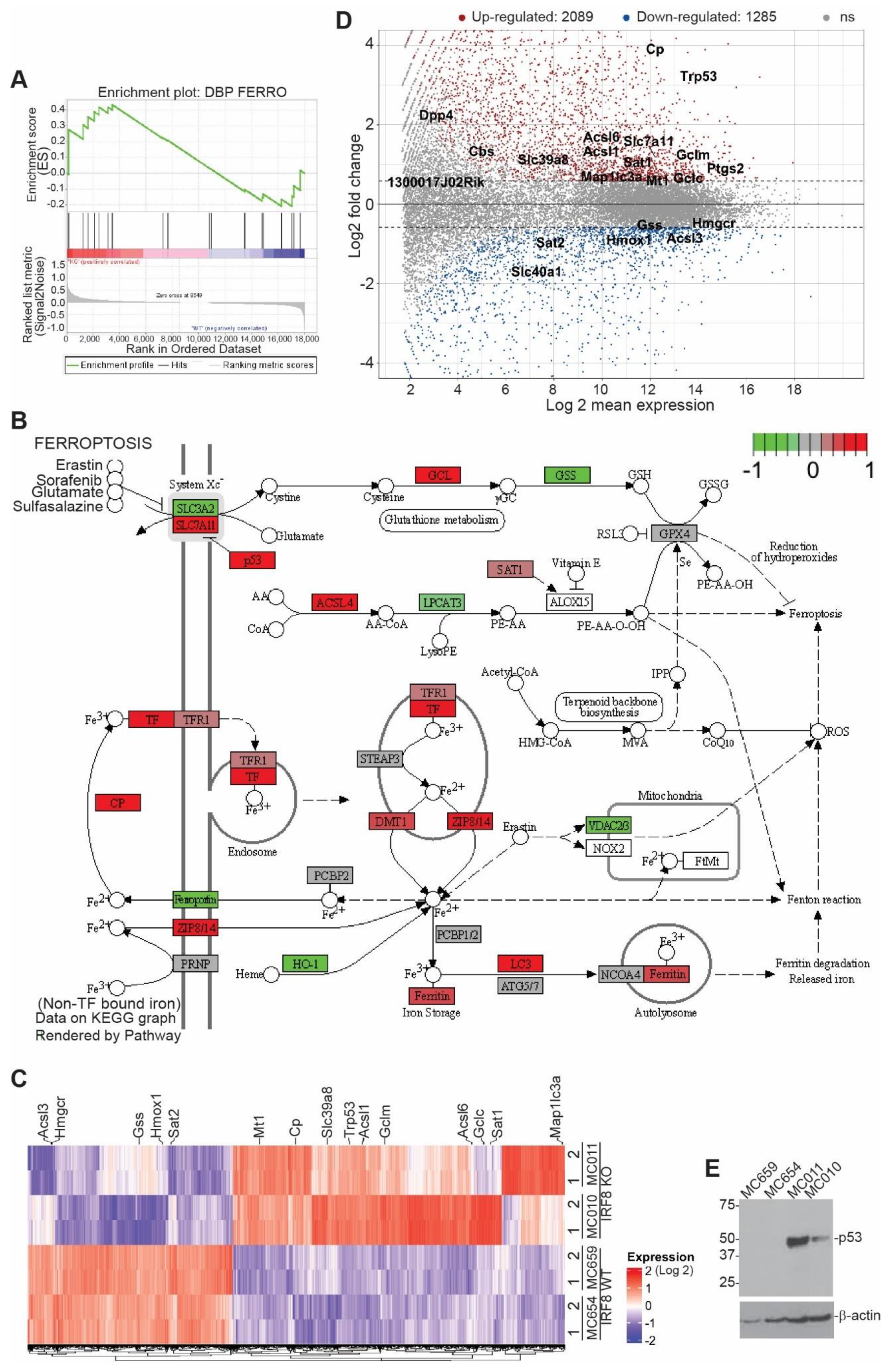
3.2. IRF8 Deficiency Leads to Dysregulated Expression of Genes in the Ferroptosis Pathway
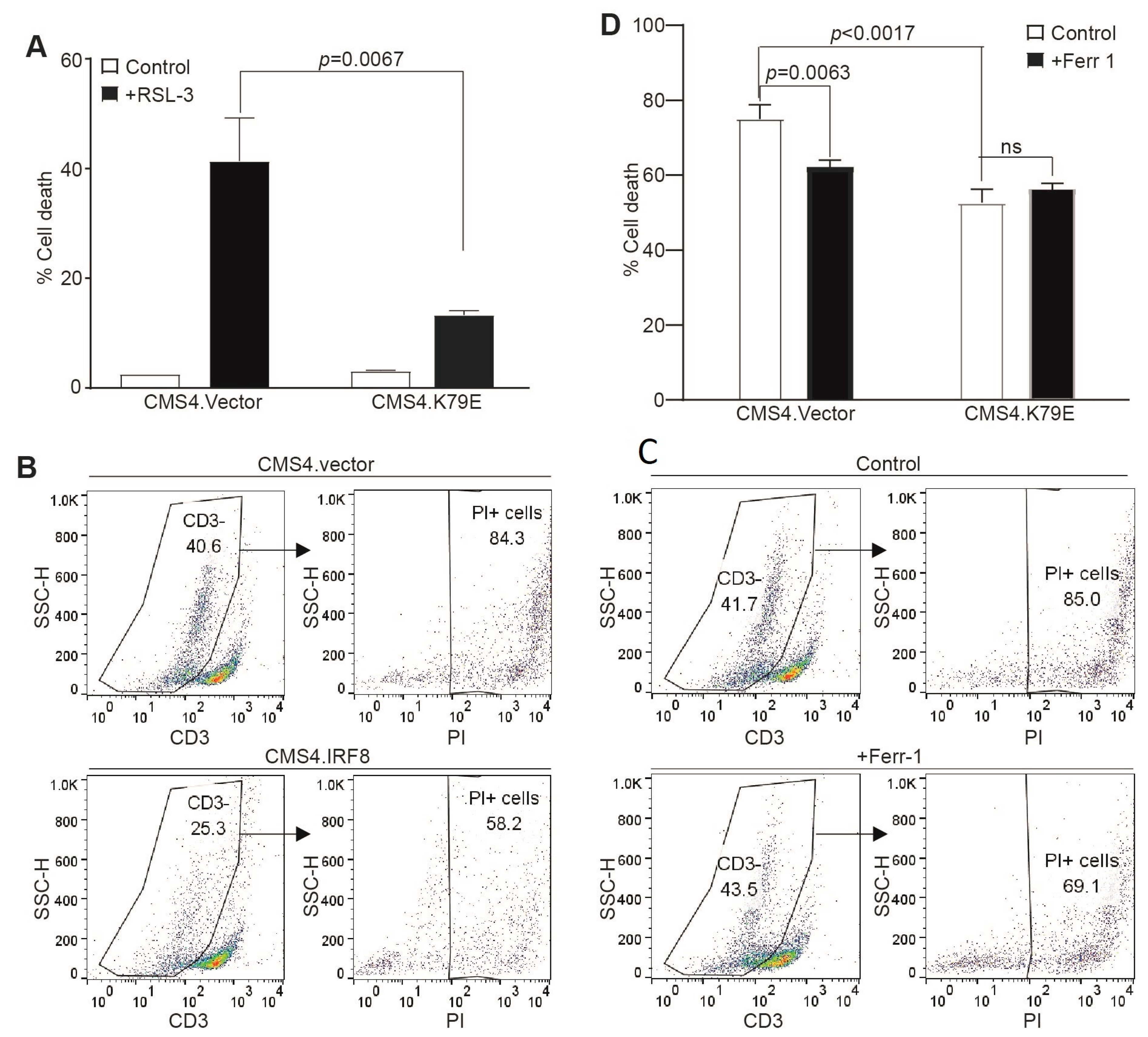
3.3. IRF8 Regulates Intrinsic Ferroptosis through Repressing p53 Expression
3.4. IRF8 Controls Tumor Cell Sensitivity to Tumor-Specific CTL-Induced Ferroptosis
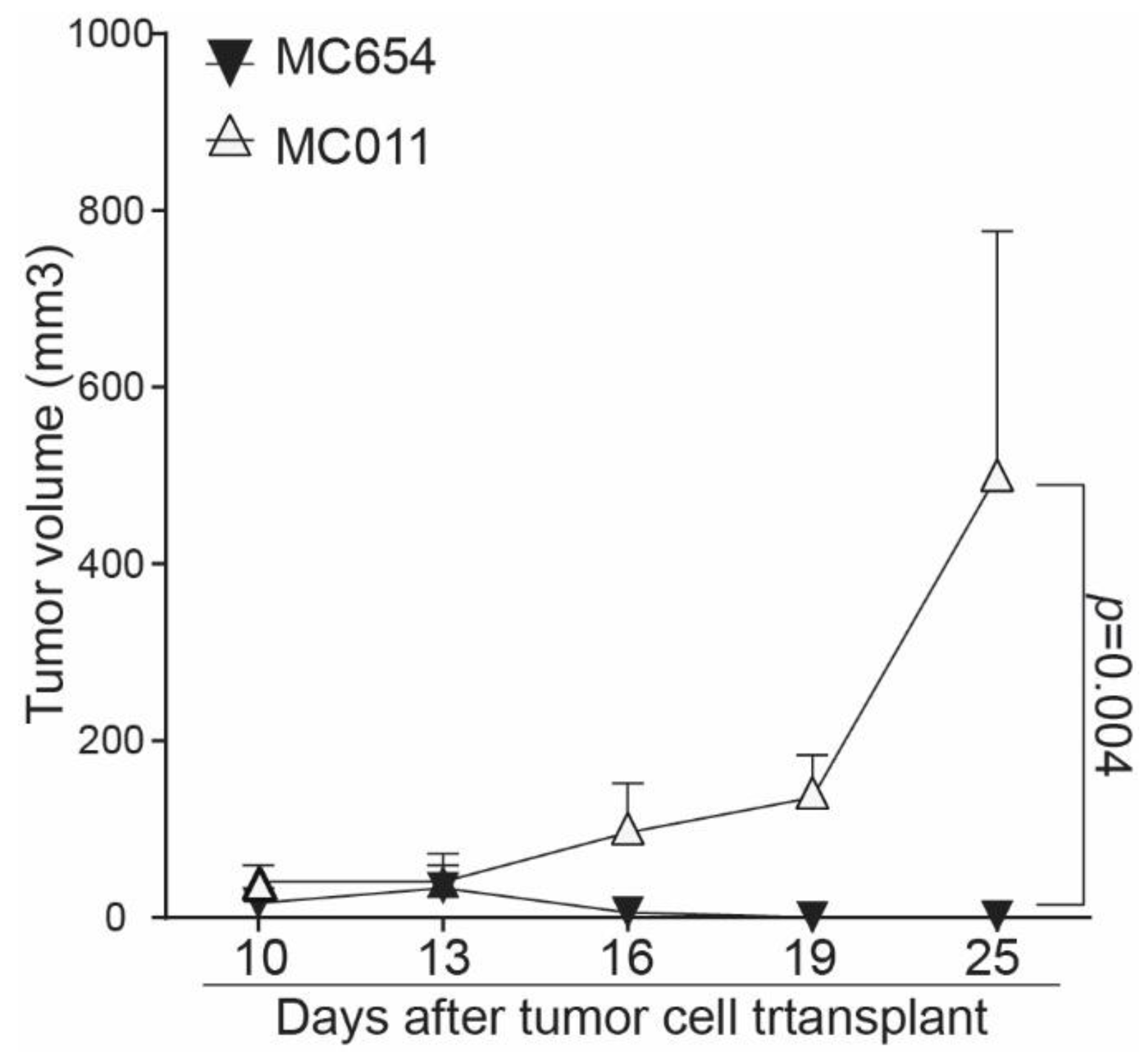
3.5. Loss of IRF8 Expression Results in Increased Tumor Growth In Vivo
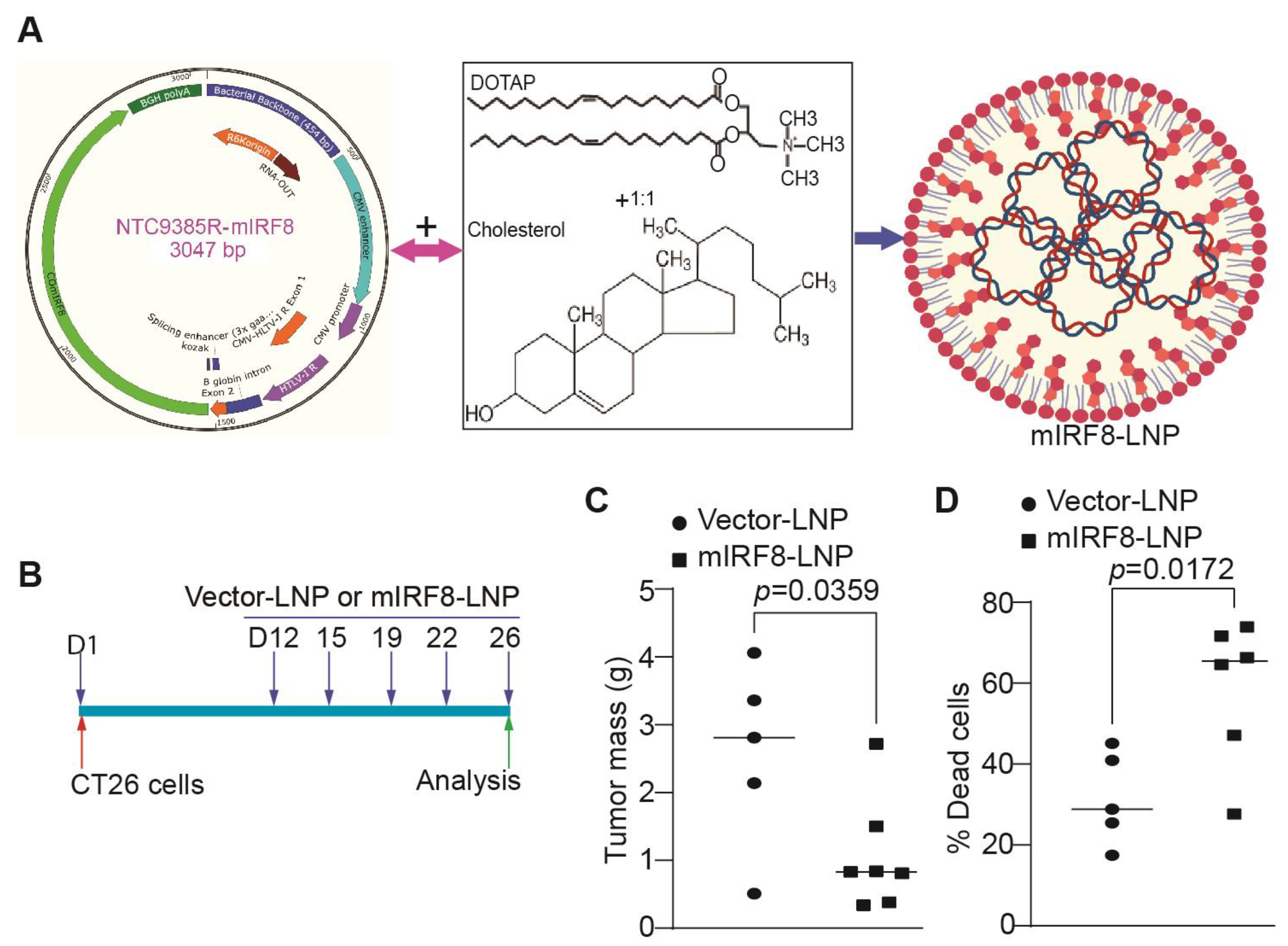
3.6. IRF8-Encoding Plasmid DNA Nanoparticle Therapy Suppresses Tumor Growth In Vitro
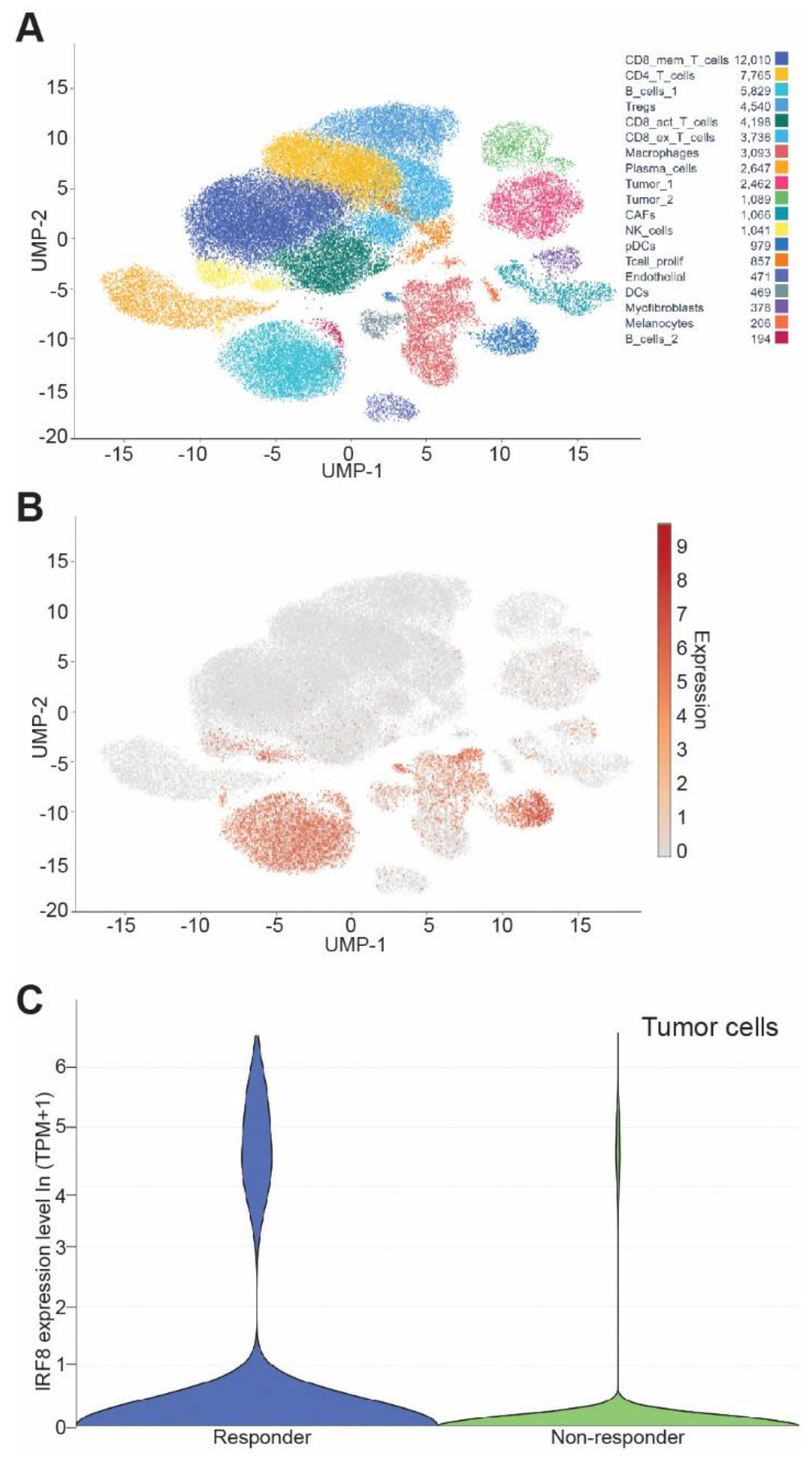
3.7. IRF8 Expression Level in Tumor Cells Correlates Patient Response to ICI Immunotherapy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, A.C.; Zappasodi, R. A decade of checkpoint blockade immunotherapy in melanoma: Understanding the molecular basis for immune sensitivity and resistance. Nat. Immunol. 2022, 23, 660–670. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Yang, D.; Klement, J.D.; Oh, I.K.; Savage, N.M.; Waller, J.L.; Colby, A.H.; Grinstaff, M.W.; Oberlies, N.H.; Pearce, C.J.; et al. SUV39H1 Represses the Expression of Cytotoxic T-Lymphocyte Effector Genes to Promote Colon Tumor Immune Evasion. Cancer Immunol. Res. 2019, 7, 414–427. [Google Scholar] [CrossRef]
- Lu, C.; Talukder, A.; Savage, N.M.; Singh, N.; Liu, K. JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. Oncoimmunology 2017, 6, e1291106. [Google Scholar] [CrossRef]
- Liu, X.; Pu, Y.; Cron, K.; Deng, L.; Kline, J.; Frazier, W.A.; Xu, H.; Peng, H.; Fu, Y.X.; Xu, M.M. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat. Med. 2015, 21, 1209–1215. [Google Scholar] [CrossRef]
- Klement, J.D.; Poschel, D.B.; Lu, C.; Merting, A.D.; Yang, D.; Redd, P.S.; Liu, K. Osteopontin Blockade Immunotherapy Increases Cytotoxic T Lymphocyte Lytic Activity and Suppresses Colon Tumor Progression. Cancers 2021, 13, 1006. [Google Scholar] [CrossRef]
- Klement, J.D.; Paschall, A.V.; Redd, P.S.; Ibrahim, M.L.; Lu, C.; Yang, D.; Celis, E.; Abrams, S.I.; Ozato, K.; Liu, K. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J. Clin. Investig. 2018, 128, 5549–5560. [Google Scholar] [CrossRef]
