

Astrocytes Are a Key Target for Neurotropic Viral Infection




Abstract
:1. Introduction
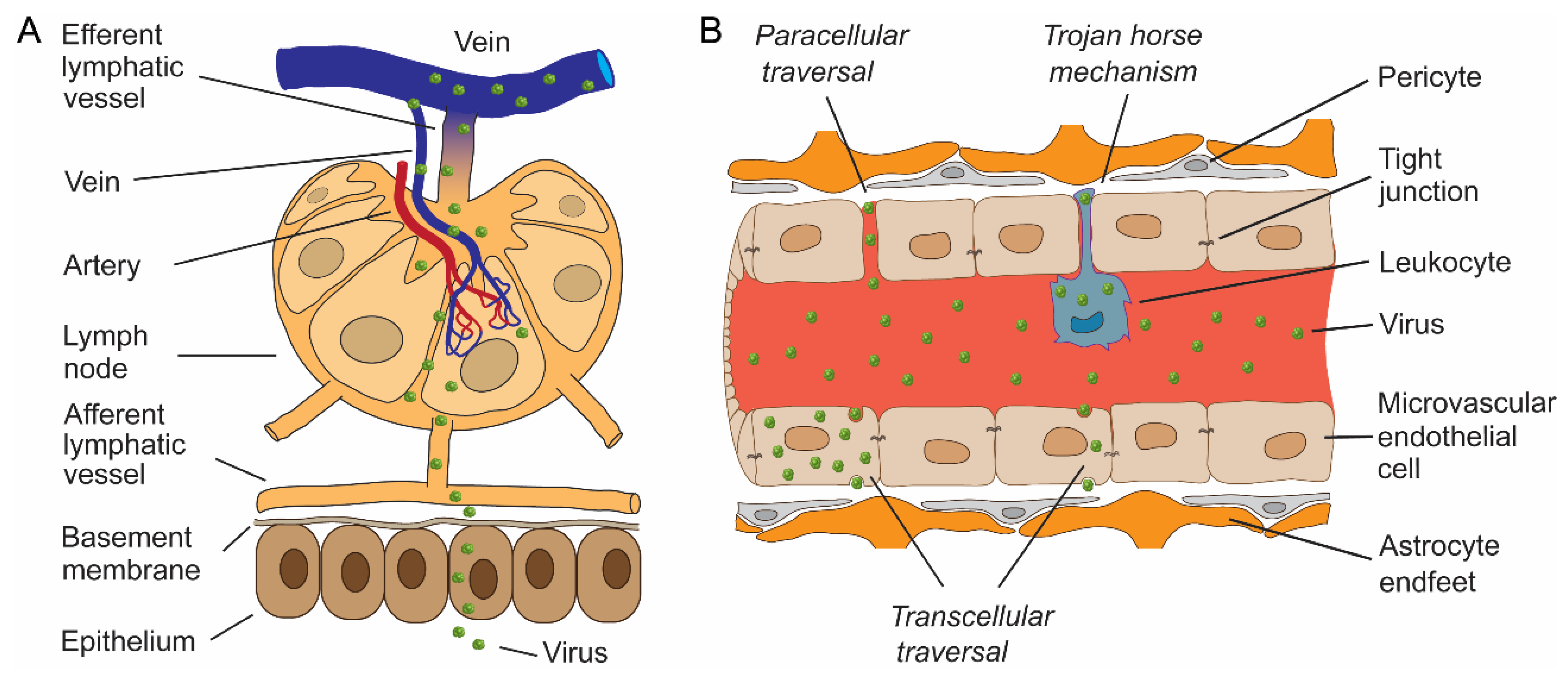
2. Virus Spread and Infection of the CNS
3. Astrocytes as the Key Cell Type of CNS Viral Infections
4. Viral Attachment to Entry Receptors in Astrocytes
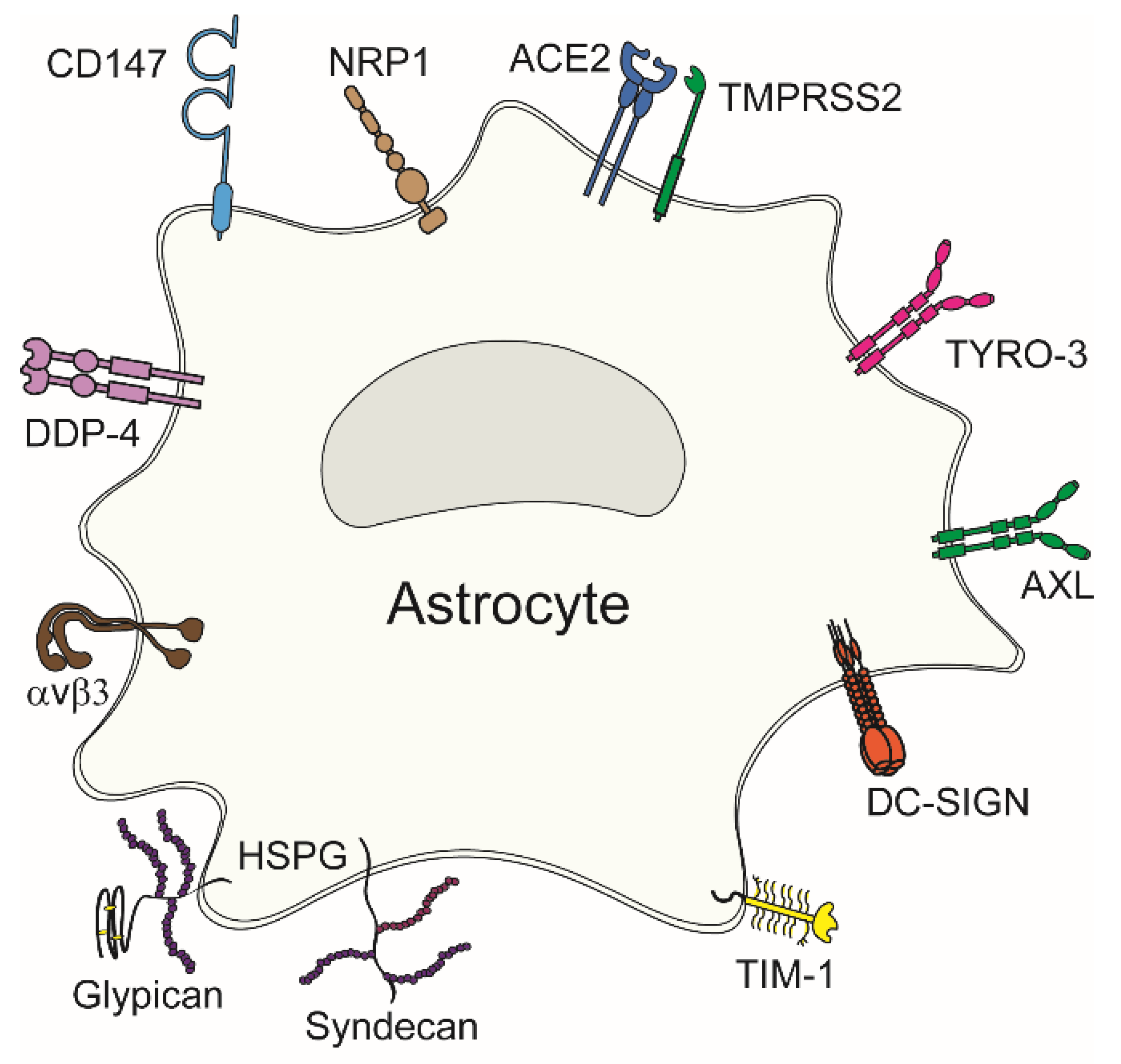
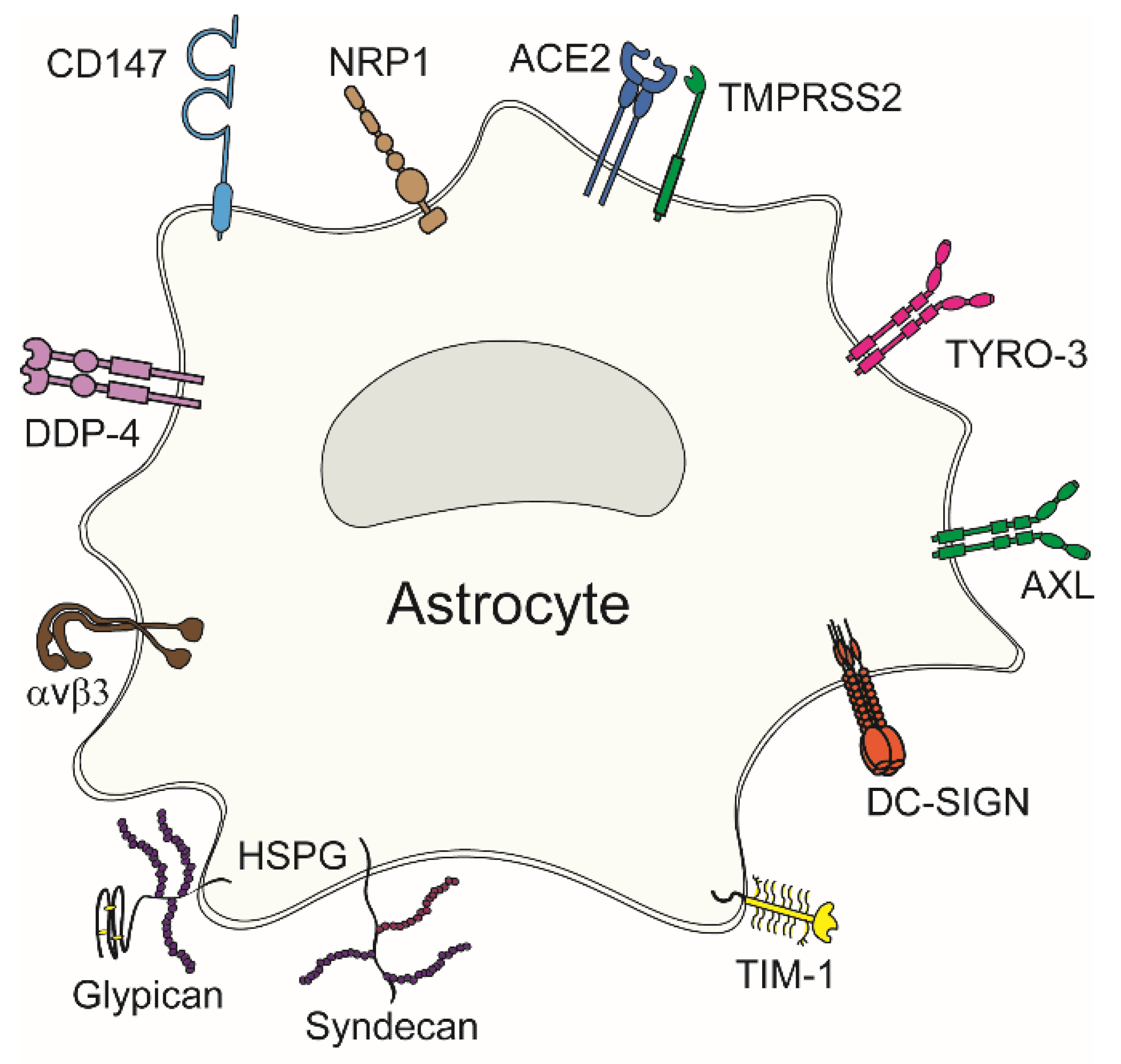
4.1. Confirmed Viral Entry Receptors in Astrocytes
4.1.1. Heparan Sulphate Proteoglycans
4.1.2. Angiotensin Converting Enzyme 2
4.1.3. Neuropilin-1
4.1.4. TIM and TAM Phosphatidylserine Receptors
4.1.5. Integrins
4.1.6. Cluster of Differentiation 147 and Dipeptidylpeptidase 4
5. Viral Entry and Remodelling of Intracellular Organelle Traffic in Infected Astrocytes
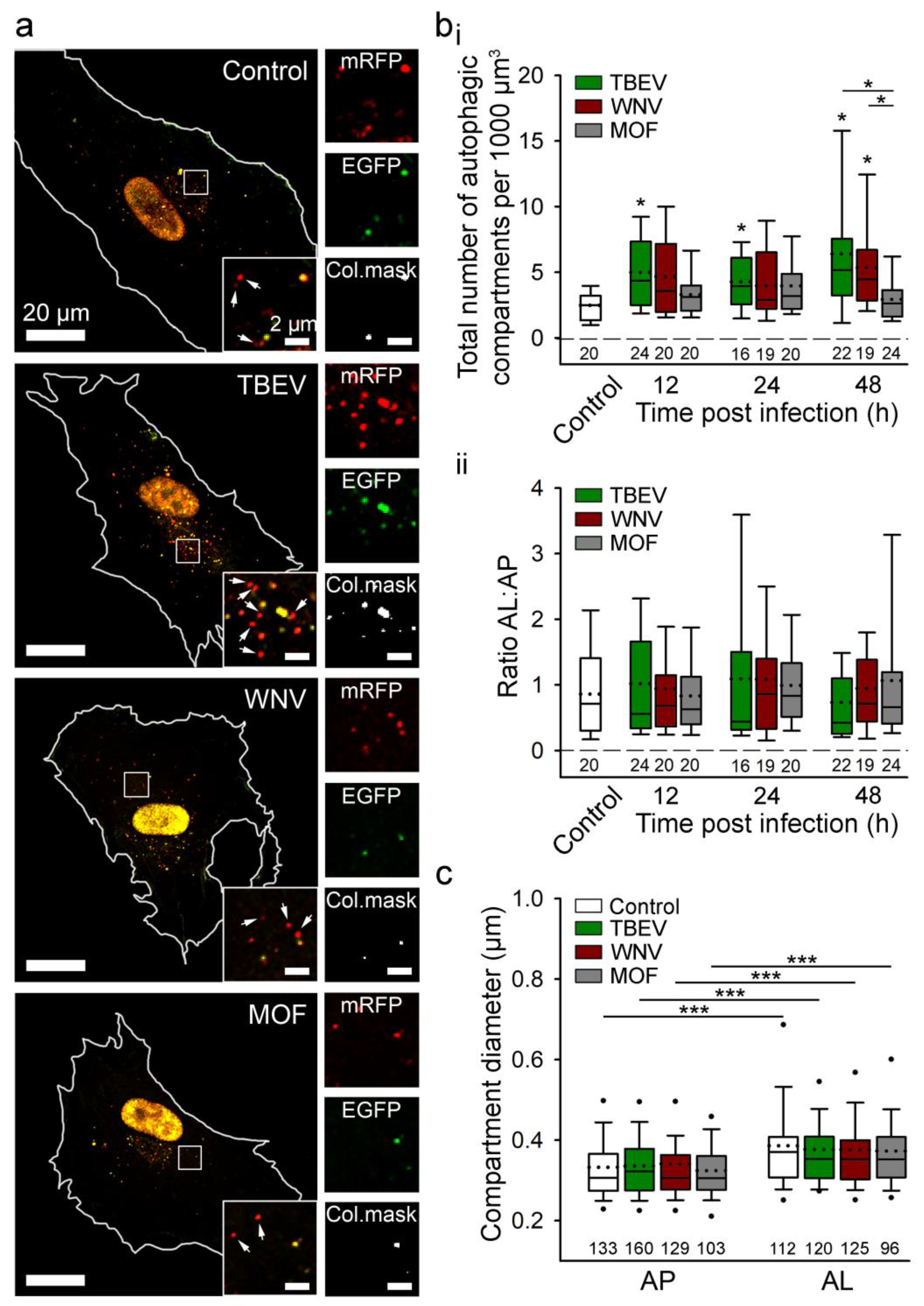
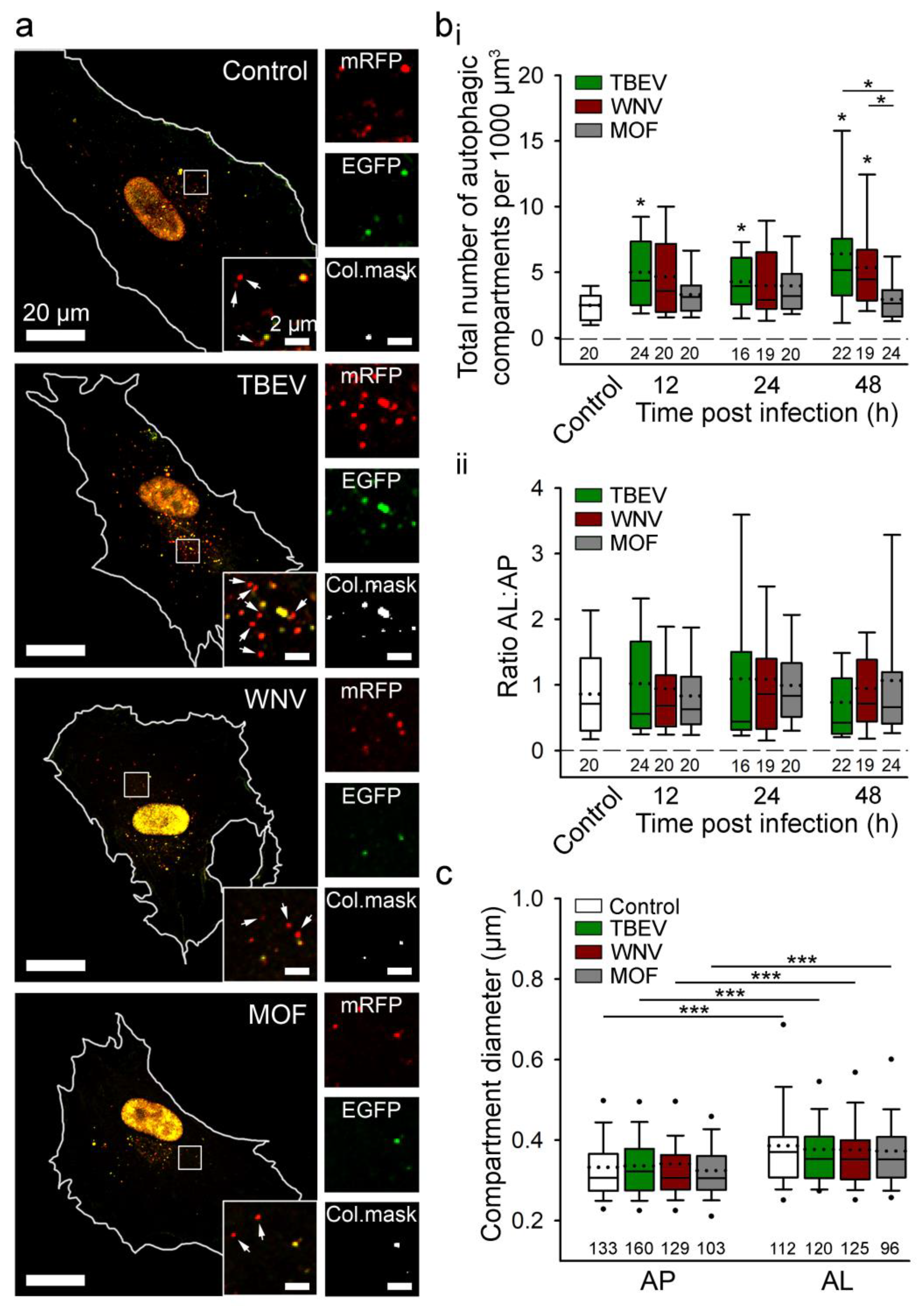
6. Autophagy in Viral Infection of Astrocytes
6.1. Autophagy of Cellular Organelles in Viral Infection
6.2. Biogenesis of Lipid Droplets and Lipophagy in Viral Infection
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Romero, J.R.; Newland, J.G. Viral meningitis and encephalitis: Traditional and emerging viral agents. Semin. Pediatr. Infect. Dis. 2003, 14, 72–82. [Google Scholar] [CrossRef]
- Gora, H.; Smith, S.; Wilson, I.; Preston-Thomas, A.; Ramsamy, N.; Hanson, J. The epidemiology and outcomes of central nervous system infections in Far North Queensland, tropical Australia; 2000–2019. PLoS ONE 2022, 17, e0265410. [Google Scholar] [CrossRef]
- Ludlow, M.; Kortekaas, J.; Herden, C.; Hoffmann, B.; Tappe, D.; Trebst, C.; Griffin, D.E.; Brindle, H.E.; Solomon, T.; Brown, A.S.; et al. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol. 2016, 131, 159–184. [Google Scholar] [CrossRef]
- Carroll, D.; Daszak, P.; Wolfe, N.D.; Gao, G.F.; Morel, C.M.; Morzaria, S.; Pablos-Mendez, A.; Tomori, O.; Mazet, J.A.K. The Global Virome Project. Science 2018, 359, 872–874. [Google Scholar] [CrossRef]
- Swanson, P.A., II; McGavern, D.B. Viral diseases of the central nervous system. Curr. Opin. Virol. 2015, 11, 44–54. [Google Scholar] [CrossRef]
- Valerio, F.; Whitehouse, D.P.; Menon, D.K.; Newcombe, V.F.J. The neurological sequelae of pandemics and epidemics. J. Neurol. 2021, 268, 2629–2655. [Google Scholar] [CrossRef]
- Burrell, C.J.; Howard, C.R.; Murphy, F.A. Pathogenesis of Virus Infections. In Fenner and White’s Medical Virology; Academic Press: Cambridge, MA, USA, 2017; pp. 77–104. [Google Scholar] [CrossRef]
- Wu, S.J.; Grouard-Vogel, G.; Sun, W.; Mascola, J.R.; Brachtel, E.; Putvatana, R.; Louder, M.K.; Filgueira, L.; Marovich, M.A.; Wong, H.K.; et al. Human skin Langerhans cells are targets of dengue virus infection. Nat. Med. 2000, 6, 816–820. [Google Scholar] [CrossRef]
- Byrne, S.N.; Halliday, G.M.; Johnston, L.J.; King, N.J. Interleukin-1beta but not tumor necrosis factor is involved in West Nile virus-induced Langerhans cell migration from the skin in C57BL/6 mice. J. Investig. Dermatol. 2001, 117, 702–709. [Google Scholar] [CrossRef]
- Iwasaki, A.; Foxman, E.F.; Molony, R.D. Early local immune defences in the respiratory tract. Nat. Rev. Immunol. 2017, 17, 7–20. [Google Scholar] [CrossRef]
- Cain, M.D.; Salimi, H.; Diamond, M.S.; Klein, R.S. Mechanisms of Pathogen Invasion into the Central Nervous System. Neuron 2019, 103, 771–783. [Google Scholar] [CrossRef]
- Mori, I.; Nishiyama, Y.; Yokochi, T.; Kimura, Y. Olfactory transmission of neurotropic viruses. J. Neurovirol. 2005, 11, 129–137. [Google Scholar] [CrossRef]
- Ren, R.; Racaniello, V.R. Poliovirus spreads from muscle to the central nervous system by neural pathways. J. Infect. Dis. 1992, 166, 747–752. [Google Scholar] [CrossRef]
