

Complement C3 Reduces Apoptosis via Interaction with the Intrinsic Apoptotic Pathway
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mouse Model of Myocardial I/R Injury
2.2. Comparative Proteomics
2.3. ELISA Measurement of Cytochrome c in the C3-Immunocomplex
2.4. Western Blotting of Cytochrome c
2.5. Cell Lines, Abs, Molecular Cloning Construct, and Proteins
2.6. qPCR for C3’s mRNA Expression
2.7. H2O2-Induced Apoptosis in Human Cardiomyocytes
2.8. Cell-Free Apoptosis Analyses
2.9. Pull-Down Assay to Detect Apoptotic Factor(s) Interacted with C3
2.9.1. Transfection and Overexpression of Human C3 in 293 T Cell Line
2.9.2. Apoptotic Cell Lysate Preparation
2.9.3. Identification of Apoptotic Factor(s) Binding to C3
2.10. Statistical Analysis
3. Results
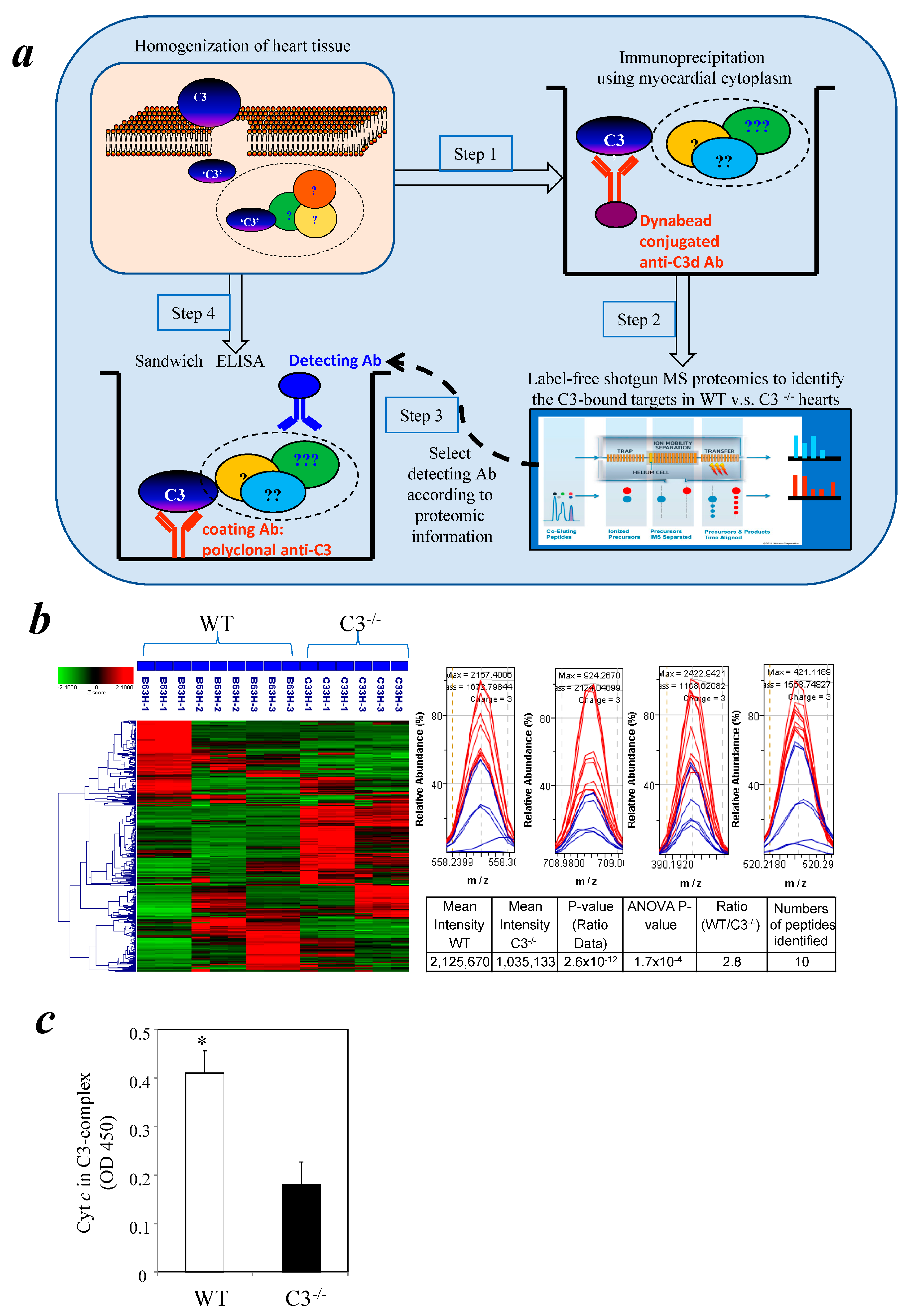
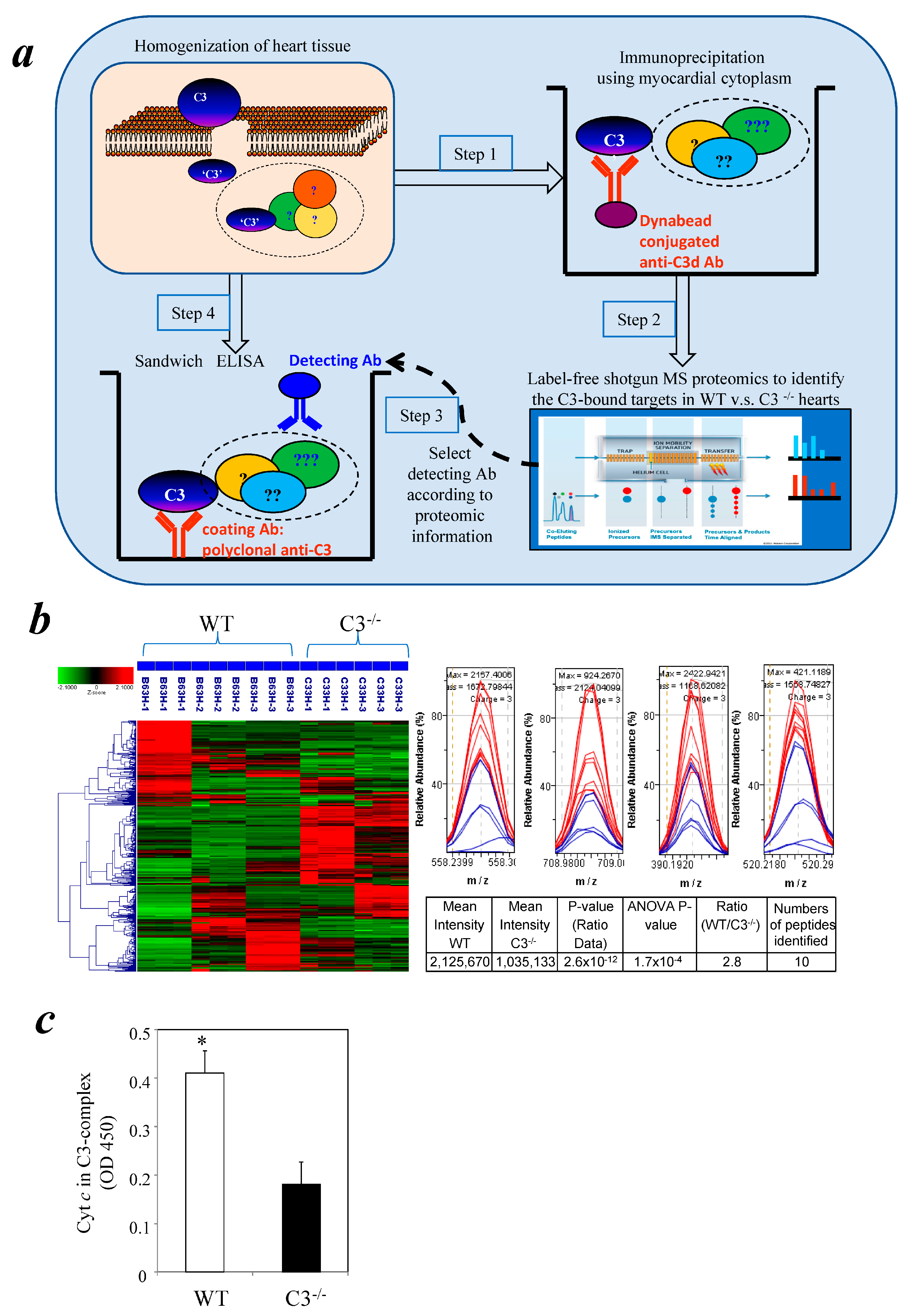
3.1. Identification of Cytochrome c in a Myocardial C3 Complex Following I/R Using Comparative Proteomics
3.2. Detection of Cytochrome c in a Cytosolic C3 Complex Following I/R by Immunoassay
3.3. Similar Levels of Cytochrome c in Myocardial Cytosol of C3−/− and WT Mice after I/R
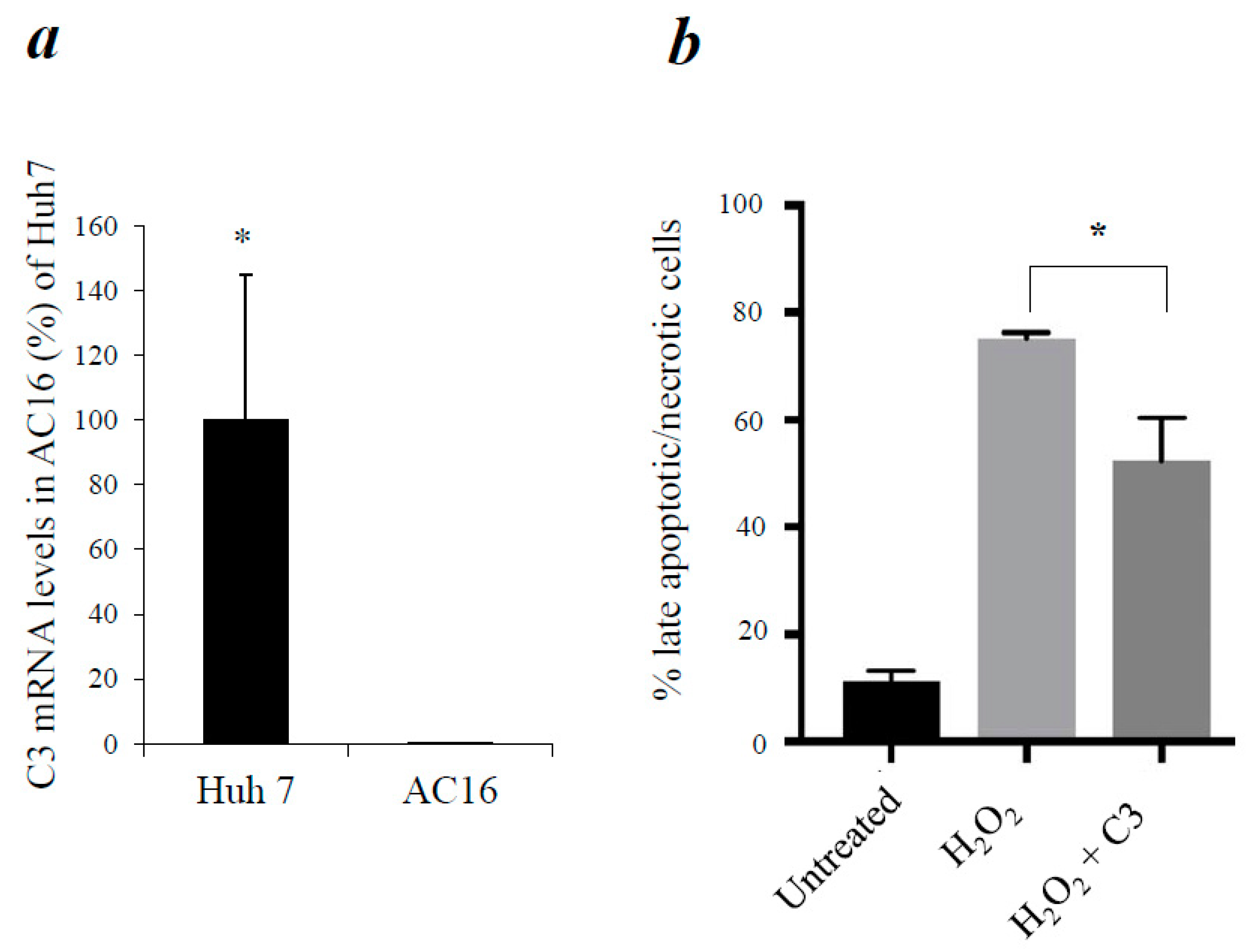
3.4. Reduction of Apoptosis in AC16 Cardiomyocytes by Exogenous C3
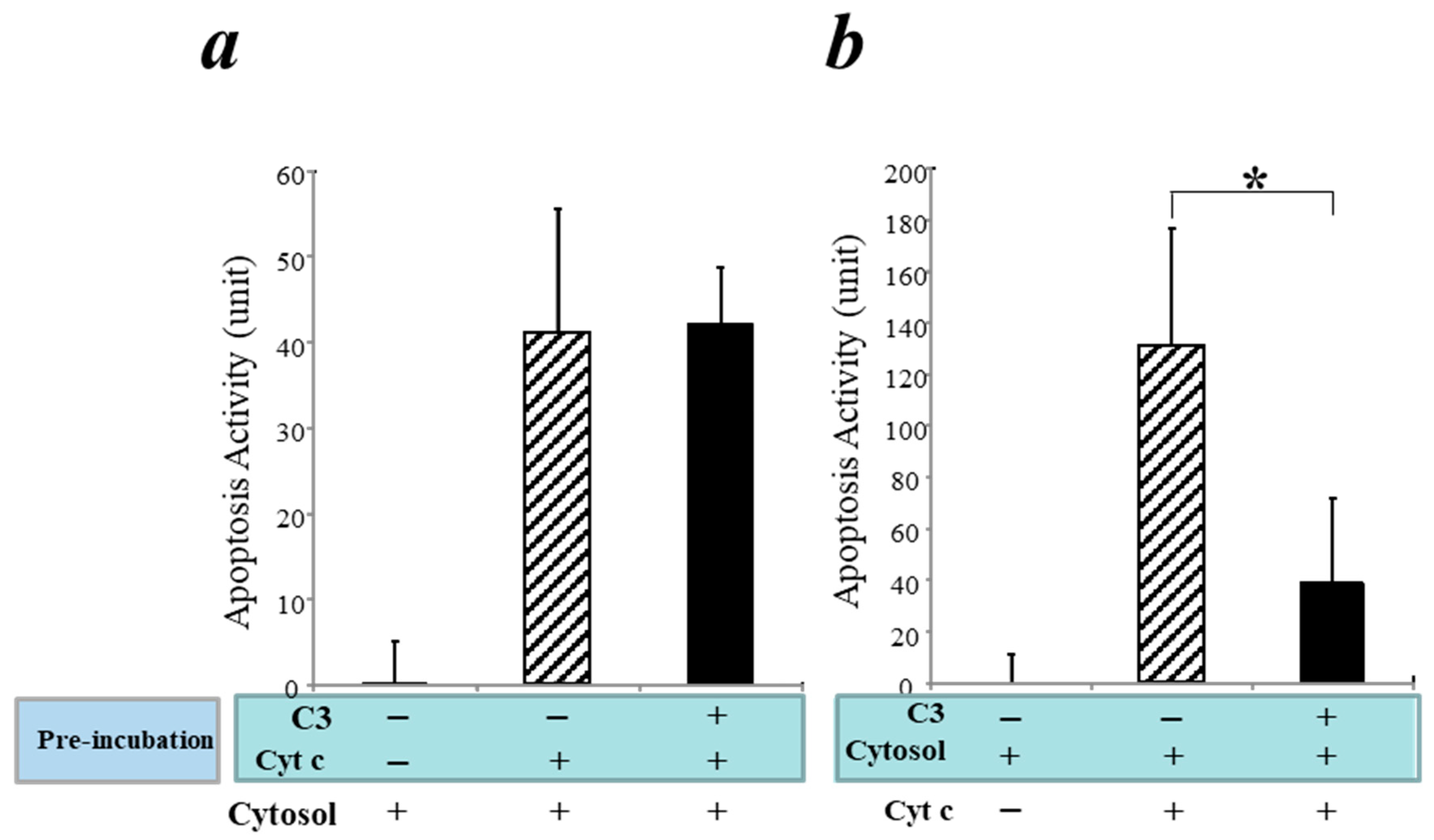
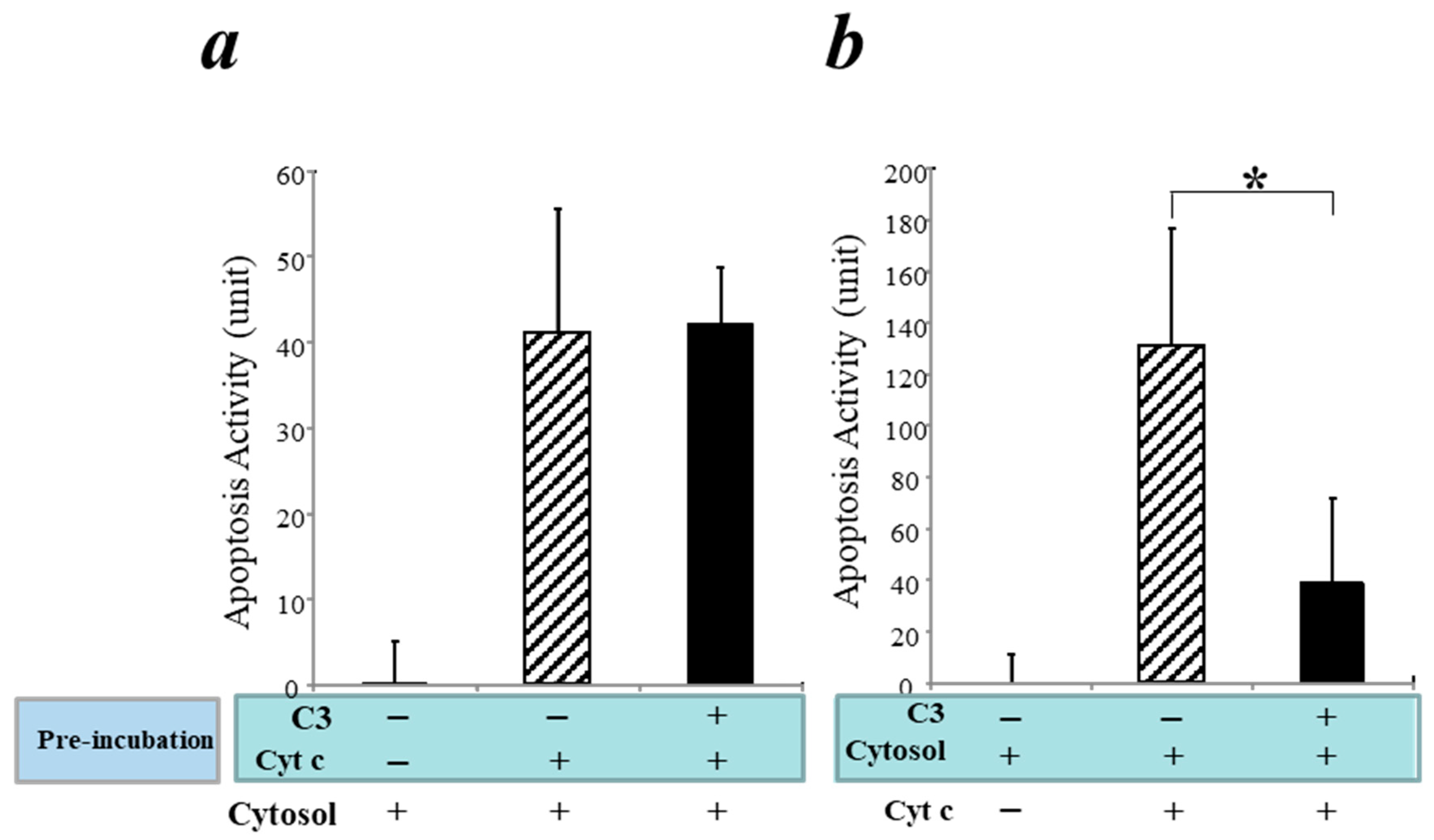
3.5. Inhibition of the Intrinsic Apoptosis Pathway by C3 in a Cell-Free System
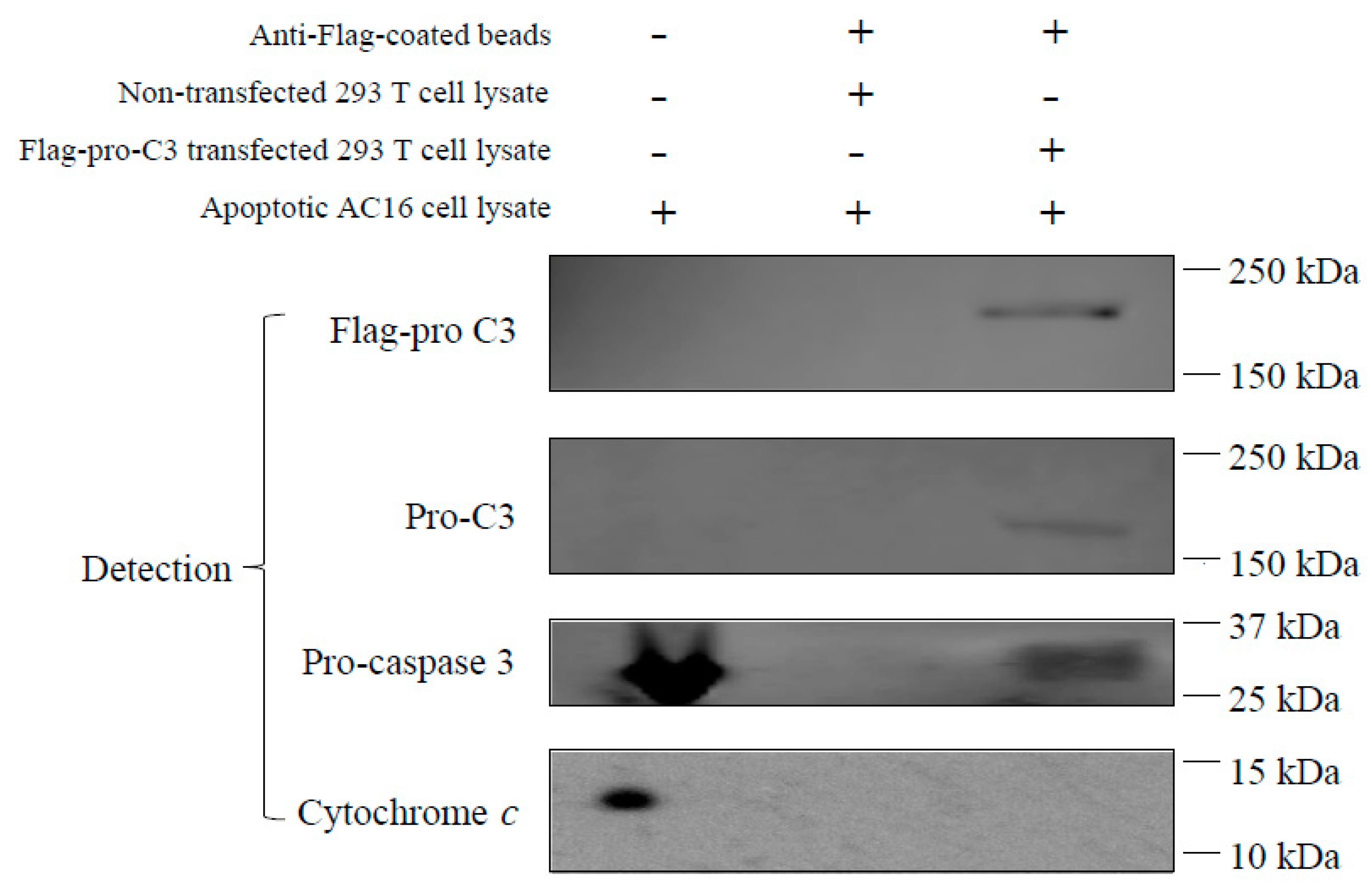
3.6. Pro-C3 Binding with Pro-Caspase 3 in a Cell-Free System
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geisbrecht, B.V.; Lambris, J.D.; Gros, P. Complement component C3: A structural perspective and potential therapeutic implications. Semin. Immunol. 2022, 59, 101627. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.C.; Isenman, D.E. Regulation of humoral immunity by complement. Immunity 2012, 37, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.V.; Sim, R.B. Complement in health and disease. Adv. Drug Deliv. Rev. 2011, 63, 965–975. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Gorsuch, W.B.; Chrysanthou, E.; Schwaeble, W.J.; Stahl, G.L. The complement system in ischemia-reperfusion injuries. Immunobiology 2012, 217, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Lambris, J.D. Progress and trends in complement therapeutics. Adv. Exp. Med. Biol. 2013, 735, 1–22. [Google Scholar]
