
Canagliflozin, an Inhibitor of the Na+-Coupled D-Glucose Cotransporter, SGLT2, Inhibits Astrocyte Swelling and Brain Swelling in Cerebral Ischemia

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
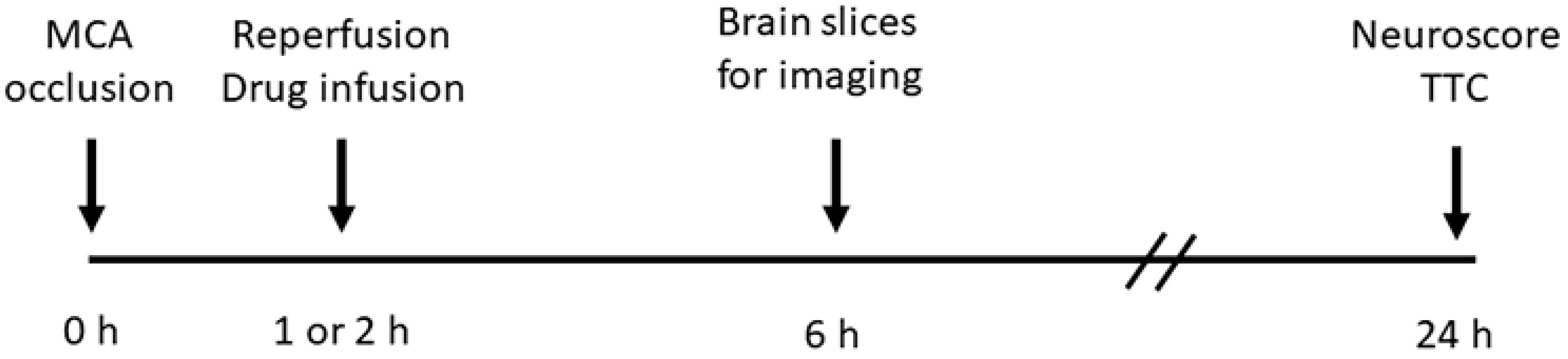
2. Materials and Methods
3. Results
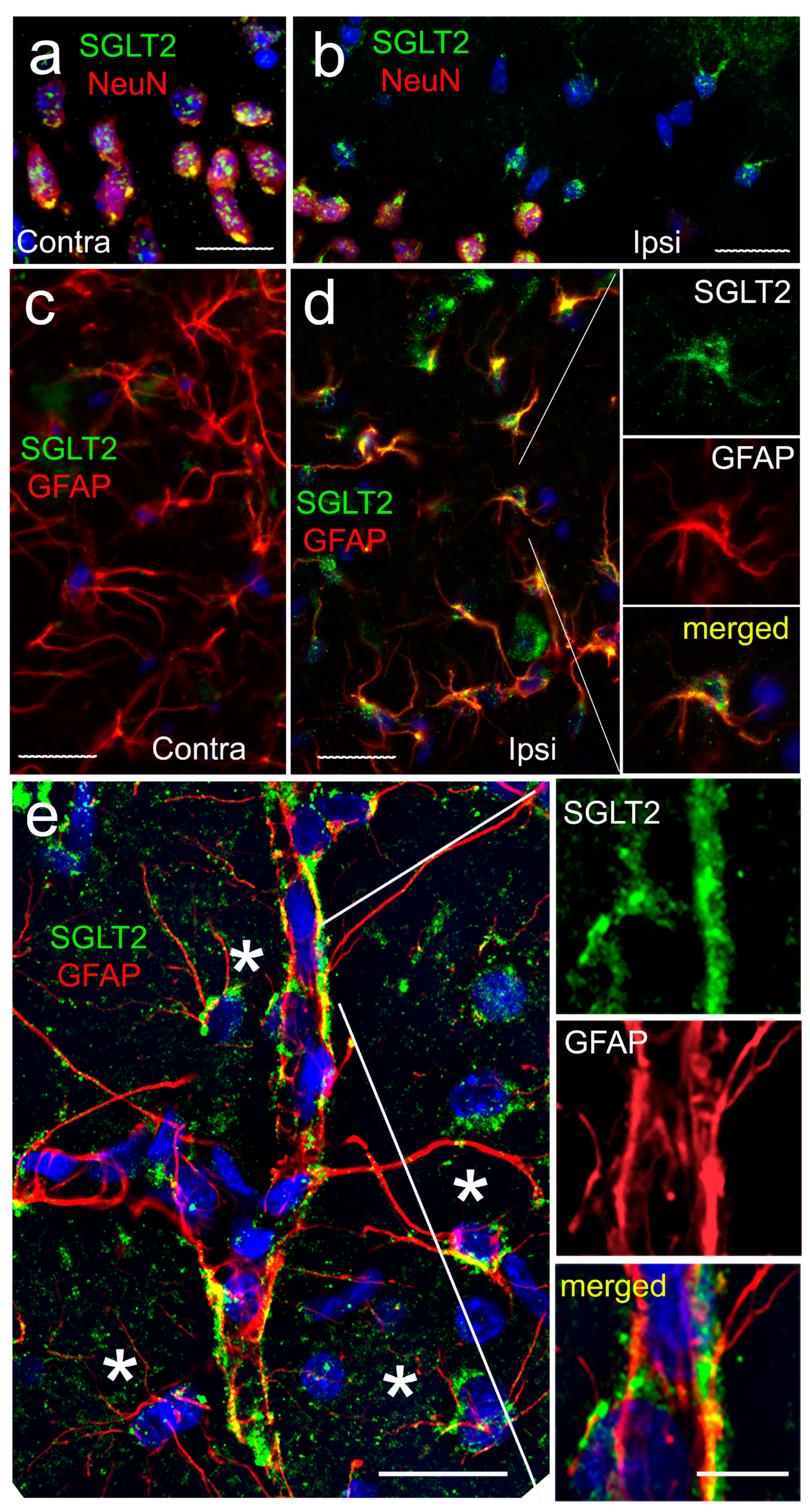
3.1. SGLT2 Expression
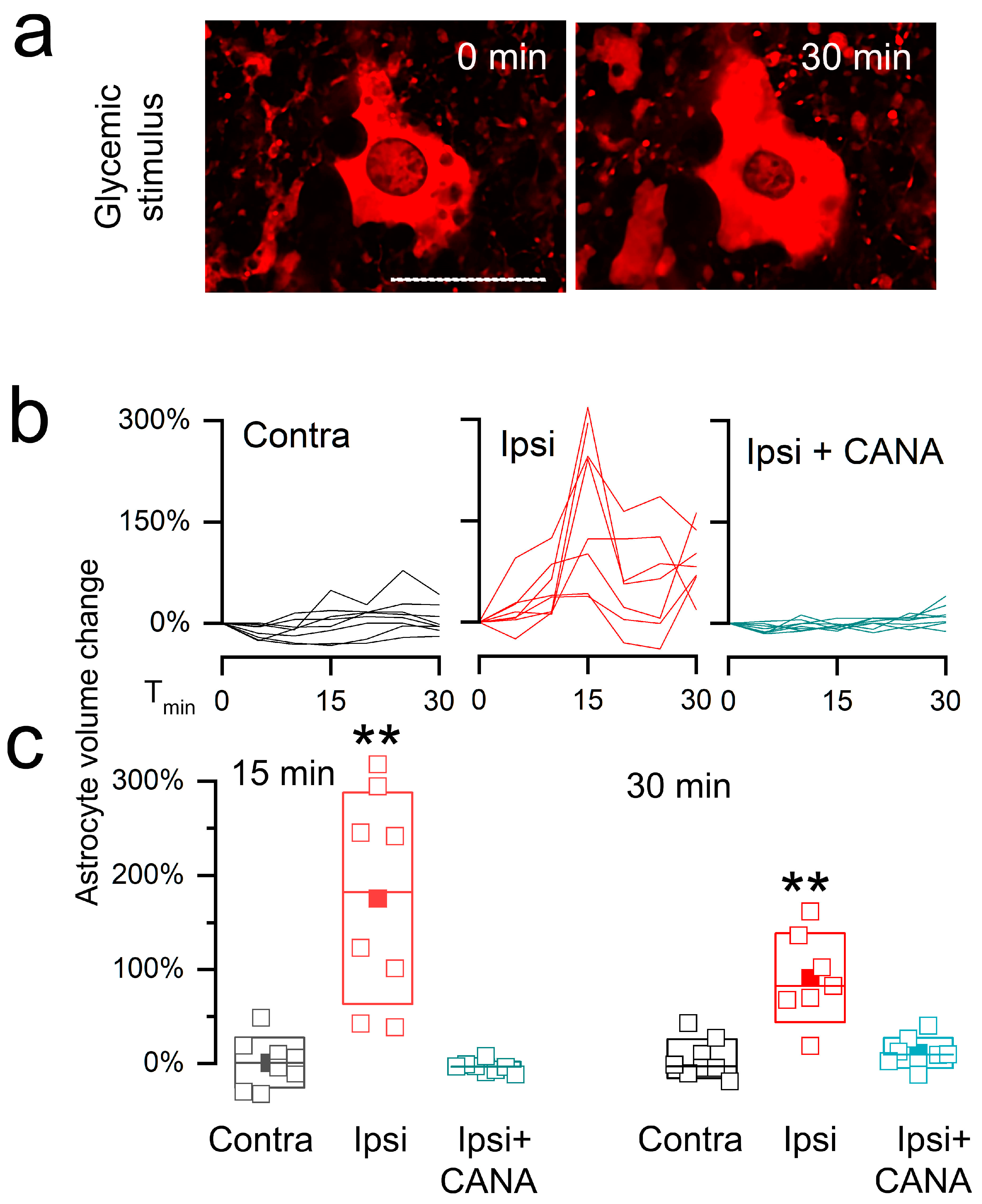
3.2. Live Cell Imaging of Astrocytes in Post-MCAo/R Brain Slices
3.2.1. Na+ Imaging
3.2.2. Cell Swelling
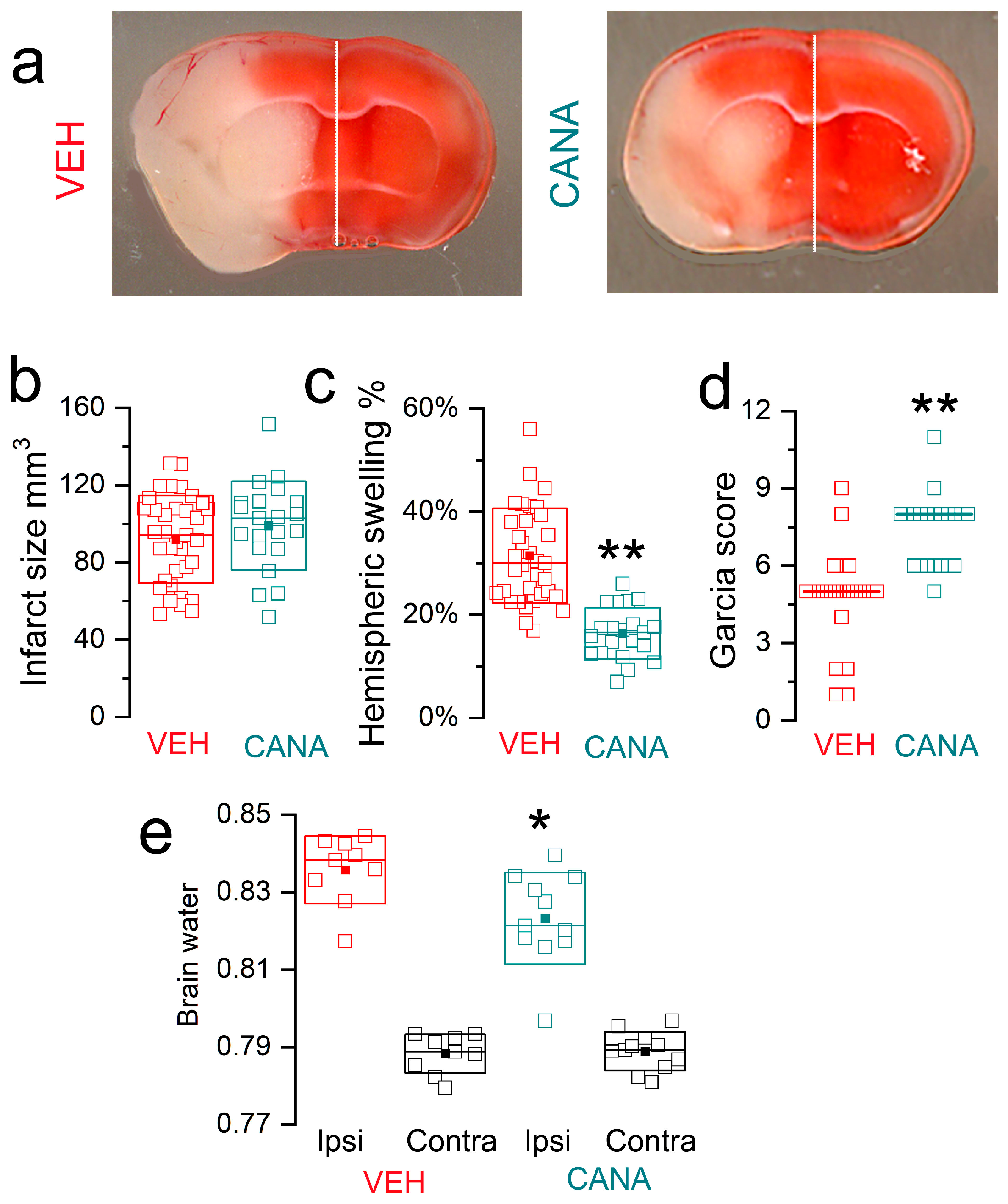
3.3. Canagliflozin in MCAo/R Models
3.3.1. MCAo/R (1/24 h) in T2DM Mice
3.3.2. MCAo/R (1/24 h) in Non-Diabetic Mice
3.3.3. MCAo/R (2/24 h) in Non-Diabetic Mice
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Katan, M.; Luft, A. Global Burden of Stroke. Semin. Neurol. 2018, 38, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Battey, T.W.; Karki, M.; Singhal, A.B.; Wu, O.; Sadaghiani, S.; Campbell, B.C.; Davis, S.M.; Donnan, G.A.; Sheth, K.N.; Kimberly, W.T. Brain edema predicts outcome after nonlacunar ischemic stroke. Stroke 2014, 45, 3643–3648. [Google Scholar] [CrossRef] [PubMed]
- Arch, A.E.; Sheth, K.N. Malignant cerebral edema after large anterior circulation infarction: A review. Curr. Treat. Options Cardiovasc. Med. 2014, 16, 275. [Google Scholar] [CrossRef] [PubMed]
- Kurland, D.B.; Khaladj-Ghom, A.; Stokum, J.A.; Carusillo, B.; Karimy, J.K.; Gerzanich, V.; Sahuquillo, J.; Simard, J.M. Complications Associated with Decompressive Craniectomy: A Systematic Review. Neurocrit. Care 2015, 23, 292–304. [Google Scholar] [CrossRef]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflugers. Arch. 2020, 472, 1299–1343. [Google Scholar] [CrossRef]
- Wright, E.M.; Loo, D.D.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef]
- Poppe, R.; Karbach, U.; Gambaryan, S.; Wiesinger, H.; Lutzenburg, M.; Kraemer, M.; Witte, O.W.; Koepsell, H. Expression of the Na+-D-glucose cotransporter SGLT1 in neurons. J. Neurochem. 1997, 69, 84–94. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Harada, S.; Tokuyama, S. Post-ischemic hyperglycemia exacerbates the development of cerebral ischemic neuronal damage through the cerebral sodium-glucose transporter. Brain Res. 2012, 1489, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Ogihara, S.; Harada, S.; Tokuyama, S. Activation of cerebral sodium-glucose transporter type 1 function mediated by post-ischemic hyperglycemia exacerbates the development of cerebral ischemia. Neuroscience 2015, 310, 674–685. