
Acute Kidney Injury-Induced Circulating TNFR1/2 Elevations Correlate with Persistent Kidney Injury and Progression to Fibrosis
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Samples
2.2. Animal Experiments
2.3. Renal Function
2.4. ELISA
2.5. Single-Cell RNA Sequencing (scRNAseq)
2.6. qPCR
2.7. Immunofluorescence Staining
2.8. Histology
2.9. Statistics
3. Results
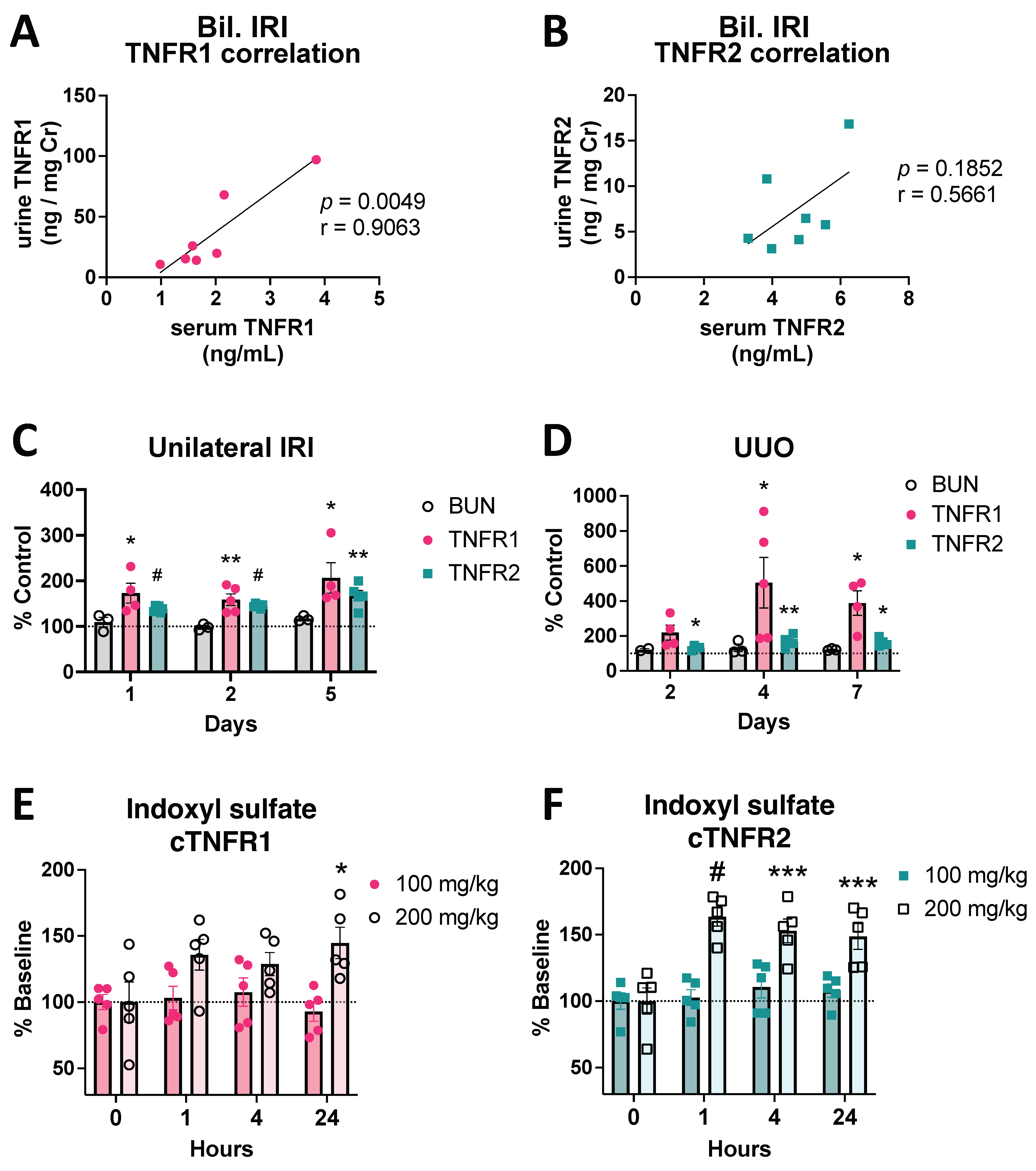
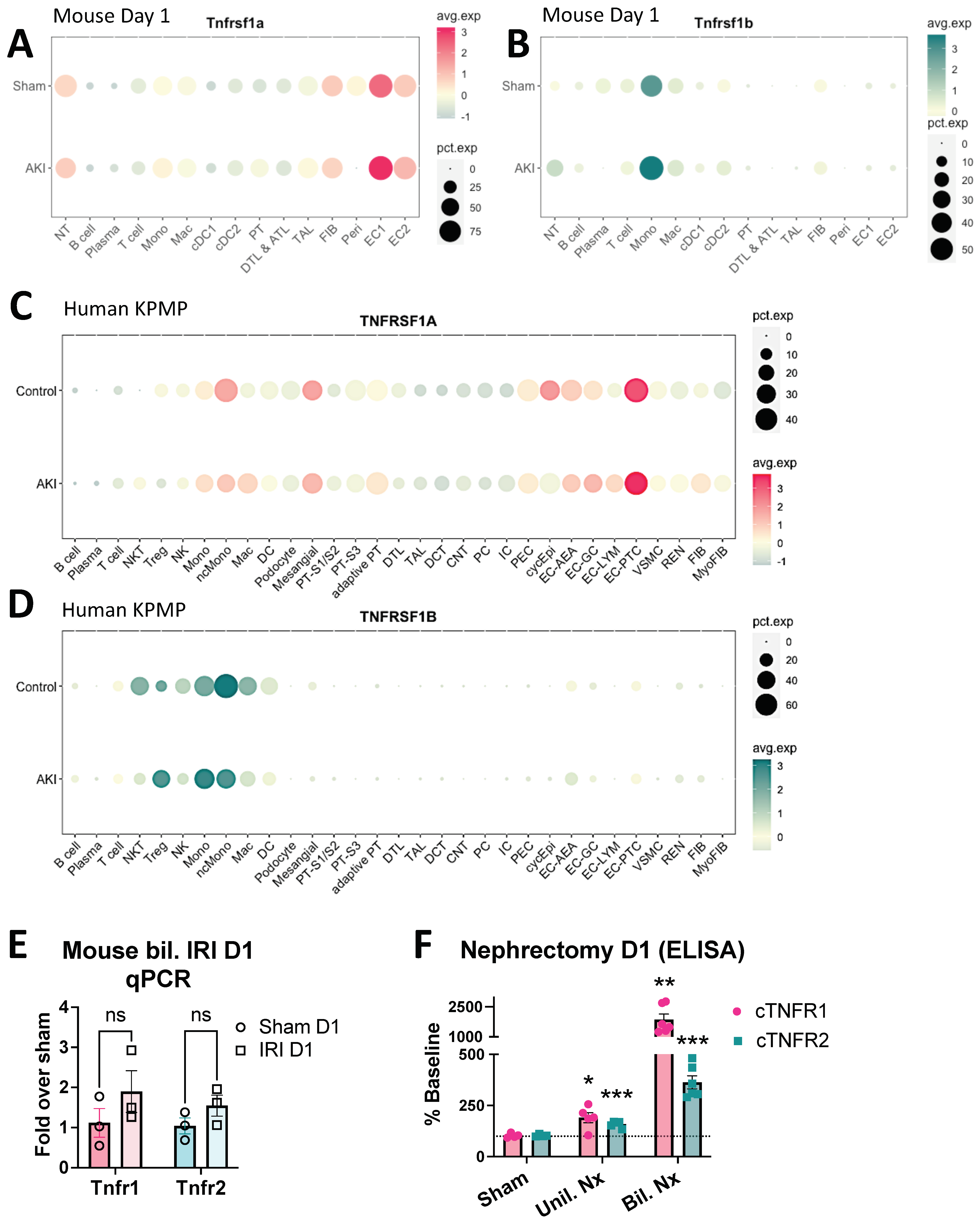
3.1. Circulating TNFR1/2 Levels Are Increased in Mouse Models of AKI and Correlate with Kidney Injury Levels
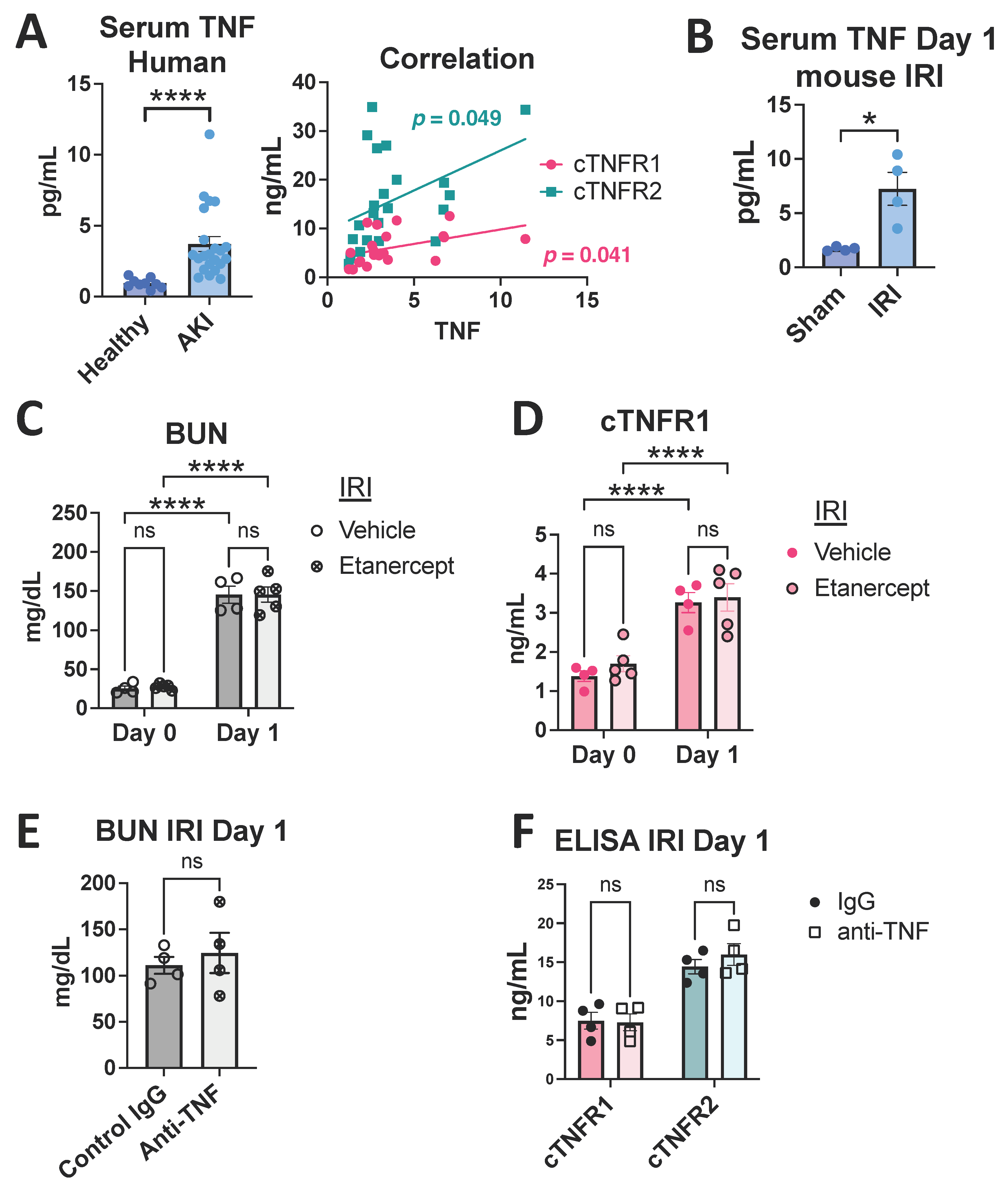
3.2. Human AKI Leads to Increased cTNFR1/2 Levels That Correlate with AKI Severity
3.3. TNF Neutralization Does Not Affect Early cTNFR1/2 Serum Elevations after Kidney Injury
3.4. Early cTNFR1/2 Elevations after AKI Can Be Attributed to Extrarenal Sources
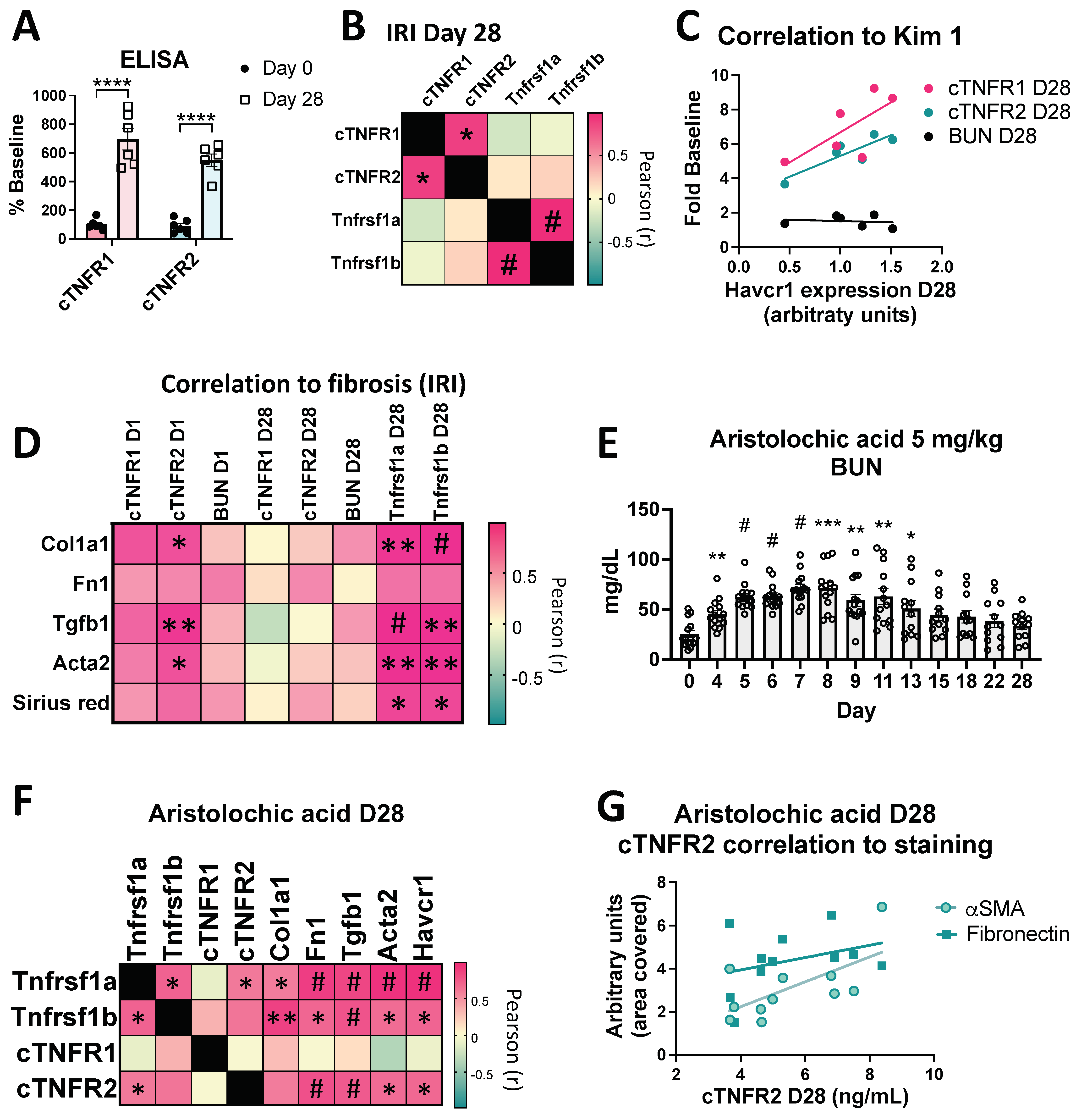
3.5. cTNFR1/2 Levels and Their Kidney Expression Levels Remain Elevated for Weeks Post Kidney Injury