- Fooks, A.R.; Cliquet, F.; Finke, S.; Freuling, C.; Hemachudha, T.; Mani, R.S.; Muller, T.; Nadin-Davis, S.; Picard-Meyer, E.; Wilde, H.; et al. Rabies. Nat. Rev. Dis. Primers 2017, 3, 17091. [Google Scholar] [CrossRef]
- Nathanson, N. The pathogenesis of poliomyelitis: What we don’t know. Adv. Virus Res. 2008, 71, 1–50. [Google Scholar] [CrossRef]
- Cho, H.; Diamond, M.S. Immune responses to West Nile virus infection in the central nervous system. Viruses 2012, 4, 3812–3830. [Google Scholar] [CrossRef]
- Saunders, N.R.; Dreifuss, J.J.; Dziegielewska, K.M.; Johansson, P.A.; Habgood, M.D.; Mollgard, K.; Bauer, H.C. The rights and wrongs of blood-brain barrier permeability studies: A walk through 100 years of history. Front. Neurosci. 2014, 8, 404. [Google Scholar] [CrossRef]
- Brown, P.D.; Davies, S.L.; Speake, T.; Millar, I.D. Molecular mechanisms of cerebrospinal fluid production. Neuroscience 2004, 129, 957–970. [Google Scholar] [CrossRef]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Sarikcioglu, L. Lina Stern (1878–1968): An outstanding scientist of her time. Childs Nerv. Syst. 2017, 33, 1027–1029. [Google Scholar] [CrossRef]
- Dahm, T.; Rudolph, H.; Schwerk, C.; Schroten, H.; Tenenbaum, T. Neuroinvasion and Inflammation in Viral Central Nervous System Infections. Mediat. Inflamm. 2016, 2016, 8562805. [Google Scholar] [CrossRef]
- Nag, S.; Begley David, J. Blood Brain Barrier, Exchange of metabolites and gases. In Pathology and Genetics: Cerebrovascular Diseases; ISN Neuropath Press: Basel, Switzerland, 2005. [Google Scholar]
- Zhao, Y.; Xin, Y.; He, Z.; Hu, W. Function of Connexins in the Interaction between Glial and Vascular Cells in the Central Nervous System and Related Neurological Diseases. Neural Plast. 2018, 2018, 6323901. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Katayama, T.; Prat, A. Glial influence on the blood brain barrier. Glia 2013, 61, 1939–1958. [Google Scholar] [CrossRef]
- Nikolakopoulou, A.M.; Zhao, Z.; Montagne, A.; Zlokovic, B.V. Regional early and progressive loss of brain pericytes but not vascular smooth muscle cells in adult mice with disrupted platelet-derived growth factor receptor-beta signaling. PLoS ONE 2017, 12, e0176225. [Google Scholar] [CrossRef]
- Kim, K.S. Mechanisms of microbial traversal of the blood-brain barrier. Nat. Rev. Microbiol. 2008, 6, 625–634. [Google Scholar] [CrossRef]
- Coyne, C.B.; Kim, K.S.; Bergelson, J.M. Poliovirus entry into human brain microvascular cells requires receptor-induced activation of SHP-2. EMBO J. 2007, 26, 4016–4028. [Google Scholar] [CrossRef]
- Verma, S.; Lo, Y.; Chapagain, M.; Lum, S.; Kumar, M.; Gurjav, U.; Luo, H.; Nakatsuka, A.; Nerurkar, V.R. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: Transmigration across the in vitro blood-brain barrier. Virology 2009, 385, 425–433. [Google Scholar] [CrossRef]
- Arsenio-Nunes, M.L.; Cerutti, I.; Farkas-Bargeton, E. Vascular and neuroglial changes in experimental herpes simplex encephalitis: Ultrastructural study. Acta Neuropathol. 1975, 33, 245–256. [Google Scholar] [CrossRef]
- Daniels, B.P.; Holman, D.W.; Cruz-Orengo, L.; Jujjavarapu, H.; Durrant, D.M.; Klein, R.S. Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. mBio 2014, 5, e01476-14. [Google Scholar] [CrossRef]
- Tabor-Godwin, J.M.; Ruller, C.M.; Bagalso, N.; An, N.; Pagarigan, R.R.; Harkins, S.; Gilbert, P.E.; Kiosses, W.B.; Gude, N.A.; Cornell, C.T.; et al. A novel population of myeloid cells responding to coxsackievirus infection assists in the dissemination of virus within the neonatal CNS. J. Neurosci. 2010, 30, 8676–8691. [Google Scholar] [CrossRef]
