
PLAC8-Mediated Activation of NOX4 Signalling Restores Angiogenic Function of Endothelial Colony-Forming Cells in Experimental Hypoxia

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. CB-ECFC Culture and Hypoxia Exposure
2.2. Cell Transfection
2.3. Reverse Transcription and Quantitative RT-PCR
2.4. Protein Extraction and Western Blotting
2.5. Cell Metabolic Activity
2.6. Tubulogenesis Assay
2.7. Immunocytochemistry
2.8. Cellular ROS Generation
2.9. Microarray Analysis
2.10. Human Phospho-Kinase Proteome Profiler Array
2.11. Statistical Analysis
3. Results
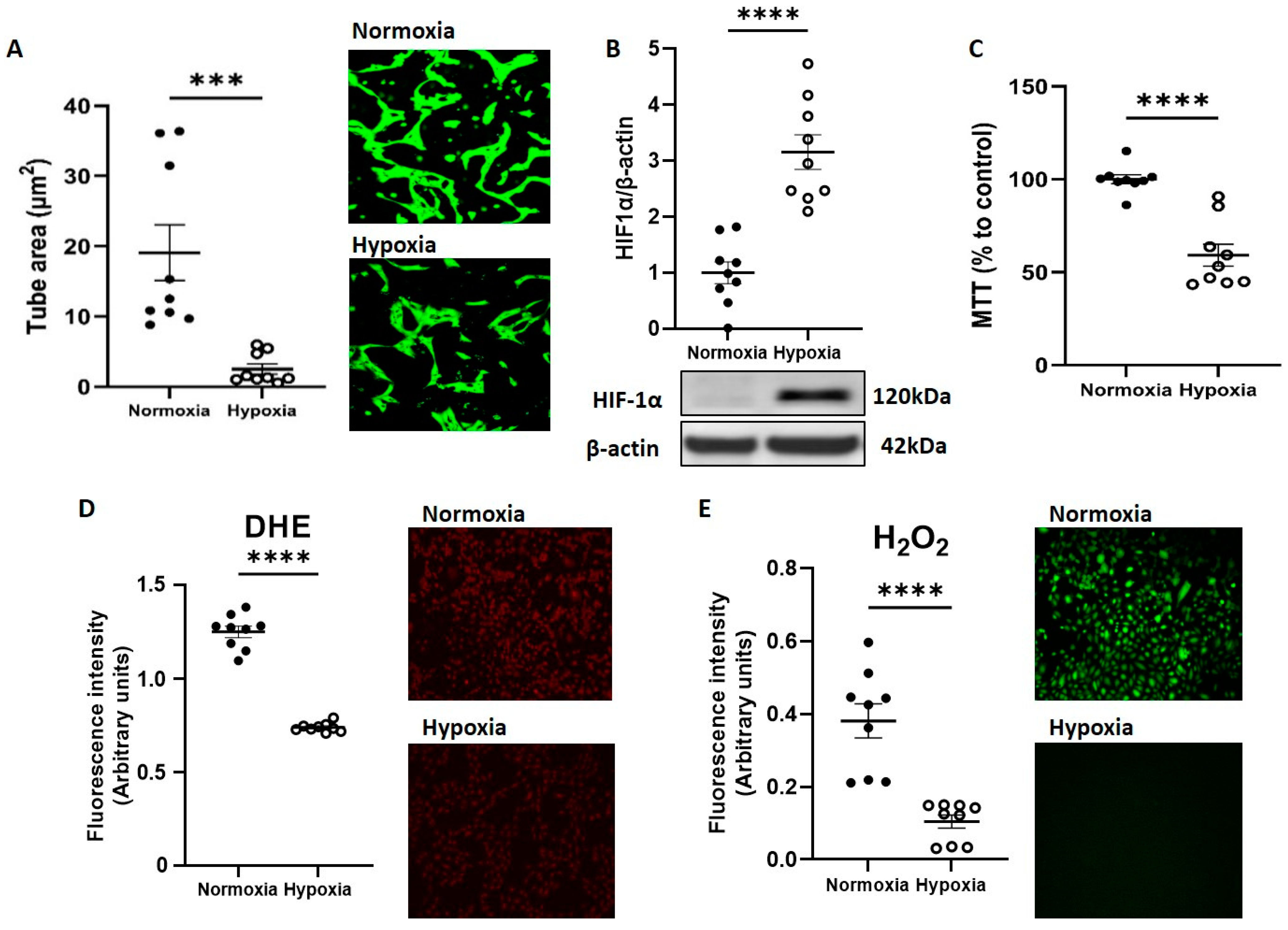
3.1. Hypoxia Promotes CB-ECFC Angiogenic Dysfunction and Reduced ROS Production
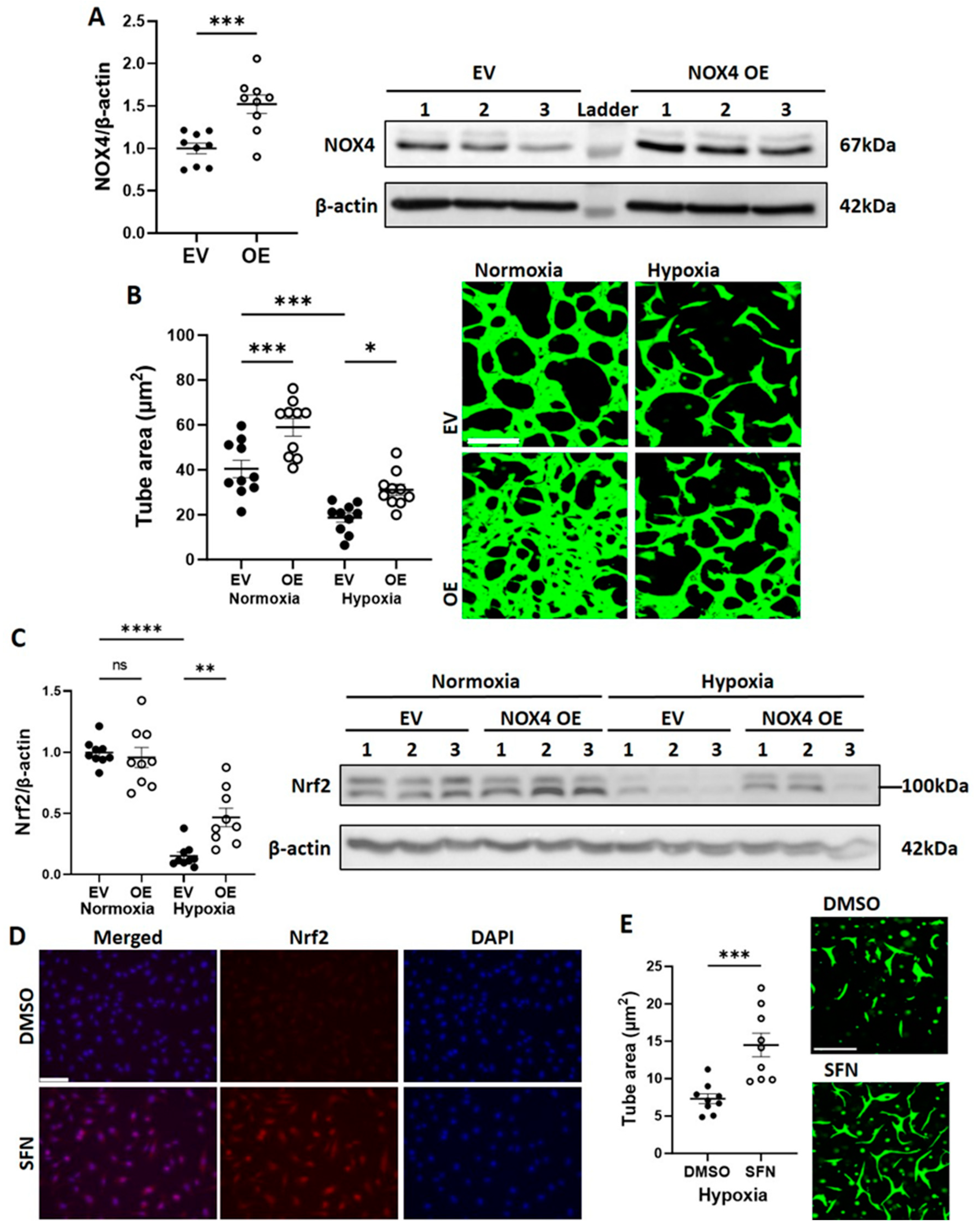
3.2. CB-ECFC NOX4 Signalling Is Suppressed by Hypoxia
3.3. NOX4 Overexpression Partially Restores CB-ECFC Angiogenic Function in Hypoxia via Activation of Nrf2
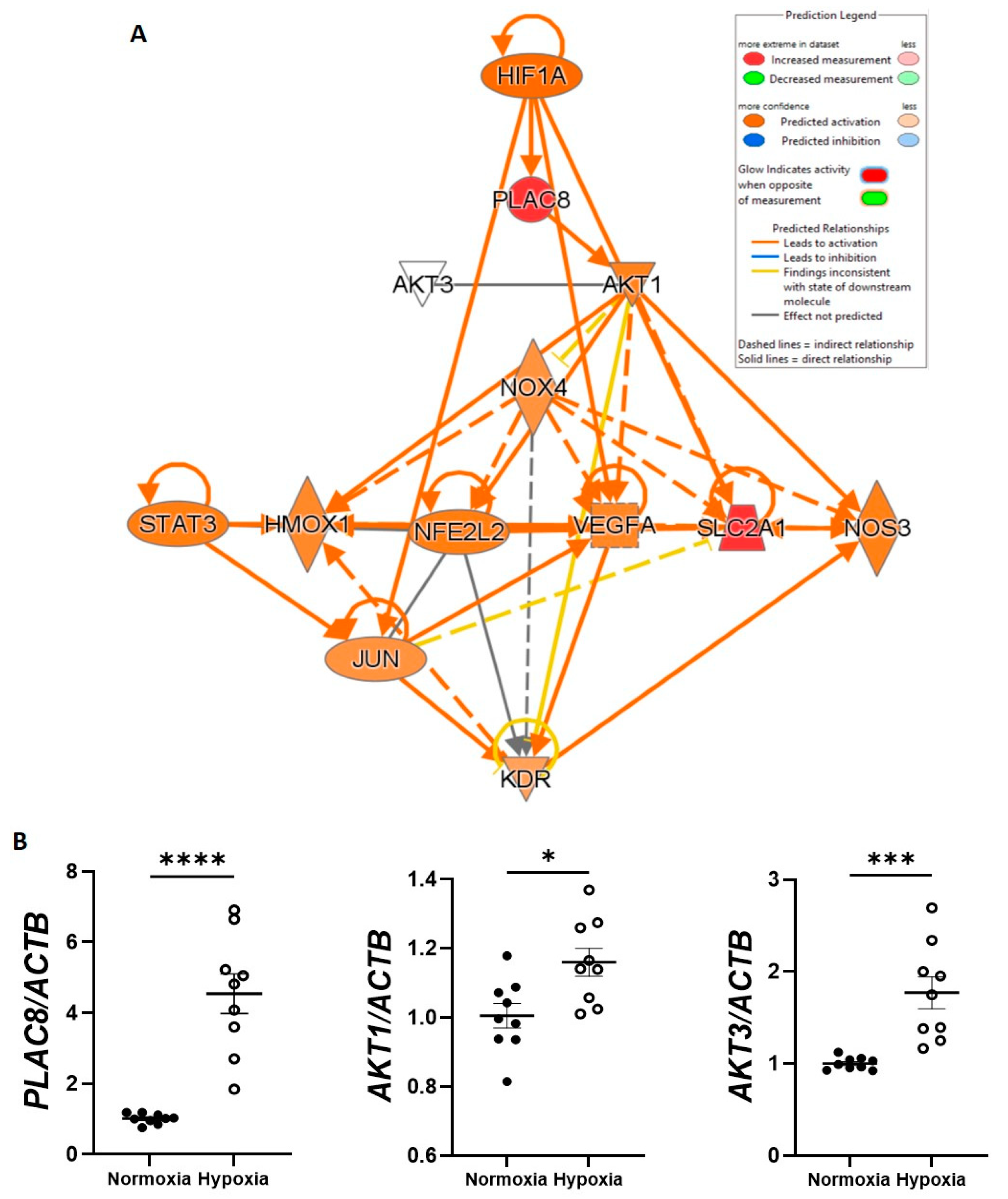
3.4. CB-ECFC Gene Expression Is Differentially Regulated in Hypoxia
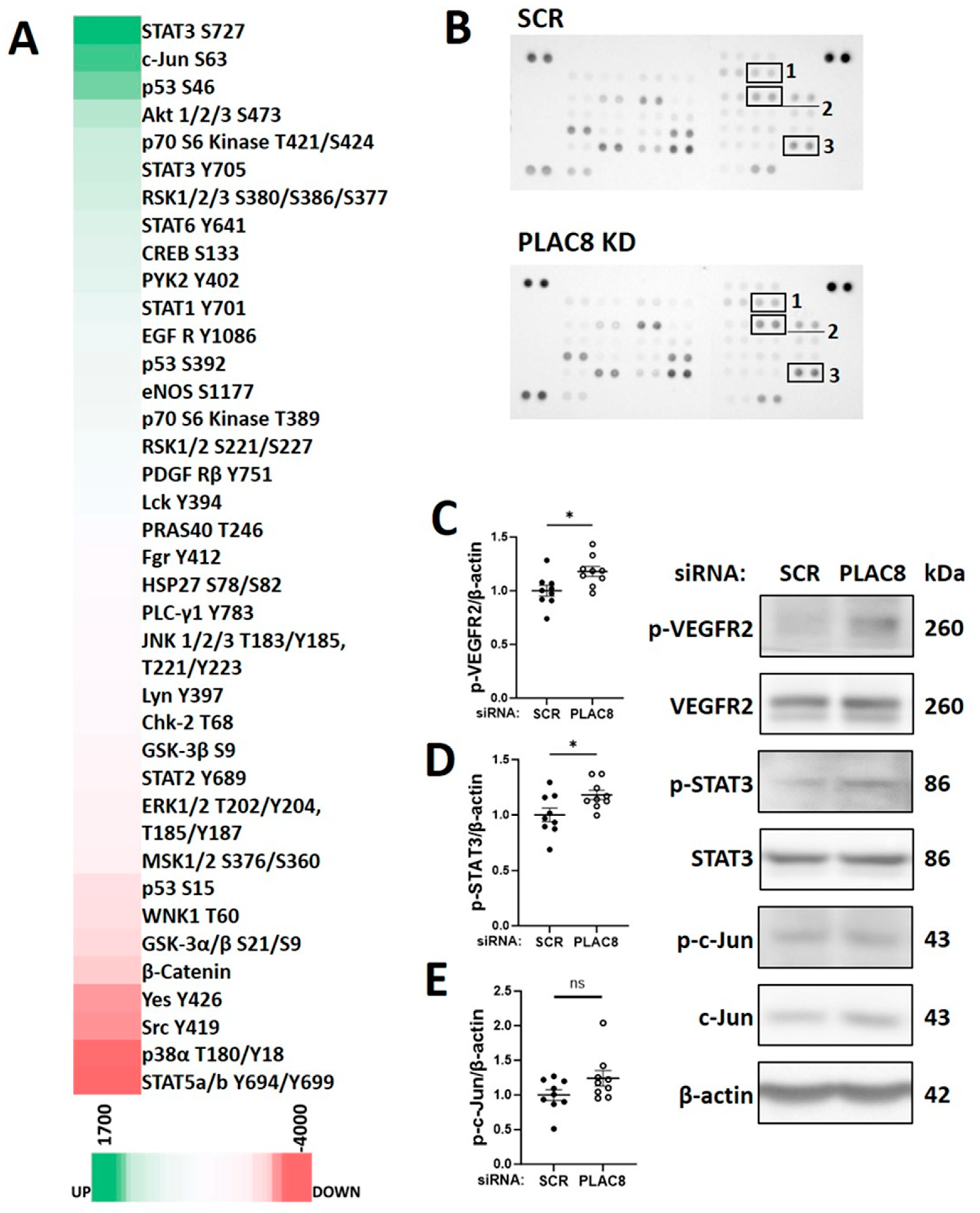
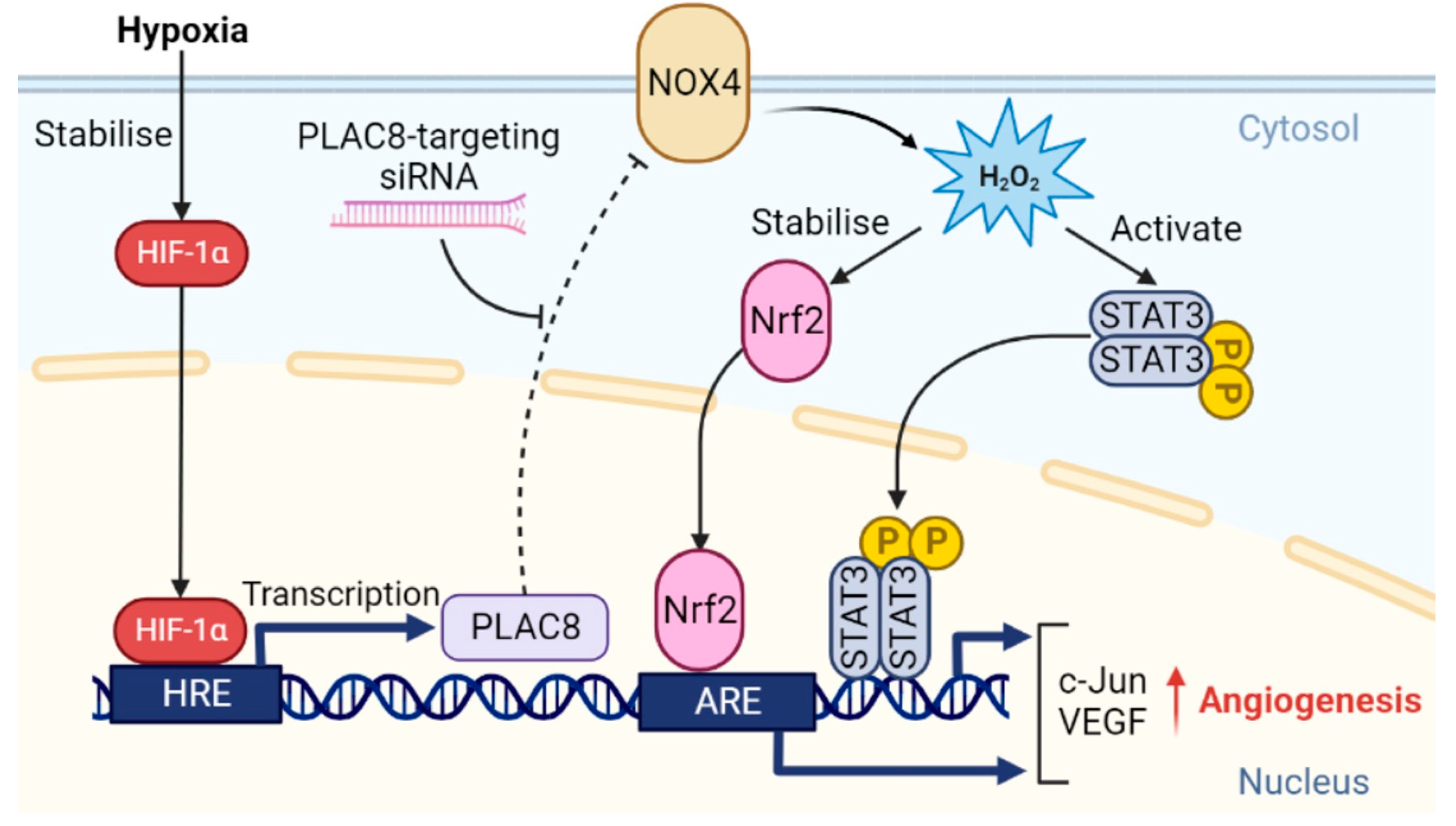
3.5. PLAC8 Is a Key Upstream Negative Regulator of CB-ECFC NOX4 Signalling in Hypoxia
3.6. PLAC8 Is a Key Upstream Negative Regulator of CB-ECFC NOX4 Signalling in Hypoxia
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Palasubramaniam, J.; Wang, X.; Peter, K. Myocardial Infarction-From Atherosclerosis to thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, e176–e185. [Google Scholar] [CrossRef] [PubMed]
- Freisinger, E.; Malyar, N.M.; Reinecke, H.; Lawall, H. Impact of diabetes on outcome in critical limb ischemia with tissue loss: A large-scaled routine data analysis. Cardiovasc. Diabetol. 2017, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Gogiraju, R.; Bochenek, M.L.; Schafer, K. Angiogenic Endothelial cell signaling in cardiac hypertrophy and heart failure. Front. Cardiovasc. Med. 2019, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Emini Veseli, B.; Perrotta, P.; De Meyer, G.R.A.; Roth, L.; Van der Donckt, C.; Martinet, W.; De Meyer, G.R.Y. Animal models of atherosclerosis. Eur. J. Pharmacol. 2017, 816, 3–13. [Google Scholar] [CrossRef]