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Harada, S.; Wada, T.; Hagiwara, T.; Yoshida, S.; Tokuyama, S. Sodium influx through cerebral sodium-glucose transporter type 1 exacerbates the development of cerebral ischemic neuronal damage. Eur. J. Pharmacol. 2017, 799, 103–110. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Arita, K.; Harada, S.; Tokuyama, S. Activation of c-Jun N-terminal kinase and p38 after cerebral ischemia upregulates cerebral sodium-glucose transporter type 1. J. Pharmacol. Sci. 2018, 138, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.P. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat. Med. 2011, 17, 439–447. [Google Scholar] [CrossRef]
- Lemale, C.L.; Luckl, J.; Horst, V.; Reiffurth, C.; Major, S.; Hecht, N.; Woitzik, J.; Dreier, J.P. Migraine Aura, Transient Ischemic Attacks, Stroke, and Dying of the Brain Share the Same Key Pathophysiological Process in Neurons Driven by Gibbs-Donnan Forces, Namely Spreading Depolarization. Front. Cell Neurosci. 2022, 16, 837650. [Google Scholar] [CrossRef] [PubMed]
- Mascolo, A.; Scavone, C.; Scisciola, L.; Chiodini, P.; Capuano, A.; Paolisso, G. SGLT-2 inhibitors reduce the risk of cerebrovascular/cardiovascular outcomes and mortality: A systematic review and meta-analysis of retrospective cohort studies. Pharmacol. Res. 2021, 172, 105836. [Google Scholar] [CrossRef] [PubMed]
- Grempler, R.; Thomas, L.; Eckhardt, M.; Himmelsbach, F.; Sauer, A.; Sharp, D.E.; Bakker, R.A.; Mark, M.; Klein, T.; Eickelmann, P. Empagliflozin, a novel selective sodium glucose cotransporter-2 (SGLT-2) inhibitor: Characterisation and comparison with other SGLT-2 inhibitors. Diabetes Obes. Metab. 2012, 14, 83–90. [Google Scholar] [CrossRef]
- Kakinuma, H.; Oi, T.; Hashimoto-Tsuchiya, Y.; Arai, M.; Kawakita, Y.; Fukasawa, Y.; Iida, I.; Hagima, N.; Takeuchi, H.; Chino, Y.; et al. (1S)-1,5-anhydro-1-[5-(4-ethoxybenzyl)-2-methoxy-4-methylphenyl]-1-thio-D-glucitol (TS-071) is a potent, selective sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for type 2 diabetes treatment. J. Med. Chem. 2010, 53, 3247–3261. [Google Scholar] [CrossRef]
- Abdel-Latif, R.G.; Rifaai, R.A.; Amin, E.F. Empagliflozin alleviates neuronal apoptosis induced by cerebral ischemia/reperfusion injury through HIF-1alpha/VEGF signaling pathway. Arch. Pharm. Res. 2020, 43, 514–525. [Google Scholar] [CrossRef]