- Clay, C.C.; Rodrigues, D.S.; Ho, Y.S.; Fallert, B.A.; Janatpour, K.; Reinhart, T.A.; Esser, U. Neuroinvasion of fluorescein-positive monocytes in acute simian immunodeficiency virus infection. J. Virol. 2007, 81, 12040–12048. [Google Scholar] [CrossRef]
- Suen, W.W.; Prow, N.A.; Hall, R.A.; Bielefeldt-Ohmann, H. Mechanism of West Nile virus neuroinvasion: A critical appraisal. Viruses 2014, 6, 2796–2825. [Google Scholar] [CrossRef] [PubMed]
- Bauer, L.; Laksono, B.M.; de Vrij, F.M.S.; Kushner, S.A.; Harschnitz, O.; van Riel, D. The neuroinvasiveness, neurotropism, and neurovirulence of SARS-CoV-2. Trends Neurosci. 2022, 45, 358–368. [Google Scholar] [CrossRef]
- Zorec, R.; Verkhratsky, A. Astrocytes in the pathophysiology of neuroinfection. Essays Biochem. 2022, 67, 131–145. [Google Scholar] [CrossRef]
- Zhang, Y.; Archie, S.R.; Ghanwatkar, Y.; Sharma, S.; Nozohouri, S.; Burks, E.; Mdzinarishvili, A.; Liu, Z.; Abbruscato, T.J. Potential role of astrocyte angiotensin converting enzyme 2 in the neural transmission of COVID-19 and a neuroinflammatory state induced by smoking and vaping. Fluids Barriers CNS 2022, 19, 46. [Google Scholar] [CrossRef] [PubMed]
- Tavcar, P.; Potokar, M.; Kolenc, M.; Korva, M.; Avsic-Zupanc, T.; Zorec, R.; Jorgacevski, J. Neurotropic Viruses, Astrocytes, and COVID-19. Front. Cell Neurosci. 2021, 15, 662578. [Google Scholar] [CrossRef]
- Jorgačevski, J.; Korva, M.; Potokar, M.; Lisjak, M.; Avšič-Županc, T.; Zorec, R. ZIKV Strains Differentially Affect Survival of Human Fetal Astrocytes versus Neurons and Traffic of ZIKV-Laden Endocytotic Compartments. Sci. Rep. 2019, 9, 8069. [Google Scholar] [CrossRef]
- Potokar, M.; Jorgacevski, J.; Zorec, R. Astrocytes in Flavivirus Infections. Int. J. Mol. Sci. 2019, 20, 691. [Google Scholar] [CrossRef]
- Potokar, M.; Korva, M.; Jorgacevski, J.; Avsic-Zupanc, T.; Zorec, R. Tick-borne encephalitis virus infects rat astrocytes but does not affect their viability. PLoS ONE 2014, 9, e86219. [Google Scholar] [CrossRef]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar] [CrossRef]
- Cali, C.; Agus, M.; Kare, K.; Boges, D.J.; Lehvaslaiho, H.; Hadwiger, M.; Magistretti, P.J. 3D cellular reconstruction of cortical glia and parenchymal morphometric analysis from Serial Block-Face Electron Microscopy of juvenile rat. Prog. Neurobiol. 2019, 183, 101696. [Google Scholar] [CrossRef]
- Prebil, M.; Vardjan, N.; Jensen, J.; Zorec, R.; Kreft, M. Dynamic monitoring of cytosolic glucose in single astrocytes. Glia 2011, 59, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Prebil, M.; Jensen, J.; Zorec, R.; Kreft, M. Astrocytes and energy metabolism. Arch. Physiol. Biochem. 2011, 117, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.T.; Alves, L.R. Extracellular Vesicles in Viral Infections: Two Sides of the Same Coin? Front. Cell Infect. Microbiol. 2020, 10, 593170. [Google Scholar] [CrossRef] [PubMed]
- Tavcar Verdev, P.; Potokar, M.; Korva, M.; Resman Rus, K.; Kolenc, M.; Avsic Zupanc, T.; Zorec, R.; Jorgacevski, J. In human astrocytes neurotropic flaviviruses increase autophagy, yet their replication is autophagy-independent. Cell Mol. Life Sci. 2022, 79, 566. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Nicholson, C.; Hrabetova, S. Brain Extracellular Space: The Final Frontier of Neuroscience. Biophys. J. 2017, 113, 2133–2142. [Google Scholar] [CrossRef]
- Li Puma, D.D.; Marcocci, M.E.; Lazzarino, G.; De Chiara, G.; Tavazzi, B.; Palamara, A.T.; Piacentini, R.; Grassi, C. Ca2+-dependent release of ATP from astrocytes affects herpes simplex virus type 1 infection of neurons. Glia 2021, 69, 201–215. [Google Scholar] [CrossRef]