- Johnson, T.; Zhao, L.; Manuel, G.; Taylor, H.; Liu, D. Approaches to therapeutic angiogenesis for ischemic heart disease. J. Mol. Med. Berl. 2019, 97, 141–151. [Google Scholar] [CrossRef]
- Al-Latayfeh, M.; Silva, P.S.; Sun, J.K.; Aiello, L.P. Antiangiogenic therapy for ischemic retinopathies. Cold Spring Harb. Perspect. Med. 2012, 2, a006411. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lu, K.; Zhu, J.; Wang, J. Stem cell therapy for ischemic heart diseases. Br. Med. Bull. 2017, 121, 135–154. [Google Scholar] [CrossRef]
- Ingram, D.A.; Mead, L.E.; Tanaka, H.; Meade, V.; Fenoglio, A.; Mortell, K.; Pollok, K.; Ferkowicz, M.J.; Gilley, D.; Yoder, M.C. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 2004, 104, 2752–2760. [Google Scholar] [CrossRef]
- Medina, R.J.; Barber, C.L.; Sabatier, F.; Dignat-George, F.; Melero-Martin, J.M.; Khosrotehrani, K.; Ohneda, O.; Randi, A.M.; Chan, J.K.Y.; Yamaguchi, T.; et al. Endothelial Progenitors: A consensus statement on nomenclature. Stem Cells Transl. Med. 2017, 6, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Au, P.; Daheron, L.M.; Duda, D.G.; Cohen, K.S.; Tyrrell, J.A.; Lanning, R.M.; Fukumura, D.; Scadden, D.T.; Jain, R. Differential in vivo potential of endothelial progenitor cells from human umbilical cord blood and adult peripheral blood to form functional long-lasting vessels. Blood 2008, 111, 1302–1305. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, T.; Naito, H.; Suehiro, J.I.; Lin, Y.; Kawaji, H.; Iba, T.; Kuono, T.; Ishikawa-Kato, S.; Furuno, M.; Takara, K.; et al. CD157 marks tissue-resident endothelial stem cells with homeostatic and regenerative properties. Cell Stem Cell 2018, 22, 384–397.e6. [Google Scholar] [CrossRef] [PubMed]