- Amin, E.F.; Rifaai, R.A.; Abdel-Latif, R.G. Empagliflozin attenuates transient cerebral ischemia/reperfusion injury in hyperglycemic rats via repressing oxidative-inflammatory-apoptotic pathway. Fundam. Clin. Pharmacol. 2020, 34, 548–558. [Google Scholar] [CrossRef]
- Takashima, M.; Nakamura, K.; Kiyohara, T.; Wakisaka, Y.; Hidaka, M.; Takaki, H.; Yamanaka, K.; Shibahara, T.; Wakisaka, M.; Ago, T.; et al. Low-dose sodium-glucose cotransporter 2 inhibitor ameliorates ischemic brain injury in mice through pericyte protection without glucose-lowering effects. Commun. Biol. 2022, 5, 653. [Google Scholar] [CrossRef]
- Vallon, V. The mechanisms and therapeutic potential of SGLT2 inhibitors in diabetes mellitus. Annu. Rev. Med. 2015, 66, 255–270. [Google Scholar] [CrossRef]
- Bruno, A.; Williams, L.S.; Kent, T.A. How important is hyperglycemia during acute brain infarction? Neurologist 2004, 10, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Mandava, P.; Martini, S.R.; Munoz, M.; Dalmeida, W.; Sarma, A.K.; Anderson, J.A.; Fabian, R.H.; Kent, T.A. Hyperglycemia worsens outcome after rt-PA primarily in the large-vessel occlusive stroke subtype. Transl. Stroke Res. 2014, 5, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Cannarsa, G.J.; Wessell, A.P.; Chryssikos, T.; Stokum, J.A.; Kim, K.; De Paula Carvalho, H.; Miller, T.R.; Morris, N.; Badjatia, N.; Chaturvedi, S.; et al. Initial Stress Hyperglycemia Is Associated with Malignant Cerebral Edema, Hemorrhage, and Poor Functional Outcome After Mechanical Thrombectomy. Neurosurgery 2022, 90, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Terajima, T.; Ogata, T.; Ueno, K.; Hashimoto, N.; Ono, K.; Yano, S. Establishment and pathophysiological characterization of type 2 diabetic mouse model produced by streptozotocin and nicotinamide. Biol. Pharm. Bull. 2006, 29, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Zuloaga, K.L.; Krasnow, S.M.; Zhu, X.; Zhang, W.; Jouihan, S.A.; Shangraw, R.E.; Alkayed, N.J.; Marks, D.L. Mechanism of protection by soluble epoxide hydrolase inhibition in type 2 diabetic stroke. PLoS ONE 2014, 9, e97529. [Google Scholar] [CrossRef]
- Schreiner, B.; Romanelli, E.; Liberski, P.; Ingold-Heppner, B.; Sobottka-Brillout, B.; Hartwig, T.; Chandrasekar, V.; Johannssen, H.; Zeilhofer, H.U.; Aguzzi, A.; et al. Astrocyte Depletion Impairs Redox Homeostasis and Triggers Neuronal Loss in the Adult CNS. Cell Rep. 2015, 12, 1377–1384. [Google Scholar] [CrossRef]