- Sacks, S.H.; Zhou, W. The role of complement in the early immune response to transplantation. Nat. Rev. Immunol. 2012, 12, 431–442. [Google Scholar] [CrossRef]
- Sarma, J.V.; Ward, P.A. The complement system. Cell Tissue Res. 2011, 343, 227–235. [Google Scholar] [CrossRef]
- Trouw, L.A.; Daha, M.R. Role of complement in innate immunity and host defense. Immunol. Lett. 2011, 138, 35–37. [Google Scholar] [CrossRef]
- Cheng, T.H.; Puskarich, M.; Li, X.; Fang, Z.; Xu, F.; Chen, Y.; Jiang, X.C.; Worah, S.; Jones, A.E.; Zhang, M. Circulating Complement C3-Alpha Chain Levels Predict Survival of Septic Shock Patients. Shock 2020, 54, 190–197. [Google Scholar] [CrossRef]
- Carroll, M.C. The role of complement and complement receptors in induction and regulation of immunity. Annu. Rev. Immunol. 1998, 16, 545–568. [Google Scholar] [CrossRef] [PubMed]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Harboe, M.; Mollnes, T.E. The alternative complement pathway revisited. J. Cell Mol. Med. 2008, 12, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sha, Y.; Wang, H.; He, L.; Li, L.; Wen, S.; Sheng, L.; Hu, W.; Zhou, H. Intracellular C3 prevents hepatic steatosis by promoting autophagy and very-low-density lipoprotein secretion. FASEB J. 2021, 35, e22037. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Walz, T.; Springer, T.A. Structural transitions of complement component C3 and its activation products. Proc. Natl. Acad. Sci. USA 2006, 103, 19737–19742. [Google Scholar] [CrossRef]
- Brade, V.; Hall, R.E.; Colten, H.R. Biosynthesis of pro-C3, a precursor of the third component of complement. J. Exp. Med. 1977, 146, 759–765. [Google Scholar] [CrossRef]
- Bokisch, V.A.; Dierich, M.P.; Mūller-Eberhard, H.J. Third component of complement (C3): Structural properties in relation to functions. Proc. Natl. Acad. Sci. USA 1975, 72, 1989–1993. [Google Scholar] [CrossRef]