- Reid, E.; Guduric-Fuchs, J.; O’Neill, C.L.; Allen, L.D.; Chambers, S.E.J.; Stitt, A.W.; Medina, R.J. Preclinical evaluation and optimization of a cell therapy using human cord blood-derived endothelial colony-forming cells for ischemic retinopathies. Stem Cells Transl. Med. 2018, 7, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Nuzzolo, E.R.; Capodimonti, S.; Martini, M.; Iachininoto, M.G.; Bianchi, M.; Cocomazzi, A.; Zini, G.; Leone, G.; Larocca, L.M.; Teofili, L. Adult and cord blood endothelial progenitor cells have different gene expression profiles and immunogenic potential. Blood Transfus. 2014, 12 (Suppl. S1), s367–s374. [Google Scholar] [PubMed]
- Zhang, Y.; Fisher, N.; Newey, S.E.; Smythe, J.; Tatton, L.; Tsaknakis, G.; Forde, S.P.; Carpenter, L.; Athanassopoulos, T.; Hale, S.J.; et al. The impact of proliferative potential of umbilical cord-derived endothelial progenitor cells and hypoxia on vascular tubule formation in vitro. Stem Cells Dev. 2009, 18, 359–375. [Google Scholar] [CrossRef]
- Nisimoto, Y.; Jackson, H.M.; Ogawa, H.; Kawahara, T.; Lambeth, J.D. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry 2010, 49, 2433–2442. [Google Scholar] [CrossRef]
- O’Neill, K.M.; Campbell, D.C.; Edgar, K.S.; Gill, E.K.; Moez, A.; McLoughlin, K.J.; O’Neill, C.L.; Dellett, M.; Hargey, C.J.; Abudalo, R.A.; et al. NOX4 is a major regulator of cord blood-derived endothelial colony-forming cells which promotes post-ischaemic revascularization. Cardiovasc. Res. 2020, 116, 393–405. [Google Scholar] [CrossRef]
- Decaris, M.L.; Lee, C.I.; Yoder, M.C.; Tarantal, A.F.; Leach, J.K. Influence of the oxygen microenvironment on the proangiogenic potential of human endothelial colony forming cells. Angiogenesis 2009, 12, 303–311. [Google Scholar] [CrossRef][Green Version]