- Lu, C.; Liu, Z.; Klement, J.D.; Yang, D.; Merting, A.D.; Poschel, D.; Albers, T.; Waller, J.L.; Shi, H.; Liu, K. WDR5-H3K4me3 epigenetic axis regulates OPN expression to compensate PD-L1 function to promote pancreatic cancer immune escape. J. Immunother. Cancer 2021, 9, e002624. [Google Scholar] [CrossRef]
- Kim, M.; Min, Y.K.; Jang, J.; Park, H.; Lee, S.; Lee, C.H. Single-cell RNA sequencing reveals distinct cellular factors for response to immunotherapy targeting CD73 and PD-1 in colorectal cancer. J. Immunother. Cancer 2021, 9, e002503. [Google Scholar] [CrossRef]
- Golstein, P.; Griffiths, G.M. An early history of T cell-mediated cytotoxicity. Nat. Rev. Immunol. 2018, 18, 527–535. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Moller, P.; Koretz, K.; Leithauser, F.; Bruderlein, S.; Henne, C.; Quentmeier, A.; Krammer, P.H. Expression of APO-1 (CD95), a member of the NGF/TNF receptor superfamily, in normal and neoplastic colon epithelium. Int. J. Cancer 1994, 57, 371–377. [Google Scholar] [CrossRef]
- Al Subeh, Z.Y.; Poschel, D.B.; Redd, P.S.; Klement, J.D.; Merting, A.D.; Yang, D.; Mehta, M.; Shi, H.; Colson, Y.L.; Oberlies, N.H.; et al. Lipid Nanoparticle Delivery of Fas Plasmid Restores Fas Expression to Suppress Melanoma Growth In Vivo. ACS Nano 2022, 16, 12695–12710. [Google Scholar] [CrossRef] [PubMed]
- Merting, A.D.; Poschel, D.B.; Lu, C.; Klement, J.D.; Yang, D.; Li, H.; Shi, H.; Chapdelaine, E.; Montgomery, M.; Redman, M.T.; et al. Restoring FAS Expression via Lipid-Encapsulated FAS DNA Nanoparticle Delivery Is Sufficient to Suppress Colon Tumor Growth In Vivo. Cancers 2022, 14, 361. [Google Scholar] [CrossRef] [PubMed]
- Bullani, R.R.; Wehrli, P.; Viard-Leveugle, I.; Rimoldi, D.; Cerottini, J.C.; Saurat, J.H.; Tschopp, J.; French, L.E. Frequent downregulation of Fas (CD95) expression and function in melanoma. Melanoma Res. 2002, 12, 263–270. [Google Scholar] [CrossRef]
- Xiao, W.; Ibrahim, M.L.; Redd, P.S.; Klement, J.D.; Lu, C.; Yang, D.; Savage, N.M.; Liu, K. Loss of Fas Expression and Function Is Coupled with Colon Cancer Resistance to Immune Checkpoint Inhibitor Immunotherapy. Mol. Cancer Res. 2019, 17, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Owen-Schaub, L.B.; van Golen, K.L.; Hill, L.L.; Price, J.E. Fas and Fas ligand interactions suppress melanoma lung metastasis. J. Exp. Med. 1998, 188, 1717–1723. [Google Scholar] [CrossRef]