- Parkes, C.; DiScipio, R.G.; Kerr, M.A.; Prohaska, R. The separation of functionally distinct forms of the third component of human complement (C3). Biochem. J. 1981, 193, 963–970. [Google Scholar] [CrossRef]
- Fontaine, M.; Rivat, C. A study of the breakdown of the third component of human complement (C3). Ann. Immunol. 1979, 130C, 349–366. [Google Scholar]
- Minta, J.O.; Man, D.; Movat, H.Z. Kinetic studies on the fragmentation of the third component of complement (C3) by trypsin. J. Immunol. 1977, 118, 2192–2198. [Google Scholar] [CrossRef]
- Fearon, D.T.; Wong, W.W. Complement ligand-receptor interactions that mediate biological responses. Annu. Rev. Immunol. 1983, 1, 243–271. [Google Scholar] [CrossRef] [PubMed]
- Fromell, K.; Adler, A.; Åman, A.; Manivel, V.A.; Huang, S.; Dührkop, C.; Sandholm, K.; Ekdahl, K.N.; Nilsson, B. Assessment of the Role of C3(H2O) in the Alternative Pathway. Front. Immunol. 2020, 11, 530. [Google Scholar] [CrossRef] [PubMed]
- Liszewski, M.K.; Kolev, M.; Le Friec, G.; Leung, M.; Bertram, P.G.; Fara, A.F.; Subias, M.; Pickering, M.C.; Drouet, C.; Meri, S.; et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 2013, 39, 1143–1157. [Google Scholar] [CrossRef] [PubMed]
- Palikhe, A.; Sinisalo, J.; Seppänen, M.; Haario, H.; Meri, S.; Valtonen, V.; Nieminen, M.S.; Lokki, M.L. Serum complement C3/C4 ratio, a novel marker for recurrent cardiovascular events. Am. J. Cardiol. 2007, 99, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Celik, T.; Iyisoy, A.; Yuksel, U.C.; Jata, B.; Ozkan, M. The impact of admission C-reactive protein levels on the development of no-reflow phenomenon after primary PCI in patients with acute myocardial infarction: The role of inflammation. Int. J. Cardiol. 2009, 136, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Cannon, R.O., III. Mechanisms, management and future directions for reperfusion injury after acute myocardial infarction. Nat. Clin. Pract. Cardiovasc. Med. 2005, 2, 88–94. [Google Scholar] [CrossRef]