- Kim, Y.M.; Kim, S.J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Ushio-Fukai, M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Cell Physiol. 2017, 312, C749–C764. [Google Scholar] [CrossRef]
- Nakayama, M.; Takahashi, K.; Kitamuro, T.; Yasumoto, K.; Katayose, D.; Shirato, K.; Fujii-Kuriyama, Y.; Shibahara, S. Repression of heme oxygenase-1 by hypoxia in vascular endothelial cells. Biochem. Biophys. Res. Commun. 2000, 271, 665–671. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; You, L.; Nepovimova, E.; Heger, Z.; Wu, W.; Kuca, K.; Adam, V. Hypoxia-inducible factors: Master regulators of hypoxic tumor immune escape. J. Hematol. Oncol. 2022, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, H.; Kuang, C.Y.; Zhu, J.K.; Yu, Y.; Qin, Z.X.; Liu, J.; Huang, L. An essential role for the Id1/PI3K/Akt/NFkB/survivin signalling pathway in promoting the proliferation of endothelial progenitor cells in vitro. Mol. Cell Biochem. 2012, 363, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Q.; Zhang, Z.; Jiang, F.; Meng, X.; Yan, H. Interleukin-10 overexpression improves the function of endothelial progenitor cells stimulated with TNF-alpha through the activation of the STAT3 signaling pathway. Int. J. Mol. Med. 2015, 35, 471–477. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, L.; Cash, T.P.; Jones, R.G.; Keith, B.; Thompson, C.B.; Simon, M.C. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell. 2006, 21, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Outtara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef]
- Liang, X.; Arullampalam, P.; Yang, Z.; Ming, X.F. Hypoxia enhances endothelial intercellular adhesion molecule 1 protein level through upregulation of arginase type II and mitochondrial oxidative stress. Front. Physiol. 2019, 10, 1003. [Google Scholar] [CrossRef]