- Kong, W.; Montano, M.; Corley, M.J.; Helmy, E.; Kobayashi, H.; Kinisu, M.; Suryawanshi, R.; Luo, X.; Royer, L.A.; Roan, N.R.; et al. Neuropilin-1 Mediates SARS-CoV-2 Infection of Astrocytes in Brain Organoids, Inducing Inflammation Leading to Dysfunction and Death of Neurons. mBio 2022, 13, e0230822. [Google Scholar] [CrossRef]
- Xiao, L.; Sakagami, H.; Miwa, N. ACE2: The key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel? Viruses 2020, 12, 491. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Y.F.; Yang, Y.; Zou, P.; Chen, J.; He, Y.; Shui, S.L.; Cui, Y.R.; Bai, R.; Liang, Y.J.; et al. AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat. Microbiol. 2018, 3, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.L.; Nemerow, G.R. Cell integrins: Commonly used receptors for diverse viral pathogens. Trends Microbiol. 2007, 15, 500–507. [Google Scholar] [CrossRef]
- Chauhan, A.; Khandkar, M. Endocytosis of human immunodeficiency virus 1 (HIV-1) in astrocytes: A fiery path to its destination. Microb. Pathog. 2015, 78, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Cagno, V.; Tseligka, E.D.; Jones, S.T.; Tapparel, C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias? Viruses 2019, 11, 596. [Google Scholar] [CrossRef] [PubMed]
- Hacker, U.; Nybakken, K.; Perrimon, N. Heparan sulphate proteoglycans: The sweet side of development. Nat. Rev. Mol. Cell Biol. 2005, 6, 530–541. [Google Scholar] [CrossRef]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef]
- Bernfield, M.; Gotte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef]
- Kroschewski, H.; Allison, S.L.; Heinz, F.X.; Mandl, C.W. Role of heparan sulfate for attachment and entry of tick-borne encephalitis virus. Virology 2003, 308, 92–100. [Google Scholar] [CrossRef]
- Properzi, F.; Lin, R.; Kwok, J.; Naidu, M.; van Kuppevelt, T.H.; Ten Dam, G.B.; Camargo, L.M.; Raha-Chowdhury, R.; Furukawa, Y.; Mikami, T.; et al. Heparan sulphate proteoglycans in glia and in the normal and injured CNS: Expression of sulphotransferases and changes in sulphation. Eur. J. Neurosci. 2008, 27, 593–604. [Google Scholar] [CrossRef]
- Alfhili, M.A.; Alsughayyir, J.; McCubrey, J.A.; Akula, S.M. GSK-3-associated signaling is crucial to virus infection of cells. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118767. [Google Scholar] [CrossRef]
- Liu, X.; Verma, A.; Garcia, G., Jr.; Ramage, H.; Lucas, A.; Myers, R.L.; Michaelson, J.J.; Coryell, W.; Kumar, A.; Charney, A.W.; et al. Targeting the coronavirus nucleocapsid protein through GSK-3 inhibition. Proc. Natl. Acad. Sci. USA 2021, 118, e2113401118. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.A.; Harris, E.A. Molecular Mechanisms for Herpes Simplex Virus Type 1 Pathogenesis in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.; Liu, J.; Blaiklock, P.; Shworak, N.W.; Bai, X.; Esko, J.D.; Cohen, G.H.; Eisenberg, R.J.; Rosenberg, R.D.; Spear, P.G. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 1999, 99, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef]
- Evans, J.P.; Liu, S.L. Role of host factors in SARS-CoV-2 entry. J. Biol. Chem. 2021, 297, 100847. [Google Scholar] [CrossRef]
- Unger, T.; Steckelings, U.M.; Santos, R.A.S.d. The Protective Arm of the Renin Angiotensin System: Functional Aspects and Therapeutic Implications; Academic Press: Amsterdam, The Netherlands, 2015; 312p. [Google Scholar]