- Maecker, H.L.; Yun, Z.; Maecker, H.T.; Giaccia, A.J. Epigenetic changes in tumor Fas levels determine immune escape and response to therapy. Cancer Cell 2002, 2, 139–148. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ma, Y.; Li, Q.; Ling, Y.; Zhou, Y.; Chu, K.; Xue, L.; Tao, S. STAT6 inhibits ferroptosis and alleviates acute lung injury via regulating P53/SLC7A11 pathway. Cell Death Dis. 2022, 13, 530. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef]
- Zhu, H.; Klement, J.D.; Lu, C.; Redd, P.S.; Yang, D.; Smith, A.D.; Poschel, D.B.; Zou, J.; Liu, D.; Wang, P.G.; et al. Asah2 Represses the p53-Hmox1 Axis to Protect Myeloid-Derived Suppressor Cells from Ferroptosis. J. Immunol. 2021, 206, 1395–1404. [Google Scholar] [CrossRef]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, Y.; Zhang, J.; Yan, W.; Jung, Y.S.; Chen, M.; Huang, E.; Lloyd, K.; Duan, Y.; Wang, J.; et al. Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev. 2017, 31, 1243–1256. [Google Scholar] [CrossRef]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef] [PubMed]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Holtschke, T.; Lohler, J.; Kanno, Y.; Fehr, T.; Giese, N.; Rosenbauer, F.; Lou, J.; Knobeloch, K.P.; Gabriele, L.; Waring, J.F.; et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell 1996, 87, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Sasaki, H.; Nishiyama, A.; Kurotaki, D.; Kawase, W.; Ban, T.; Nakabayashi, J.; Kanzaki, S.; Sekita, Y.; Nakajima, H.; et al. A RUNX-CBFbeta-driven enhancer directs the Irf8 dose-dependent lineage choice between DCs and monocytes. Nat. Immunol. 2021, 22, 301–311. [Google Scholar] [CrossRef]
- Kim, S.H.; Burton, J.; Yu, C.R.; Sun, L.; He, C.; Wang, H.; Morse, H.C., 3rd; Egwuagu, C.E. Dual Function of the IRF8 Transcription Factor in Autoimmune Uveitis: Loss of IRF8 in T Cells Exacerbates Uveitis, Whereas Irf8 Deletion in the Retina Confers Protection. J. Immunol. 2015, 195, 1480–1488. [Google Scholar] [CrossRef]
- Dambuza, I.M.; He, C.; Choi, J.K.; Yu, C.R.; Wang, R.; Mattapallil, M.J.; Wingfield, P.T.; Caspi, R.R.; Egwuagu, C.E. IL-12p35 induces expansion of IL-10 and IL-35-expressing regulatory B cells and ameliorates autoimmune disease. Nat. Commun. 2017, 8, 719. [Google Scholar] [CrossRef]