- De Pascali, F.; Hemann, C.; Samons, K.; Chen, C.A.; Zweier, J.L. Hypoxia and reoxygenation induce endothelial nitric oxide synthase uncoupling in endothelial cells through tetrahydrobiopterin depletion and S-glutathionylation. Biochemistry 2014, 53, 3679–3688. [Google Scholar] [CrossRef]
- Fish, J.E.; Yan, M.S.; Matouk, C.C.; St Bernard, R.; Ho, J.J.; Gavryushova, A.; Srivastava, D.; Marsden, P.A. Hypoxic repression of endothelial nitric-oxide synthase transcription is coupled with eviction of promoter histones. J. Biol. Chem. 2010, 285, 810–826. [Google Scholar] [CrossRef]
- Olszewska-Pazdrak, B.; Hein, T.W.; Olszewska, P.; Carney, D.H. Chronic hypoxia attenuates VEGF signaling and angiogenic responses by downregulation of KDR in human endothelial cells. Am. J. Physiol. Cell Physiol. 2009, 296, C1162–C1170. [Google Scholar] [CrossRef]
- Liao, J.K.; Zulueta, J.J.; Yu, F.S.; Peng, H.B.; Cote, C.G.; Hassoun, P.M. Regulation of bovine endothelial constitutive nitric oxide synthase by oxygen. J. Clin. Investig. 1995, 96, 2661–2666. [Google Scholar] [CrossRef]
- Kitamuro, T.; Takahashi, K.; Ogawa, K.; Udono-Fujimori, R.; Takeda, K.; Furuyama, K.; Nakayama, M.; Sun, J.; Fujita, H.; Hida, W.; et al. Bach1 functions as a hypoxia-inducible repressor for the heme oxygenase-1 gene in human cells. J. Biol. Chem. 2003, 278, 9125–9133. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Wang, X.; Chang, Q.; Hitron, A.; Zhang, Z.; Xu, M.; Chen, G.; Luo, J.; Jiang, B.; Fang, J.; et al. Arsenic promotes angiogenesis in vitro via a heme oxygenase-1-dependent mechanism. Toxicol. Appl. Pharmacol. 2010, 244, 291–299. [Google Scholar] [CrossRef]
- Kuang, L.; Feng, J.; He, G.; Jing, T. Knockdown of Nrf2 inhibits the angiogenesis of rat cardiac micro-vascular endothelial cells under hypoxic conditions. Int. J. Biol. Sci. 2013, 9, 656–665. [Google Scholar] [CrossRef]