- Crunfli, F.; Carregari, V.C.; Veras, F.P.; Silva, L.S.; Nogueira, M.H.; Antunes, A.; Vendramini, P.H.; Valenca, A.G.F.; Brandao-Teles, C.; Zuccoli, G.D.S.; et al. Morphological, cellular, and molecular basis of brain infection in COVID-19 patients. Proc. Natl. Acad. Sci. USA 2022, 119, e2200960119. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.K.; Huang, I.C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Torices, S.; Cabrera, R.; Stangis, M.; Naranjo, O.; Fattakhov, N.; Teglas, T.; Adesse, D.; Toborek, M. Expression of SARS-CoV-2-related receptors in cells of the neurovascular unit: Implications for HIV-1 infection. J. Neuroinflamm. 2021, 18, 167. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, V.S.; Zetter, M.A.; Guerra, E.C.; Hernandez-Araiza, I.; Karuzin, N.; Hernandez-Perez, O.R.; Eiden, L.E.; Zhang, L. ACE2 expression in rat brain: Implications for COVID-19 associated neurological manifestations. Exp. Neurol. 2021, 345, 113837. [Google Scholar] [CrossRef]
- Pellet-Many, C.; Frankel, P.; Jia, H.; Zachary, I. Neuropilins: Structure, function and role in disease. Biochem. J. 2008, 411, 211–226. [Google Scholar] [CrossRef]
- Graziani, G.; Lacal, P.M. Neuropilin-1 as Therapeutic Target for Malignant Melanoma. Front. Oncol. 2015, 5, 125. [Google Scholar] [CrossRef]
- Lee, J.; Chong, K.; Lee, J.; Kim, C.; Kim, J.H.; Choi, K.; Choi, C. Differential dependency of human glioblastoma cells on vascular endothelial growth factor-A signaling via neuropilin-1. Int. J. Oncol. 2022, 61, 122. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Chapoval, S.P.; Keegan, A.D. Perspectives and potential approaches for targeting neuropilin 1 in SARS-CoV-2 infection. Mol. Med. 2021, 27, 162. [Google Scholar] [CrossRef]
- Pang, H.B.; Braun, G.B.; Friman, T.; Aza-Blanc, P.; Ruidiaz, M.E.; Sugahara, K.N.; Teesalu, T.; Ruoslahti, E. An endocytosis pathway initiated through neuropilin-1 and regulated by nutrient availability. Nat. Commun. 2014, 5, 4904. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, G.; Yamauchi, Y.; Teesalu, T. A widespread viral entry mechanism: The C-end Rule motif-neuropilin receptor interaction. Proc. Natl. Acad. Sci. USA 2021, 118, e2112457118. [Google Scholar] [CrossRef] [PubMed]
- Gudowska-Sawczuk, M.; Mroczko, B. The Role of Neuropilin-1 (NRP-1) in SARS-CoV-2 Infection: Review. J. Clin. Med. 2021, 10, 2772. [Google Scholar] [CrossRef] [PubMed]
- De Vlaeminck, Y.; Bonelli, S.; Awad, R.M.; Dewilde, M.; Rizzolio, S.; Lecocq, Q.; Bolli, E.; Santos, A.R.; Laoui, D.; Schoonooghe, S.; et al. Targeting Neuropilin-1 with Nanobodies Reduces Colorectal Carcinoma Development. Cancers 2020, 12, 3582. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Anton-Plagaro, C.; Shoemark, D.K.; Simon-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Welsch, S.; Muller, B.; Krausslich, H.G. More than one door—Budding of enveloped viruses through cellular membranes. FEBS Lett. 2007, 581, 2089–2097. [Google Scholar] [CrossRef]
- Moller-Tank, S.; Maury, W. Phosphatidylserine receptors: Enhancers of enveloped virus entry and infection. Virology 2014, 468–470, 565–580. [Google Scholar] [CrossRef]
- Tsuji, T.; Cheng, J.; Tatematsu, T.; Ebata, A.; Kamikawa, H.; Fujita, A.; Gyobu, S.; Segawa, K.; Arai, H.; Taguchi, T.; et al. Predominant localization of phosphatidylserine at the cytoplasmic leaflet of the ER, and its TMEM16K-dependent redistribution. Proc. Natl. Acad. Sci. USA 2019, 116, 13368–13373. [Google Scholar] [CrossRef]
- Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Sandoval-Espinosa, C.; Bershteyn, M.; Kriegstein, A.R. Expression Analysis Highlights AXL as a Candidate Zika Virus Entry Receptor in Neural Stem Cells. Cell Stem Cell 2016, 18, 591–596. [Google Scholar] [CrossRef]
- Miner, J.J.; Daniels, B.P.; Shrestha, B.; Proenca-Modena, J.L.; Lew, E.D.; Lazear, H.M.; Gorman, M.J.; Lemke, G.; Klein, R.S.; Diamond, M.S. The TAM receptor Mertk protects against neuroinvasive viral infection by maintaining blood-brain barrier integrity. Nat. Med. 2015, 21, 1464–1472. [Google Scholar] [CrossRef]