- Bertrand, L.; Dygert, L.; Toborek, M. Induction of Ischemic Stroke and Ischemia-reperfusion in Mice Using the Middle Artery Occlusion Technique and Visualization of Infarct Area. J. Vis. Exp. 2017, 120, e54805. [Google Scholar] [CrossRef]
- Stokum, J.A.; Shim, B.; Negoita, S.; Tsymbalyuk, N.; Tsymbalyuk, O.; Ivanova, S.; Keledjian, K.; Bryan, J.; Blaustein, M.P.; Jha, R.M.; et al. Cation flux through SUR1-TRPM4 and NCX1 in astrocyte endfeet induces water influx through AQP4 and brain swelling after ischemic stroke. Sci. Signal. 2023, 16, eadd6364. [Google Scholar] [CrossRef]
- Devineni, D.; Murphy, J.; Wang, S.S.; Stieltjes, H.; Rothenberg, P.; Scheers, E.; Mamidi, R.N. Absolute oral bioavailability and pharmacokinetics of canagliflozin: A microdose study in healthy participants. Clin. Pharmacol. Drug Dev. 2015, 4, 295–304. [Google Scholar] [CrossRef]
- Tahara, A.; Takasu, T.; Yokono, M.; Imamura, M.; Kurosaki, E. Characterization and comparison of sodium-glucose cotransporter 2 inhibitors in pharmacokinetics, pharmacodynamics, and pharmacologic effects. J. Pharmacol. Sci. 2016, 130, 159–169. [Google Scholar] [CrossRef]
- Liang, Y.; Arakawa, K.; Ueta, K.; Matsushita, Y.; Kuriyama, C.; Martin, T.; Du, F.; Liu, Y.; Xu, J.; Conway, B.; et al. Effect of canagliflozin on renal threshold for glucose, glycemia, and body weight in normal and diabetic animal models. PLoS ONE 2012, 7, e30555. [Google Scholar] [CrossRef]
- Shimamura, N.; Matchett, G.; Tsubokawa, T.; Ohkuma, H.; Zhang, J. Comparison of silicon-coated nylon suture to plain nylon suture in the rat middle cerebral artery occlusion model. J. Neurosci. Methods 2006, 156, 161–165. [Google Scholar] [CrossRef]
- Friedlander, F.; Bohmann, F.; Brunkhorst, M.; Chae, J.H.; Devraj, K.; Kohler, Y.; Kraft, P.; Kuhn, H.; Lucaciu, A.; Luger, S.; et al. Reliability of infarct volumetry: Its relevance and the improvement by a software-assisted approach. J. Cereb. Blood Flow. Metab. 2017, 37, 3015–3026. [Google Scholar] [CrossRef] [PubMed]
- Keep, R.F.; Hua, Y.; Xi, G. Brain water content. A misunderstood measurement? Transl. Stroke Res. 2012, 3, 263–265. [Google Scholar] [CrossRef]
- Chiba, Y.; Sugiyama, Y.; Nishi, N.; Nonaka, W.; Murakami, R.; Ueno, M. Sodium/glucose cotransporter 2 is expressed in choroid plexus epithelial cells and ependymal cells in human and mouse brains. Neuropathology 2020, 40, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, C.; Calmettes, G.; Morand, P.; Ribalet, B.; John, S. Real-time imaging of sodium glucose transporter (SGLT1) trafficking and activity in single cells. Physiol. Rep. 2017, 5, e13062. [Google Scholar] [CrossRef] [PubMed]