- Hakami, N.Y.; Ranjan, A.K.; Hardikar, A.A.; Dusting, G.J.; Peshavariya, H.M. Role of NADPH oxidase-4 in human endothelial progenitor cells. Front. Physiol. 2017, 8, 150. [Google Scholar] [CrossRef]
- Blue, E.K.; Sheehan, B.M.; Nuss, Z.V.; Boyle, F.A.; Hocutt, C.M.; Gohn, C.R.; Varberg, K.M.; McClintick, J.N.; Haneline, L.S. Epigenetic regulation of placenta-specific 8 contributes to altered function of endothelial colony-forming cells exposed to intrauterine gestational diabetes mellitus. Diabetes 2015, 64, 2664–2675. [Google Scholar] [CrossRef]
- Jimenez-Preitner, M.; Berney, X.; Uldry, M.; Vitali, A.; Cinti, S.; Ledford, J.G.; Thorens, B. Plac8 is an inducer of C/EBPbeta required for brown fat differentiation, thermoregulation, and control of body weight. Cell Metab. 2011, 14, 658–670. [Google Scholar] [CrossRef]
- Galaviz-Hernandez, C.; Stagg, C.; de Ridder, G.; Tanaka, T.S.; Ko, M.S.; Schlessinger, D.; Nagaraja, R. Plac8 and Plac9, novel placental-enriched genes identified through microarray analysis. Gene 2003, 309, 81–89. [Google Scholar] [CrossRef]
- Sugimura, S.; Kobayashi, S.; Hashiyada, Y.; Ohtake, M.; Kaneda, M.; Yamanouchi, T.; Matsuda, H.; Aikawa, Y.; Watanabe, S.; Nagai, T.; et al. Follicular growth-stimulated cows provide favorable oocytes for producing cloned embryos. Cell Reprogram. 2012, 14, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Hikasa, K.; Umesono, Y.; Hayashi, T.; Agata, K.; Shibata, N. Loss of plac8 expression rapidly leads pluripotent stem cells to enter active state during planarian regeneration. Development 2022, 149, dev199449. [Google Scholar] [CrossRef]
- Jia, Y.; Ying, X.; Zhou, J.; Chen, Y.; Luo, X.; Xie, S.; Wang, Q.c.; Hu, W.; Wang, L. The novel KLF4/PLAC8 signaling pathway regulates lung cancer growth. Cell Death Dis. 2018, 9, 603. [Google Scholar] [CrossRef]