- Sun, L.; St Leger, A.J.; Yu, C.R.; He, C.; Mahdi, R.M.; Chan, C.C.; Wang, H.; Morse, H.C., 3rd; Egwuagu, C.E. Interferon Regulator Factor 8 (IRF8) Limits Ocular Pathology during HSV-1 Infection by Restraining the Activation and Expansion of CD8+ T Cells. PLoS ONE 2016, 11, e0155420. [Google Scholar] [CrossRef]
- Choi, J.K.; Yu, C.R.; Bing, S.J.; Jittayasothorn, Y.; Mattapallil, M.J.; Kang, M.; Park, S.B.; Lee, H.S.; Dong, L.; Shi, G.; et al. IL-27-producing B-1a cells suppress neuroinflammation and CNS autoimmune diseases. Proc. Natl. Acad. Sci. USA 2021, 118, e2109548118. [Google Scholar] [CrossRef]
- Kim, S.; Bagadia, P.; Anderson, D.A., 3rd; Liu, T.T.; Huang, X.; Theisen, D.J.; O’Connor, K.W.; Ohara, R.A.; Iwata, A.; Murphy, T.L.; et al. High Amount of Transcription Factor IRF8 Engages AP1-IRF Composite Elements in Enhancers to Direct Type 1 Conventional Dendritic Cell Identity. Immunity 2020, 53, 759–774. [Google Scholar] [CrossRef]
- Lanca, T.; Ungerback, J.; Da Silva, C.; Joeris, T.; Ahmadi, F.; Vandamme, J.; Svensson-Frej, M.; Mowat, A.M.; Kotarsky, K.; Sigvardsson, M.; et al. IRF8 deficiency induces the transcriptional, functional, and epigenetic reprogramming of cDC1 into the cDC2 lineage. Immunity 2022, 55, 1431–1447. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, C.; Pandit, H.; Nagy, B.A.; Stellas, D.; Jensen, S.M.; Bear, J.; Cam, M.; Valentin, A.; Fox, B.A.; Felber, B.K.; et al. Heterodimeric IL-15 delays tumor growth and promotes intratumoral CTL and dendritic cell accumulation by a cytokine network involving XCL1, IFN-gamma, CXCL9 and CXCL10. J. Immunother. Cancer 2020, 8, e000599. [Google Scholar] [CrossRef]
- Jiang, D.S.; Wei, X.; Zhang, X.F.; Liu, Y.; Zhang, Y.; Chen, K.; Gao, L.; Zhou, H.; Zhu, X.H.; Liu, P.P.; et al. IRF8 suppresses pathological cardiac remodelling by inhibiting calcineurin signalling. Nat. Commun. 2014, 5, 3303. [Google Scholar] [CrossRef]