- Tsymbalyuk, O.; Gerzanich, V.; Mumtaz, A.; Andhavarapu, S.; Ivanova, S.; Makar, T.K.; Sansur, C.A.; Keller, A.; Nakamura, Y.; Bryan, J.; et al. SUR1, newly expressed in astrocytes, mediates neuropathic pain in a mouse model of peripheral nerve injury. Mol. Pain 2021, 17, 6603. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.A.; Shim, B.; Huang, W.; Kane, M.; Smith, J.A.; Gerzanich, V.; Simard, J.M. A large portion of the astrocyte proteome is dedicated to perivascular endfeet, including critical components of the electron transport chain. J. Cereb. Blood Flow. Metab. 2021, 41, 2546–2560. [Google Scholar] [CrossRef]
- Kurland, D.B.; Gerzanich, V.; Karimy, J.K.; Woo, S.K.; Vennekens, R.; Freichel, M.; Nilius, B.; Bryan, J.; Simard, J.M. The Sur1-Trpm4 channel regulates NOS2 transcription in TLR4-activated microglia. J. Neuroinflamm. 2016, 13, 130. [Google Scholar] [CrossRef]
- Meyer, J.; Gerkau, N.J.; Kafitz, K.W.; Patting, M.; Jolmes, F.; Henneberger, C.; Rose, C.R. Rapid Fluorescence Lifetime Imaging Reveals That TRPV4 Channels Promote Dysregulation of Neuronal Na(+) in Ischemia. J. Neurosci. 2022, 42, 552–566. [Google Scholar] [CrossRef]
- Shigetomi, E.; Bushong, E.A.; Haustein, M.D.; Tong, X.; Jackson-Weaver, O.; Kracun, S.; Xu, J.; Sofroniew, M.V.; Ellisman, M.H.; Khakh, B.S. Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J. Gen. Physiol. 2013, 141, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Wen, S.; Gong, M.; Yuan, X.; Xu, D.; Wang, C.; Jin, J.; Zhou, L. Dapagliflozin Activates Neurons in the Central Nervous System and Regulates Cardiovascular Activity by Inhibiting SGLT-2 in Mice. Diabetes Metab. Syndr. Obes. 2020, 13, 2781–2799. [Google Scholar] [CrossRef] [PubMed]
- Stokum, J.A.; Mehta, R.I.; Ivanova, S.; Yu, E.; Gerzanich, V.; Simard, J.M. Heterogeneity of aquaporin-4 localization and expression after focal cerebral ischemia underlies differences in white versus grey matter swelling. Acta Neuropathol. Commun. 2015, 3, 61. [Google Scholar] [CrossRef] [PubMed]
- Finan, J.D.; Guilak, F. The effects of osmotic stress on the structure and function of the cell nucleus. J. Cell Biochem. 2010, 109, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Loots, T. Experimental rodent models of type 2 diabetes: A review. Methods Find. Exp. Clin. Pharmacol. 2009, 31, 249–261. [Google Scholar] [CrossRef]