- Mao, M.; Chen, Y.; Yang, J.; Cheng, Y.; Xu, L.; Ji, F.; Zhou, J.; Zhang, X.; Li, Z.; Chen, C.; et al. Modification of PLAC8 by UFM1 affects tumorous proliferation and immune response by impacting PD-L1 levels in triple-negative breast cancer. J. Immunother. Cancer 2022, 10, e005668. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.R.; Hatzistergos, K.E.; Addicott, B.; McCall, F.; Carvalho, D.; Suncion, V.; Morales, A.R.; Da Silva, J.; Sussman, M.A.; Heldman, A.W.; et al. Enhanced effect of combining human cardiac stem cells and bone marrow mesenchymal stem cells to reduce infarct size and to restore cardiac function after myocardial infarction. Circulation 2013, 127, 213–223. [Google Scholar] [CrossRef]
- Wilson, A.J.; Gill, E.K.; Abudalo, R.A.; Edgar, K.S.; Watson, C.J.; Grieve, D.J. Reactive oxygen species signalling in the diabetic heart: Emerging prospect for therapeutic targeting. Heart 2018, 104, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.R.; Yang, S. Hydrogen peroxide: A signaling messenger. Antioxid. Redox Signal. 2006, 8, 243–270. [Google Scholar] [CrossRef]
- Takac, I.; Schroder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, J.; Duan, H.; Li, R.; Peng, W.; Wu, C. Activation of Nrf2/HO-1 signaling: An important molecular mechanism of herbal medicine in the treatment of atherosclerosis via the protection of vascular endothelial cells from oxidative stress. J. Adv. Res. 2021, 34, 43–63. [Google Scholar] [CrossRef]
- Tian, Y.; Liu, H.; Wang, M.; Wang, R.; Yi, G.; Zhang, M.; Chen, R. Role of STAT3 and NRF2 in tumors: Potential targets for antitumor therapy. Molecules 2022, 27, 8768. [Google Scholar] [CrossRef]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett. 2017, 387, 95–105. [Google Scholar] [CrossRef]
- Guduric-Fuchs, J.; Pedrini, E.; Lechner, J.; Chambers, S.E.J.; O’Neill, C.L.; Mendes Lopes de Melo, J.; Pathak, V.; Church, R.H.; McKeown, S.; Bojdo, J.; et al. miR-130a activates the VEGFR2/STAT3/HIF1alpha axis to potentiate the vasoregenerative capacity of endothelial colony-forming cells in hypoxia. Mol. Ther.-Nucleic Acids 2021, 23, 968–981. [Google Scholar] [CrossRef]