- Crawford, M.H.; Grover, F.L.; Kolb, W.P.; McMahan, C.A.; O’rourke, R.A.; McManus, L.M.; Pinckard, R.N. Complement and neutrophil activation in the pathogenesis of ischemic myocardial injury. Circulation 1988, 78, 1449–1458. [Google Scholar] [CrossRef]
- Shandelya, S.M.; Kuppusamy, P.; Herskowitz, A.; Weisfeldt, M.L.; Zweier, J.L. complement receptor type 1 inhibits the complement pathway and prevents contractile failure in the postischemic heart. Evidence that complement activation is required for neutrophil-mediated reperfusion injury. Circulation 1933, 88, 2812–2826. [Google Scholar] [CrossRef]
- Weisman, H.F.; Bartow, T.; Leppo, M.K.; Marsh, H.C., Jr.; Carson, G.R.; Concino, M.F.; Boyle, M.P.; Roux, K.H.; Weisfeldt, M.L.; Fearon, D.T. Soluble human complement receptor type 1: In vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science 1990, 249, 146–151. [Google Scholar] [CrossRef]
- Vakeva, A.P.; Agah, A.; Rollins, S.A.; Matis, L.A.; Li, L.; Stahl, G.L. Myocardial infarction and apoptosis after myocardial ischemia and reperfusion: Role of the terminal complement components and inhibition by anti-C5 therapy. Circulation 1998, 97, 2259–2267. [Google Scholar] [CrossRef]
- Ündar, A.; Eichstaedt, H.C.; Clubb, F.J., Jr.; Fung, M.; Lu, M.; E Bigley, J.; Vaughn, W.K.; Fraser, C.D., Jr. Novel anti-factor D monoclonal antibody inhibits complement and leukocyte activation in a baboon model of cardiopulmonary bypass. Ann. Thorac. Surg. 2002, 74, 355–362; discussion 362. [Google Scholar] [CrossRef] [PubMed]
- Verrier, E.D.; Shernan, S.K.; Taylor, K.M.; Van de Werf, F.; Newman, M.F.; Chen, J.C.; Carrier, M.; Haverich, A.; Malloy, K.J.; Adams, P.X.; et al. Terminal complement blockade with pexelizumab during coronary artery bypass graft surgery requiring cardiopulmonary bypass: A randomized trial. JAMA 2004, 291, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, P.W.; Granger, C.B.; Adams, P.X.; Hamm, C.; Holmes, D., Jr.; O’Neill, W.W.; Todaro, T.G.; Vahanian, A.; Van de Werf, F. Pexelizumab for acute ST-elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: A randomized controlled trial. JAMA 2007, 297, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Jordan, J.E.; Montalto, M.C.; Stahl, G.L. Inhibition of mannose-binding lectin reduces postischemic myocardial reperfusion injury. Circulation 2001, 104, 1413–1418. [Google Scholar] [CrossRef]