- Kusakabe, T.; Tanioka, H.; Ebihara, K.; Hirata, M.; Miyamoto, L.; Miyanaga, F.; Hige, H.; Aotani, D.; Fujisawa, T.; Masuzaki, H.; et al. Beneficial effects of leptin on glycaemic and lipid control in a mouse model of type 2 diabetes with increased adiposity induced by streptozotocin and a high-fat diet. Diabetologia 2009, 52, 675–683. [Google Scholar] [CrossRef]
- Glastras, S.J.; Chen, H.; Teh, R.; McGrath, R.T.; Chen, J.; Pollock, C.A.; Wong, M.G.; Saad, S. Mouse Models of Diabetes, Obesity and Related Kidney Disease. PLoS ONE 2016, 11, e0162131. [Google Scholar] [CrossRef]
- Cividini, F.; Scott, B.T.; Suarez, J.; Casteel, D.E.; Heinz, S.; Dai, A.; Diemer, T.; Suarez, J.A.; Benner, C.W.; Ghassemian, M.; et al. Ncor2/PPARalpha-Dependent Upregulation of MCUb in the Type 2 Diabetic Heart Impacts Cardiac Metabolic Flexibility and Function. Diabetes 2021, 70, 665–679. [Google Scholar] [CrossRef]
- Vemula, S.; Roder, K.E.; Yang, T.; Bhat, G.J.; Thekkumkara, T.J.; Abbruscato, T.J. A functional role for sodium-dependent glucose transport across the blood-brain barrier during oxygen glucose deprivation. J. Pharmacol. Exp. Ther. 2009, 328, 487–495. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Harada, S.; Tokuyama, S. Relationship between cerebral sodium-glucose transporter and hyperglycemia in cerebral ischemia. Neurosci. Lett. 2015, 604, 134–139. [Google Scholar] [CrossRef]
- Cinti, F.; Moffa, S.; Impronta, F.; Cefalo, C.M.; Sun, V.A.; Sorice, G.P.; Mezza, T.; Giaccari, A. Spotlight on ertugliflozin and its potential in the treatment of type 2 diabetes: Evidence to date. Drug. Des. Devel. Ther. 2017, 11, 2905–2919. [Google Scholar] [CrossRef]
- Kondo, T.; Reaume, A.G.; Huang, T.T.; Carlson, E.; Murakami, K.; Chen, S.F.; Hoffman, E.K.; Scott, R.W.; Epstein, C.J.; Chan, P.H. Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J. Neurosci. 1997, 17, 4180–4189. [Google Scholar] [CrossRef] [PubMed]
- Park, C.K.; Jun, S.S.; Cho, S.H.; Kang, J.K. Assessment of the relationship between ischemic damage and brain swelling in frozen brain slices. Acta. Neurochir. Suppl. 1997, 70, 17–19. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.C.; Yassi, N.; Sharma, G.; Brown, S.B.; Goyal, M.; Majoie, C.; Jovin, T.G.; Hill, M.D.; Muir, K.W.; Saver, J.L.; et al. Cerebral Edema in Patients with Large Hemispheric Infarct Undergoing Reperfusion Treatment: A HERMES Meta-Analysis. Stroke 2021, 52, 3450–3458. [Google Scholar] [CrossRef] [PubMed]