- Cirone, M.; D’Orazi, G. NRF2 in cancer: Cross-talk with oncogenic pathways and involvement in gammaherpesvirus-driven carcinogenesis. Int. J. Mol. Sci. 2022, 24, 595. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pun, S.H.; O’Neill, K.M.; Edgar, K.S.; Gill, E.K.; Moez, A.; Naderi-Meshkin, H.; Malla, S.B.; Hookham, M.B.; Alsaggaf, M.; Madishetti, V.V.; et al. PLAC8-Mediated Activation of NOX4 Signalling Restores Angiogenic Function of Endothelial Colony-Forming Cells in Experimental Hypoxia. Cells 2023, 12, 2220. https://doi.org/10.3390/cells12182220
Pun SH, O’Neill KM, Edgar KS, Gill EK, Moez A, Naderi-Meshkin H, Malla SB, Hookham MB, Alsaggaf M, Madishetti VV, et al. PLAC8-Mediated Activation of NOX4 Signalling Restores Angiogenic Function of Endothelial Colony-Forming Cells in Experimental Hypoxia. Cells. 2023; 12(18):2220. https://doi.org/10.3390/cells12182220
Chicago/Turabian StylePun, Shun Hay, Karla M. O’Neill, Kevin S. Edgar, Eleanor K. Gill, Arya Moez, Hojjat Naderi-Meshkin, Sudhir B. Malla, Michelle B. Hookham, Mohammed Alsaggaf, Vinuthna Vani Madishetti, and et al. 2023. "PLAC8-Mediated Activation of NOX4 Signalling Restores Angiogenic Function of Endothelial Colony-Forming Cells in Experimental Hypoxia" Cells 12, no. 18: 2220. https://doi.org/10.3390/cells12182220
APA StylePun, S. H., O’Neill, K. M., Edgar, K. S., Gill, E. K., Moez, A., Naderi-Meshkin, H., Malla, S. B., Hookham, M. B., Alsaggaf, M., Madishetti, V. V., Botezatu, B., King, W., Brunssen, C., Morawietz, H., Dunne, P. D., Brazil, D. P., Medina, R. J., Watson, C. J., & Grieve, D. J. (2023). PLAC8-Mediated Activation of NOX4 Signalling Restores Angiogenic Function of Endothelial Colony-Forming Cells in Experimental Hypoxia. Cells, 12(18), 2220. https://doi.org/10.3390/cells12182220