- Axelrod, H.; Pienta, K.J. Axl as a mediator of cellular growth and survival. Oncotarget 2014, 5, 8818–8852. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, M.; Knyazev, P.; Abate, A.; Reschke, M.; Maier, H.; Stefanova, N.; Knyazeva, T.; Barbieri, V.; Reindl, M.; Muigg, A.; et al. Axl and growth arrest-specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Retallack, H.; Di Lullo, E.; Arias, C.; Knopp, K.A.; Laurie, M.T.; Sandoval-Espinosa, C.; Mancia Leon, W.R.; Krencik, R.; Ullian, E.M.; Spatazza, J.; et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, O.A.; Narasipura, S.D.; Barbian, H.J.; Albalawi, Y.A.; Seaton, M.S.; Robinson, K.F.; Al-Harthi, L. beta-Catenin Restricts Zika Virus Internalization by Downregulating Axl. J. Virol. 2021, 95, e0070521. [Google Scholar] [CrossRef]
- Lagos-Cabre, R.; Alvarez, A.; Kong, M.; Burgos-Bravo, F.; Cardenas, A.; Rojas-Mancilla, E.; Perez-Nunez, R.; Herrera-Molina, R.; Rojas, F.; Schneider, P.; et al. αVβ3 Integrin regulates astrocyte reactivity. J. Neuroinflamm. 2017, 14, 194. [Google Scholar] [CrossRef]
- Milner, R.; Huang, X.; Wu, J.; Nishimura, S.; Pytela, R.; Sheppard, D.; ffrench-Constant, C. Distinct roles for astrocyte alphavbeta5 and alphavbeta8 integrins in adhesion and migration. J. Cell Sci. 1999, 112 Pt 23, 4271–4279. [Google Scholar] [CrossRef]
- Gianni, T.; Leoni, V.; Chesnokova, L.S.; Hutt-Fletcher, L.M.; Campadelli-Fiume, G. alphavbeta3-integrin is a major sensor and activator of innate immunity to herpes simplex virus-1. Proc. Natl. Acad. Sci. USA 2012, 109, 19792–19797. [Google Scholar] [CrossRef]
- Andrews, M.G.; Mukhtar, T.; Eze, U.C.; Simoneau, C.R.; Ross, J.; Parikshak, N.; Wang, S.; Zhou, L.; Koontz, M.; Velmeshev, D.; et al. Tropism of SARS-CoV-2 for human cortical astrocytes. Proc. Natl. Acad. Sci. USA 2022, 119, e2122236119. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Yang, L.; Lian, X.; Xie, Y.; Li, S.; Xin, S.; Cao, P.; Lu, J. The MERS-CoV Receptor DPP4 as a Candidate Binding Target of the SARS-CoV-2 Spike. iScience 2020, 23, 101400. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, J.; Chen, L.; Zhong, W.D.; Zhang, Z.; Mi, L.; Zhang, Y.; Liao, C.G.; Bian, H.J.; Jiang, J.L.; et al. HAb18G (CD147), a cancer-associated biomarker and its role in cancer detection. Histopathology 2009, 54, 677–687. [Google Scholar] [CrossRef]
- Kiraly, K.; Kozsurek, M.; Lukacsi, E.; Barta, B.; Alpar, A.; Balazsa, T.; Fekete, C.; Szabon, J.; Helyes, Z.; Bolcskei, K.; et al. Glial cell type-specific changes in spinal dipeptidyl peptidase 4 expression and effects of its inhibitors in inflammatory and neuropatic pain. Sci. Rep. 2018, 8, 3490. [Google Scholar] [CrossRef]
- Jin, R.; Xiao, A.Y.; Chen, R.; Granger, D.N.; Li, G. Inhibition of CD147 (Cluster of Differentiation 147) Ameliorates Acute Ischemic Stroke in Mice by Reducing Thromboinflammation. Stroke 2017, 48, 3356–3365. [Google Scholar] [CrossRef] [PubMed]
- Jorgacevski, J.; Potokar, M. Immune Functions of Astrocytes in Viral Neuroinfections. Int. J. Mol. Sci. 2023, 24, 3514. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Naghavi, M.H. Exploitation of Cytoskeletal Networks during Early Viral Infection. Trends Microbiol. 2019, 27, 39–50. [Google Scholar] [CrossRef]
- Chauhan, A.; Tikoo, A.; Patel, J.; Abdullah, A.M. HIV-1 endocytosis in astrocytes: A kiss of death or survival of the fittest? Neurosci. Res. 2014, 88, 16–22. [Google Scholar] [CrossRef]
- She, J.; Zeng, W.; Guo, J.; Chen, Q.; Bai, X.C.; Jiang, Y. Structural mechanisms of phospholipid activation of the human TPC2 channel. Elife 2019, 8, e45222. [Google Scholar] [CrossRef]
- Cang, C.; Zhou, Y.; Navarro, B.; Seo, Y.J.; Aranda, K.; Shi, L.; Battaglia-Hsu, S.; Nissim, I.; Clapham, D.E.; Ren, D. mTOR regulates lysosomal ATP-sensitive two-pore Na+ channels to adapt to metabolic state. Cell 2013, 152, 778–790. [Google Scholar] [CrossRef]