3.6. Sustained cTNFR1/2 Elevations Correlate with Persistent Injury and Progression to Fibrosis in AKI-to-CKD Models
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Niewczas, M.A.; Gohda, T.; Skupien, J.; Smiles, A.M.; Walker, W.H.; Rosetti, F.; Cullere, X.; Eckfeldt, J.H.; Doria, A.; Mayadas, T.N.; et al. Circulating TNF Receptors 1 and 2 Predict ESRD in Type 2 Diabetes. J. Am. Soc. Nephrol. 2012, 23, 507–515. [Google Scholar] [CrossRef]
- Gohda, T.; Niewczas, M.A.; Ficociello, L.H.; Walker, W.H.; Skupien, J.; Rosetti, F.; Cullere, X.; Johnson, A.C.; Crabtree, G.; Smiles, A.M.; et al. Circulating TNF Receptors 1 and 2 Predict Stage 3 CKD in Type 1 Diabetes. J. Am. Soc. Nephrol. 2012, 23, 516–524. [Google Scholar] [CrossRef]
- Kurashina, T.; Nagasaka, S.; Watanabe, N.; Yabe, D.; Sugi, N.; Nin, K.; Hosokawa, M.; Nomura, Y.; Fukushima, M.; Nakai, Y.; et al. Circulating TNF Receptor 2 Is Closely Associated with the Kidney Function in Non-Diabetic Japanese Subjects. J. Atheroscler. Thromb. 2014, 21, 730–738. [Google Scholar] [CrossRef]