- Charlagorla, P.; Liu, J.; Patel, M.; Rushbrook, J.I.; Zhang, M. Loss of plasma membrane integrity, complement response and formation of reactive oxygen species during early myocardial ischemia/reperfusion. Mol. Immunol. 2013, 56, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Li, X.; Liu, J.; Lee, H.; Salciccioli, L.; Lazar, J.; Zhang, M. The role of complement C3 in the outcome of regional myocardial infarction. Biochem. Biophys. Rep. 2023, 33, 101434. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.Q.; Tian, J.; Luo, X.J.; Peng, J. Targeting the pathways of regulated necrosis: A potential strategy for alleviation of cardio-cerebrovascular injury. Cell Mol. Life Sci. 2021, 78, 63–78. [Google Scholar] [CrossRef]
- Dong, Y.; Chen, H.; Gao, J.; Liu, Y.; Li, J.; Wang, J. Molecular machinery and interplay of apoptosis and autophagy in coronary heart disease. J. Mol. Cell. Cardiol. 2019, 136, 27–41. [Google Scholar] [CrossRef]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef]
- Zhang, M.; Michael, L.H.; Grosjean, S.A.; Kelly, R.A.; Carroll, M.C.; Entman, M.L. The role of natural IgM in myocardial ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 2006, 41, 62–67. [Google Scholar] [CrossRef]
- Zhang, G.X.; Kimura, S.; Murao, K.; Obata, K.; Matsuyoshi, H.; Takaki, M. Inhibition of cytochrome c release by 10-N-nonyl acridine orange, a cardiolipin-specific dye, during myocardial ischemia-reperfusion in the rat. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H433–H439. [Google Scholar] [CrossRef] [PubMed]
- de Bruijn, M.H.; Fey, G.H. Human complement component C3: cDNA coding sequence and derived primary structure. Proc. Natl. Acad. Sci. USA 1985, 82, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, H.S.; Elvington, M.L.; Perng, Y.-C.; Liszewski, M.K.; Byers, D.E.; Farkouh, C.; Yusen, R.D.; Lenschow, D.J.; Brody, S.L.; Atkinson, J.P. Intracellular C3 Protects Human Airway Epithelial Cells from Stress-associated Cell Death. Am. J. Respir. Cell Mol. Biol. 2019, 60, 144–157. [Google Scholar] [CrossRef] [PubMed]