- Oerter, S.; Forster, C.; Bohnert, M. Validation of sodium/glucose cotransporter proteins in human brain as a potential marker for temporal narrowing of the trauma formation. Int. J. Legal Med. 2019, 133, 1107–1114. [Google Scholar] [CrossRef]
- Cianciolo, G.; De Pascalis, A.; Gasperoni, L.; Tondolo, F.; Zappulo, F.; Capelli, I.; Cappuccilli, M.; La Manna, G. The Off-Target Effects, Electrolyte and Mineral Disorders of SGLT2i. Molecules 2020, 25, 2757. [Google Scholar] [CrossRef]
- Chen, S.; Coronel, R.; Hollmann, M.W.; Weber, N.C.; Zuurbier, C.J. Direct cardiac effects of SGLT2 inhibitors. Cardiovasc. Diabetol. 2022, 21, 45. [Google Scholar] [CrossRef]
- Begum, G.; Song, S.; Wang, S.; Zhao, H.; Bhuiyan, M.I.H.; Li, E.; Nepomuceno, R.; Ye, Q.; Sun, M.; Calderon, M.J.; et al. Selective knockout of astrocytic Na(+) /H(+) exchanger isoform 1 reduces astrogliosis, BBB damage, infarction, and improves neurological function after ischemic stroke. Glia 2018, 66, 126–144. [Google Scholar] [CrossRef]
- Chung, Y.J.; Park, K.C.; Tokar, S.; Eykyn, T.R.; Fuller, W.; Pavlovic, D.; Swietach, P.; Shattock, M.J. Off-target effects of sodium-glucose co-transporter 2 blockers: Empagliflozin does not inhibit Na+/H+ exchanger-1 or lower [Na+]i in the heart. Cardiovasc. Res. 2021, 117, 2794–2806. [Google Scholar] [CrossRef]
- Sabolic, I.; Vrhovac, I.; Eror, D.B.; Gerasimova, M.; Rose, M.; Breljak, D.; Ljubojevic, M.; Brzica, H.; Sebastiani, A.; Thal, S.C.; et al. Expression of Na+-D-glucose cotransporter SGLT2 in rodents is kidney-specific and exhibits sex and species differences. Am. J. Physiol. Cell Physiol. 2012, 302, C1174–C1188. [Google Scholar] [CrossRef]
- Jayarathne, H.S.M.; Debarba, L.K.; Jaboro, J.J.; Ginsburg, B.C.; Miller, R.A.; Sadagurski, M. Neuroprotective effects of Canagliflozin: Lessons from aged genetically diverse UM-HET3 mice. Aging Cell 2022, 21, e13653. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.E.; Ahuja, T.; Bridgeman, M.B. Renal and Cardiac Implications of Sodium Glucose Cotransporter 2 (SGLT2) Inhibitors: The State of the Science. Ann. Pharmacother. 2018, 52, 1238–1249. [Google Scholar] [CrossRef] [PubMed]