- Lee, S.M.; Yang, S.; Cha, R.; Kim, M.; An, J.N.; Paik, J.H.; Kim, D.K.; Kang, S.-W.; Lim, C.S.; Kim, Y.S.; et al. Circulating TNF Receptors Are Significant Prognostic Biomarkers for Idiopathic Membranous Nephropathy. PLoS ONE 2014, 9, e104354. [Google Scholar] [CrossRef]
- Sonoda, Y.; Gohda, T.; Suzuki, Y.; Omote, K.; Ishizaka, M.; Matsuoka, J.; Tomino, Y. Circulating TNF Receptors 1 and 2 Are Associated with the Severity of Renal Interstitial Fibrosis in IgA Nephropathy. PLoS ONE 2015, 10, e0122212. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.J.; An, J.N.; Kim, C.T.; Yang, S.H.; Lee, H.; Kim, D.K.; Joo, K.W.; Paik, J.H.; Kang, S.-W.; Park, J.T.; et al. Circulating Tumor Necrosis Factor α Receptors Predict the Outcomes of Human IgA Nephropathy: A Prospective Cohort Study. PLoS ONE 2015, 10, e0132826. [Google Scholar] [CrossRef]
- Pavkov, M.E.; Nelson, R.G.; Knowler, W.C.; Cheng, Y.; Krolewski, A.S.; Niewczas, M.A. Elevation of Circulating TNF Receptors 1 and 2 Increases the Risk of End-Stage Renal Disease in American Indians with Type 2 Diabetes. Kidney Int. 2015, 87, 812–819. [Google Scholar] [CrossRef]
- Patel, M.; Oni, L.; Midgley, A.; Smith, E.; Tullus, K.; Marks, S.D.; Jones, C.A.; Pilkington, C.; Beresford, M.W. Increased Concentration of Plasma TNFR1 and TNFR2 in Paediatric Lupus Nephritis. Lupus 2016, 25, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Niewczas, M.A.; Pavkov, M.E.; Skupien, J.; Smiles, A.; Dom, Z.I.M.; Wilson, J.M.; Park, J.; Nair, V.; Schlafly, A.; Saulnier, P.-J.; et al. A Signature of Circulating Inflammatory Proteins and Development of End-Stage Renal Disease in Diabetes. Nat. Med. 2019, 25, 805–813. [Google Scholar] [CrossRef]