- Ibrahim, M.L.; Klement, J.D.; Lu, C.; Redd, P.S.; Xiao, W.; Yang, D.; Browning, D.D.; Savage, N.M.; Buckhaults, P.J.; Morse, H.C., 3rd; et al. Myeloid-Derived Suppressor Cells Produce IL-10 to Elicit DNMT3b-Dependent IRF8 Silencing to Promote Colitis-Associated Colon Tumorigenesis. Cell Rep. 2018, 25, 3036–3046. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yan, M.; Sun, J.; Jain, S.; Yoshimi, R.; Abolfath, S.M.; Ozato, K.; Coleman, W.G., Jr.; Ng, A.P.; Metcalf, D.; et al. A reporter mouse reveals lineage-specific and heterogeneous expression of IRF8 during lymphoid and myeloid cell differentiation. J. Immunol. 2014, 193, 1766–1777. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Nagineni, C.N.; Ge, H.; Efiok, B.; Chepelinsky, A.B.; Egwuagu, C.E. Interferon consensus sequence-binding protein is constitutively expressed and differentially regulated in the ocular lens. J. Biol. Chem. 1999, 274, 9686–9691. [Google Scholar] [CrossRef] [PubMed]
- Egwuagu, C.E.; Li, W.; Yu, C.R.; Che Mei Lin, M.; Chan, C.C.; Nakamura, T.; Chepelinsky, A.B. Interferon-gamma induces regression of epithelial cell carcinoma: Critical roles of IRF-1 and ICSBP transcription factors. Oncogene 2006, 25, 3670–3679. [Google Scholar] [CrossRef]
- Mattei, F.; Schiavoni, G.; Sestili, P.; Spadaro, F.; Fragale, A.; Sistigu, A.; Lucarini, V.; Spada, M.; Sanchez, M.; Scala, S.; et al. IRF-8 controls melanoma progression by regulating the cross talk between cancer and immune cells within the tumor microenvironment. Neoplasia 2012, 14, 1223–1235. [Google Scholar] [CrossRef]
- Suzuki, M.; Ikeda, K.; Shiraishi, K.; Eguchi, A.; Mori, T.; Yoshimoto, K.; Shibata, H.; Ito, T.; Baba, Y.; Baba, H. Aberrant methylation and silencing of IRF8 expression in non-small cell lung cancer. Oncol. Lett. 2014, 8, 1025–1030. [Google Scholar] [CrossRef]
- Liang, J.; Lu, F.; Li, B.; Liu, L.; Zeng, G.; Zhou, Q.; Chen, L. IRF8 induces senescence of lung cancer cells to exert its tumor suppressive function. Cell Cycle 2019, 18, 3300–3312. [Google Scholar] [CrossRef]
- Yang, D.; Thangaraju, M.; Greeneltch, K.; Browning, D.D.; Schoenlein, P.V.; Tamura, T.; Ozato, K.; Ganapathy, V.; Abrams, S.I.; Liu, K. Repression of IFN regulatory factor 8 by DNA methylation is a molecular determinant of apoptotic resistance and metastatic phenotype in metastatic tumor cells. Cancer Res. 2007, 67, 3301–3309. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Li, Y.; Shi, G.; Du, S.; Wang, X.; Ye, W.; Zhang, Z.; Chu, Y.; Ma, S.; Wang, D.; et al. Hepatic interferon regulatory factor 8 expression suppresses hepatocellular carcinoma progression and enhances the response to anti-programmed cell death protein-1 therapy. Hepatology 2022, 76, 1602–1616. [Google Scholar] [CrossRef] [PubMed]