- Wicinski, M.; Wodkiewicz, E.; Gorski, K.; Walczak, M.; Malinowski, B. Perspective of SGLT2 Inhibition in Treatment of Conditions Connected to Neuronal Loss: Focus on Alzheimer’s Disease and Ischemia-Related Brain Injury. Pharmaceuticals 2020, 13, 379. [Google Scholar] [CrossRef] [PubMed]
- Nakhal, M.M.; Aburuz, S.; Sadek, B.; Akour, A. Repurposing SGLT2 Inhibitors for Neurological Disorders: A Focus on the Autism Spectrum Disorder. Molecules 2022, 27, 7174. [Google Scholar] [CrossRef]
- Youssef, M.E.; Yahya, G.; Popoviciu, M.S.; Cavalu, S.; Abd-Eldayem, M.A.; Saber, S. Unlocking the Full Potential of SGLT2 Inhibitors: Expanding Applications beyond Glycemic Control. Int. J. Mol. Sci. 2023, 24, 6039. [Google Scholar] [CrossRef]
- Al Hamed, F.A.; Elewa, H. Potential Therapeutic Effects of Sodium Glucose-linked Cotransporter 2 Inhibitors in Stroke. Clin. Ther. 2020, 42, e242–e249. [Google Scholar] [CrossRef]
- Pawlos, A.; Broncel, M.; Wozniak, E.; Gorzelak-Pabis, P. Neuroprotective Effect of SGLT2 Inhibitors. Molecules 2021, 26, 7213. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shim, B.; Stokum, J.A.; Moyer, M.; Tsymbalyuk, N.; Tsymbalyuk, O.; Keledjian, K.; Ivanova, S.; Tosun, C.; Gerzanich, V.; Simard, J.M. Canagliflozin, an Inhibitor of the Na+-Coupled D-Glucose Cotransporter, SGLT2, Inhibits Astrocyte Swelling and Brain Swelling in Cerebral Ischemia. Cells 2023, 12, 2221. https://doi.org/10.3390/cells12182221
Shim B, Stokum JA, Moyer M, Tsymbalyuk N, Tsymbalyuk O, Keledjian K, Ivanova S, Tosun C, Gerzanich V, Simard JM. Canagliflozin, an Inhibitor of the Na+-Coupled D-Glucose Cotransporter, SGLT2, Inhibits Astrocyte Swelling and Brain Swelling in Cerebral Ischemia. Cells. 2023; 12(18):2221. https://doi.org/10.3390/cells12182221
Chicago/Turabian StyleShim, Bosung, Jesse A. Stokum, Mitchell Moyer, Natalya Tsymbalyuk, Orest Tsymbalyuk, Kaspar Keledjian, Svetlana Ivanova, Cigdem Tosun, Volodymyr Gerzanich, and J. Marc Simard. 2023. "Canagliflozin, an Inhibitor of the Na+-Coupled D-Glucose Cotransporter, SGLT2, Inhibits Astrocyte Swelling and Brain Swelling in Cerebral Ischemia" Cells 12, no. 18: 2221. https://doi.org/10.3390/cells12182221
APA StyleShim, B., Stokum, J. A., Moyer, M., Tsymbalyuk, N., Tsymbalyuk, O., Keledjian, K., Ivanova, S., Tosun, C., Gerzanich, V., & Simard, J. M. (2023). Canagliflozin, an Inhibitor of the Na+-Coupled D-Glucose Cotransporter, SGLT2, Inhibits Astrocyte Swelling and Brain Swelling in Cerebral Ischemia. Cells, 12(18), 2221. https://doi.org/10.3390/cells12182221