- Abboud-Jarrous, G.; Atzmon, R.; Peretz, T.; Palermo, C.; Gadea, B.B.; Joyce, J.A.; Vlodavsky, I. Cathepsin L is responsible for processing and activation of proheparanase through multiple cleavages of a linker segment. J. Biol. Chem. 2008, 283, 18167–18176. [Google Scholar] [CrossRef]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.X.; Ma, J.; Parrington, J.; Galione, A.; Evans, A.M. TPCs: Endolysosomal channels for Ca2+ mobilization from acidic organelles triggered by NAADP. FEBS Lett. 2010, 584, 1966–1974. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Wang, X.; Xu, H. Pairing phosphoinositides with calcium ions in endolysosomal dynamics: Phosphoinositides control the direction and specificity of membrane trafficking by regulating the activity of calcium channels in the endolysosomes. Bioessays 2011, 33, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Khoo, U.S.; Chan, K.Y.; Chan, V.S.; Lin, C.L. DC-SIGN and L-SIGN: The SIGNs for infection. J Mol Med. 2008, 86, 861–874. [Google Scholar] [CrossRef]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef]
- Lennemann, N.J.; Coyne, C.B. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy 2017, 13, 322–332. [Google Scholar] [CrossRef]
- Echavarria-Consuegra, L.; Smit, J.M.; Reggiori, F. Role of autophagy during the replication and pathogenesis of common mosquito-borne flavi- and alphaviruses. Open Biol. 2019, 9, 190009. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, M.L.; Zhao, J. Crosstalk between Autophagy and Type I Interferon Responses in Innate Antiviral Immunity. Viruses 2019, 11, 132. [Google Scholar] [CrossRef]
- Lindqvist, R.; Mundt, F.; Gilthorpe, J.D.; Wolfel, S.; Gekara, N.O.; Kroger, A.; Overby, A.K. Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J. Neuroinflamm. 2016, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Greber, U.F. The endoplasmic reticulum unfolded protein response—Homeostasis, cell death and evolution in virus infections. FEMS Microbiol. Rev. 2021, 45, fuab016. [Google Scholar] [CrossRef] [PubMed]
- Mehla, R.; Chauhan, A. HIV-1 differentially modulates autophagy in neurons and astrocytes. J. Neuroimmunol. 2015, 285, 106–118. [Google Scholar] [CrossRef]
- Saribas, A.S.; Khalili, K.; Sariyer, I.K. Dysregulation of autophagy by HIV-1 Nef in human astrocytes. Cell Cycle 2015, 14, 2899–2904. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Ding, C.; Sun, Y. Morphology Remodeling and Selective Autophagy of Intracellular Organelles during Viral Infections. Int. J. Mol. Sci. 2020, 21, 3689. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Kim, J.K.; Jo, E.K. Mitophagy and Innate Immunity in Infection. Mol. Cells 2020, 43, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, J.; Zhou, A.; Khan, F.A.; Hu, L.; Zhang, S. Porcine reproductive and respiratory syndrome virus triggers mitochondrial fission and mitophagy to attenuate apoptosis. Oncotarget 2016, 7, 56002–56012. [Google Scholar] [CrossRef]
- Zhu, L.; Mou, C.; Yang, X.; Lin, J.; Yang, Q. Mitophagy in TGEV infection counteracts oxidative stress and apoptosis. Oncotarget 2016, 7, 27122–27141. [Google Scholar] [CrossRef]
- Ojeda, D.S.; Grasso, D.; Urquiza, J.; Till, A.; Vaccaro, M.I.; Quarleri, J. Cell Death Is Counteracted by Mitophagy in HIV-Productively Infected Astrocytes but Is Promoted by Inflammasome Activation Among Non-productively Infected Cells. Front. Immunol. 2018, 9, 2633. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pohlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef]