- McCoy, F.; Darbandi, R.; Lee, H.C.; Bharatham, K.; Moldoveanu, T.; Grace, C.R.; Dodd, K.; Lin, W.; Chen, S.I.; Tangallapally, R.P.; et al. Metabolic activation of CaMKII by coenzyme A. Mol. Cell 2013, 52, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Nutt, L.K.; Margolis, S.S.; Jensen, M.; Herman, C.E.; Dunphy, W.G.; Rathmell, J.C.; Kornbluth, S. Metabolic regulation of oocyte cell death through the CaMKII-mediated phosphorylation of caspase-2. Cell 2005, 123, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Li, F. Antibody Pull-Down Experiments in Fission Yeast. Methods Mol. Biol. 2018, 1721, 117–123. [Google Scholar] [CrossRef]
- Brymora, A.; Valova, V.A.; Robinson, P.J. Protein-protein interactions identified by pull-down experiments and mass spectrometry. Curr. Protoc. Cell Biol. 2004. Chapter 17, Unit 17.15. [Google Scholar] [CrossRef]
- Prelich, G. Gene overexpression: Uses, mechanisms, and interpretation. Genetics 2012, 190, 841–854. [Google Scholar] [CrossRef] [PubMed]
- Väkevä, A.; Morgan, B.P.; Tikkanen, I.; Helin, K.; Laurila, P.; Meri, S. Time course of complement activation and inhibitor expression after ischemic injury of rat myocardium. Am. J. Pathol. 1994, 144, 1357–1368. [Google Scholar]
- Whelan, R.S.; Kaplinskiy, V.; Kitsis, R.N. Cell death in the pathogenesis of heart disease: Mechanisms and significance. Annu. Rev. Physiol. 2010, 72, 19–44. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Wan, C.; Guo, R.; Guo, D. Is Hydrogen Peroxide a Suitable Apoptosis Inducer for All Cell Types? Biomed. Res. Int. 2016, 2016, 7343965. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Henzel, W.J.; Liu, X.; Lutschg, A.; Wang, X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell 1997, 90, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of cell death in heart disease. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Ow, Y.P.; Green, D.R.; Hao, Z.; Mak, T.W. Cytochrome c: Functions beyond respiration. Nat. Rev. Mol. Cell Biol. 2008, 9, 532–542. [Google Scholar] [CrossRef]