- Gatti, G.; Betts, C.; Rocha, D.; Nicola, M.; Grupe, V.; Ditada, C.; Nunez, N.G.; Roselli, E.; Araya, P.; Dutto, J.; et al. High IRF8 expression correlates with CD8 T cell infiltration and is a predictive biomarker of therapy response in ER-negative breast cancer. Breast Cancer Res. 2021, 23, 40. [Google Scholar] [CrossRef] [PubMed]
- Gabriele, L.; Phung, J.; Fukumoto, J.; Segal, D.; Wang, I.M.; Giannakakou, P.; Giese, N.A.; Ozato, K.; Morse, H.C. Regulation of apoptosis in myeloid cells by interferon consensus sequence-binding protein. J. Exp. Med. 1999, 190, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Bardhan, K.; Paschall, A.V.; Yang, D.; Waller, J.L.; Park, M.A.; Nayak-Kapoor, A.; Samuel, T.A.; Abrams, S.I.; Liu, K. Deregulation of apoptotic factors Bcl-xL and Bax confers apoptotic resistance to myeloid-derived suppressor cells and contributes to their persistence in cancer. J. Biol. Chem. 2013, 288, 19103–19115. [Google Scholar] [CrossRef]
- DeLeo, A.B.; Shiku, H.; Takahashi, T.; John, M.; Old, L.J. Cell surface antigens of chemically induced sarcomas of the mouse. I. Mouse leukemia virus related antigens and alloantigens on cultured fibroblasts and sarcomas: Description of a unique antigen on BALB/c Meth A sarcoma. J. Exp. Med. 1977, 146, 720–734. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef]
- Liu, F.; Bardhan, K.; Yang, D.; Thangaraju, M.; Ganapathy, V.; Waller, J.L.; Liles, G.B.; Lee, J.R.; Liu, K. NF-kappaB Directly Regulates Fas Transcription to Modulate Fas-mediated Apoptosis and Tumor Suppression. J. Biol. Chem. 2012, 287, 25530–25540. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Lu, C.; Paschall, A.V.; Shi, H.; Savage, N.; Waller, J.L.; Sabbatini, M.E.; Oberlies, N.H.; Pearce, C.; Liu, K. The MLL1-H3K4me3 Axis-Mediated PD-L1 Expression and Pancreatic Cancer Immune Evasion. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.H.; Bristol, J.A.; McDuffie, E.; Abrams, S.I. Regression of extensive pulmonary metastases in mice by adoptive transfer of antigen-specific CD8(+) CTL reactive against tumor cells expressing a naturally occurring rejection epitope. J. Immunol. 2001, 167, 4286–4292. [Google Scholar] [CrossRef] [PubMed]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997. [Google Scholar] [CrossRef]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [PubMed]