- Li, J.; Gao, E.; Xu, C.; Wang, H.; Wei, Y. ER-Phagy and Microbial Infection. Front. Cell Dev. Biol. 2021, 9, 771353. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Chen, J.; Zhang, B. Critical roles of FAM134B in ER-phagy and diseases. Cell Death Dis. 2020, 11, 983. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Tsai, B. How viruses use the endoplasmic reticulum for entry, replication, and assembly. Cold Spring Harb. Perspect. Biol. 2013, 5, a013250. [Google Scholar] [CrossRef] [PubMed]
- Ralhan, I.; Chang, C.L.; Lippincott-Schwartz, J.; Ioannou, M.S. Lipid droplets in the nervous system. J. Cell Biol. 2021, 220, e202102136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Peng, X.; Yang, S.; Li, X.; Huang, M.; Wei, S.; Liu, J.; He, G.; Zheng, H.; Yang, L.; et al. The regulation, function, and role of lipophagy, a form of selective autophagy, in metabolic disorders. Cell Death Dis. 2022, 13, 132. [Google Scholar] [CrossRef]
- Smolic, T.; Tavcar, P.; Horvat, A.; Cerne, U.; Haluzan Vasle, A.; Tratnjek, L.; Kreft, M.E.; Scholz, N.; Matis, M.; Petan, T.; et al. Astrocytes in stress accumulate lipid droplets. Glia 2021, 69, 1540–1562. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef]
- Liu, L.; MacKenzie, K.R.; Putluri, N.; Maletic-Savatic, M.; Bellen, H.J. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab. 2017, 26, 719–737.e716. [Google Scholar] [CrossRef]
- Samsa, M.M.; Mondotte, J.A.; Iglesias, N.G.; Assuncao-Miranda, I.; Barbosa-Lima, G.; Da Poian, A.T.; Bozza, P.T.; Gamarnik, A.V. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009, 5, e1000632. [Google Scholar] [CrossRef]
- Monson, E.A.; Trenerry, A.M.; Laws, J.L.; Mackenzie, J.M.; Helbig, K.J. Lipid droplets and lipid mediators in viral infection and immunity. FEMS Microbiol. Rev. 2021, 45, fuaa066. [Google Scholar] [CrossRef]
- Monson, E.A.; Whelan, D.R.; Helbig, K.J. Lipid Droplet Motility Increases Following Viral Immune Stimulation. Int. J. Mol. Sci. 2021, 22, 4418. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.W. Lipophagy: Molecular Mechanisms and Implications in Metabolic Disorders. Mol. Cells 2020, 43, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Randall, G. Multifaceted roles for lipids in viral infection. Trends Microbiol. 2011, 19, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Jordan, T.X.; Randall, G. Dengue Virus Activates the AMP Kinase-mTOR Axis To Stimulate a Proviral Lipophagy. J. Virol. 2017, 91, e02020-16. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Garcia, F.J.; Perez-Hernandez, C.A.; Rodriguez-Murillo, M.; Moreno-Altamirano, M.M.B. The Role of Tricarboxylic Acid Cycle Metabolites in Viral Infections. Front. Cell Infect. Microbiol. 2021, 11, 725043. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Entry Receptor | Virus | References |
|---|---|---|
| HSPGs, αvβ3 | HSV-1 | [50] |
| ACE2 | SARS-CoV-2 | [36] |
| NRP1 | SARS-CoV-2 | [51] |
| TMPRSS2 | SARS-CoV-2 | [52] |
| AXL | ZIKV | [53] |
| α2β1 | EV1 | [54] |
| DC-SIGN | HIV-1 | [55] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potokar, M.; Zorec, R.; Jorgačevski, J. Astrocytes Are a Key Target for Neurotropic Viral Infection. Cells 2023, 12, 2307. https://doi.org/10.3390/cells12182307
Potokar M, Zorec R, Jorgačevski J. Astrocytes Are a Key Target for Neurotropic Viral Infection. Cells. 2023; 12(18):2307. https://doi.org/10.3390/cells12182307
Chicago/Turabian StylePotokar, Maja, Robert Zorec, and Jernej Jorgačevski. 2023. "Astrocytes Are a Key Target for Neurotropic Viral Infection" Cells 12, no. 18: 2307. https://doi.org/10.3390/cells12182307
APA StylePotokar, M., Zorec, R., & Jorgačevski, J. (2023). Astrocytes Are a Key Target for Neurotropic Viral Infection. Cells, 12(18), 2307. https://doi.org/10.3390/cells12182307