- Elvington, M.; Liszewski, M.K.; Bertram, P.; Kulkarni, H.S.; Atkinson, J.P. A C3(H2O) recycling pathway is a component of the intracellular complement system. J. Clin. Invest. 2017, 127, 970–981. [Google Scholar] [CrossRef]
- Kaur, G.; Tan, L.X.; Rathnasamy, G.; La Cunza, N.; Germer, C.J.; Toops, K.A.; Fernandes, M.; Blenkinsop, T.A.; Lakkaraju, A. Aberrant early endosome biogenesis mediates complement activation in the retinal pigment epithelium in models of macular degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, 9014–9019. [Google Scholar] [CrossRef]
- Jiao, H.; Provis, J.M.; Natoli, R.; Rutar, M. Ablation of C3 modulates macrophage reactivity in the outer retina during photo-oxidative damage. Mol. Vis. 2020, 26, 679–690. [Google Scholar]
- Halbgebauer, R.; Karasu, E.; Braun, C.K.; Palmer, A.; Braumüller, S.; Schultze, A.; Schäfer, F.; Bückle, S.; Eigner, A.; Wachter, U.; et al. Thirty-Eight-Negative Kinase 1 Is a Mediator of Acute Kidney Injury in Experimental and Clinical Traumatic Hemorrhagic Shock. Front. Immunol. 2020, 11, 2081. [Google Scholar] [CrossRef]
- Dos Santos, R.S.; Marroqui, L.; Grieco, F.A.; Marselli, L.; Suleiman, M.; Henz, S.R.; Marchetti, P.; Wernersson, R.; Eizirik, D.L. Protective Role of Complement C3 Against Cytokine-Mediated β-Cell Apoptosis. Endocrinology 2017, 158, 2503–2521. [Google Scholar] [CrossRef] [PubMed]
- King, B.C.; Renström, E.; Blom, A.M. Intracellular cytosolic complement component C3 regulates cytoprotective autophagy in pancreatic beta cells by interaction with ATG16L1. Autophagy 2019, 15, 919–921. [Google Scholar] [CrossRef] [PubMed]
- King, B.C.; Kulak, K.; Krus, U.; Rosberg, R.; Golec, E.; Wozniak, K.; Gomez, M.F.; Zhang, E.; O’Connell, D.J.; Renström, E.; et al. Complement Component C3 Is Highly Expressed in Human Pancreatic Islets and Prevents β Cell Death via ATG16L1 Interaction and Autophagy Regulation. Cell Metab. 2019, 29, 202–210.e206. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Zhang, J.; Qu, J.; Rao, Y.; Li, D.; Gai, X.; Chen, Y.; Liang, Y.; Sun, Y. Complement component 3 protects human bronchial epithelial cells from cigarette smoke-induced oxidative stress and prevents incessant apoptosis. Front. Immunol. 2022, 13, 1035930. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.Y.; Liang, S.J.; Xu, F.; Yang, Y.; Feng, J.L.; Shen, F.; Zhong, Y.; Wu, S.; Shu, Y.; Sun, D.D.; et al. Complement component 3 prevents imiquimod-induced psoriatic skin inflammation by inhibiting apoptosis in mice. Int. Immunopharmacol. 2020, 85, 106692. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.M.; Tu, V.C. Apoptosis and heart failure: Mechanisms and therapeutic implications. Am. J. Cardiovasc. Drugs 2002, 2, 43–57. [Google Scholar] [CrossRef]
- Adjedj, J.; Muller, O.; Eeckhout, E.; Watson, T.J.; Ong, P.J.; Tcheng, J.E. A Handbook of Primary PCI: No-Reflow Management. In Primary Angioplasty: A Practical Guide; Springer: Singapore, 2018; Chapter 17. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, Z.; Lee, H.; Liu, J.; Wong, K.A.; Brown, L.M.; Li, X.; Xiaoli, A.M.; Yang, F.; Zhang, M. Complement C3 Reduces Apoptosis via Interaction with the Intrinsic Apoptotic Pathway. Cells 2023, 12, 2282. https://doi.org/10.3390/cells12182282
Fang Z, Lee H, Liu J, Wong KA, Brown LM, Li X, Xiaoli AM, Yang F, Zhang M. Complement C3 Reduces Apoptosis via Interaction with the Intrinsic Apoptotic Pathway. Cells. 2023; 12(18):2282. https://doi.org/10.3390/cells12182282
Chicago/Turabian StyleFang, Zhou, Haekyung Lee, Junying Liu, Karen A. Wong, Lewis M. Brown, Xiang Li, Alus M. Xiaoli, Fajun Yang, and Ming Zhang. 2023. "Complement C3 Reduces Apoptosis via Interaction with the Intrinsic Apoptotic Pathway" Cells 12, no. 18: 2282. https://doi.org/10.3390/cells12182282
APA StyleFang, Z., Lee, H., Liu, J., Wong, K. A., Brown, L. M., Li, X., Xiaoli, A. M., Yang, F., & Zhang, M. (2023). Complement C3 Reduces Apoptosis via Interaction with the Intrinsic Apoptotic Pathway. Cells, 12(18), 2282. https://doi.org/10.3390/cells12182282