- Luda, K.M.; Joeris, T.; Persson, E.K.; Rivollier, A.; Demiri, M.; Sitnik, K.M.; Pool, L.; Holm, J.B.; Melo-Gonzalez, F.; Richter, L.; et al. IRF8 Transcription-Factor-Dependent Classical Dendritic Cells Are Essential for Intestinal T Cell Homeostasis. Immunity 2016, 44, 860–874. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gu, W. p53 in ferroptosis regulation: The new weapon for the old guardian. Cell Death Differ. 2022, 29, 895–910. [Google Scholar] [CrossRef]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014, 3, e02523. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Y.; Du, T.; Yang, H.; Lei, L.; Guo, M.; Ding, H.F.; Zhang, J.; Wang, H.; Chen, X.; et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc(.). Cell Death Differ. 2020, 27, 662–675. [Google Scholar] [CrossRef]
- Laricchia-Robbio, L.; Tamura, T.; Karpova, T.; Sprague, B.L.; McNally, J.G.; Ozato, K. Partner-regulated interaction of IFN regulatory factor 8 with chromatin visualized in live macrophages. Proc. Natl. Acad. Sci. USA 2005, 102, 14368–14373. [Google Scholar] [CrossRef]
- McGough, J.M.; Yang, D.; Huang, S.; Georgi, D.; Hewitt, S.M.; Rocken, C.; Tanzer, M.; Ebert, M.P.; Liu, K. DNA methylation represses IFN-gamma-induced and signal transducer and activator of transcription 1-mediated IFN regulatory factor 8 activation in colon carcinoma cells. Mol. Cancer Res. 2008, 6, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Geng, H.; Ng, K.M.; Yu, J.; van Hasselt, A.; Cao, Y.; Zeng, Y.X.; Wong, A.H.; Wang, X.; Ying, J.; et al. Epigenetic disruption of interferon-gamma response through silencing the tumor suppressor interferon regulatory factor 8 in nasopharyngeal, esophageal and multiple other carcinomas. Oncogene 2008, 27, 5267–5276. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A. Vector Design for Improved DNA Vaccine Efficacy, Safety and Production. Vaccines 2013, 1, 225–249. [Google Scholar] [CrossRef] [PubMed]
- Crook, K.; Stevenson, B.J.; Dubouchet, M.; Porteous, D.J. Inclusion of cholesterol in DOTAP transfection complexes increases the delivery of DNA to cells in vitro in the presence of serum. Gene Ther. 1998, 5, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Templeton, N.S.; Lasic, D.D.; Frederik, P.M.; Strey, H.H.; Roberts, D.D.; Pavlakis, G.N. Improved DNA: Liposome complexes for increased systemic delivery and gene expression. Nat. Biotechnol. 1997, 15, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Stewart, D.J.; Lee, J.J.; Ji, L.; Ramesh, R.; Jayachandran, G.; Nunez, M.I.; Wistuba, I.I.; Erasmus, J.J.; Hicks, M.E.; et al. Phase I clinical trial of systemically administered TUSC2(FUS1)-nanoparticles mediating functional gene transfer in humans. PLoS ONE 2012, 7, e34833. [Google Scholar] [CrossRef]
- Kanno, Y.; Levi, B.Z.; Tamura, T.; Ozato, K. Immune cell-specific amplification of interferon signaling by the IRF-4/8-PU.1 complex. J. Interferon Cytokine Res. 2005, 25, 770–779. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poschel, D.B.; Kehinde-Ige, M.; Klement, J.D.; Yang, D.; Merting, A.D.; Savage, N.M.; Shi, H.; Liu, K. IRF8 Regulates Intrinsic Ferroptosis through Repressing p53 Expression to Maintain Tumor Cell Sensitivity to Cytotoxic T Lymphocytes. Cells 2023, 12, 310. https://doi.org/10.3390/cells12020310
Poschel DB, Kehinde-Ige M, Klement JD, Yang D, Merting AD, Savage NM, Shi H, Liu K. IRF8 Regulates Intrinsic Ferroptosis through Repressing p53 Expression to Maintain Tumor Cell Sensitivity to Cytotoxic T Lymphocytes. Cells. 2023; 12(2):310. https://doi.org/10.3390/cells12020310
Chicago/Turabian StylePoschel, Dakota B., Mercy Kehinde-Ige, John D. Klement, Dafeng Yang, Alyssa D. Merting, Natasha M. Savage, Huidong Shi, and Kebin Liu. 2023. "IRF8 Regulates Intrinsic Ferroptosis through Repressing p53 Expression to Maintain Tumor Cell Sensitivity to Cytotoxic T Lymphocytes" Cells 12, no. 2: 310. https://doi.org/10.3390/cells12020310
APA StylePoschel, D. B., Kehinde-Ige, M., Klement, J. D., Yang, D., Merting, A. D., Savage, N. M., Shi, H., & Liu, K. (2023). IRF8 Regulates Intrinsic Ferroptosis through Repressing p53 Expression to Maintain Tumor Cell Sensitivity to Cytotoxic T Lymphocytes. Cells, 12(2), 310. https://doi.org/10.3390/cells12020310