- Greenberg, J.H.; Abraham, A.G.; Xu, Y.; Schelling, J.R.; Feldman, H.I.; Sabbisetti, V.S.; Gonzalez, M.C.; Coca, S.; Schrauben, S.J.; Waikar, S.S.; et al. Plasma Biomarkers of Tubular Injury and Inflammation Are Associated with CKD Progression in Children. J. Am. Soc. Nephrol. 2020, 31, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.K.; Coca, S.G.; Estrella, M.M.; Appel, L.J.; Coresh, J.; Philbrook, H.T.; Obeid, W.; Fried, L.F.; Heerspink, H.J.L.; Ix, J.H.; et al. Longitudinal TNFR1 and TNFR2 and Kidney Outcomes: Results from AASK and VA NEPHRON-D. J. Am. Soc. Nephrol. 2022, 33, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, O.M.; Shlipak, M.G.; Katz, R.; Waikar, S.S.; Greenberg, J.H.; Schrauben, S.J.; Coca, S.; Parikh, C.R.; Vasan, R.S.; Feldman, H.I.; et al. Associations of Plasma Biomarkers of Inflammation, Fibrosis, and Kidney Tubular Injury With Progression of Diabetic Kidney Disease: A Cohort Study. Am. J. Kidney Dis. 2022, 79, 849–857.e1. [Google Scholar] [CrossRef] [PubMed]
- Gohda, T.; Maruyama, S.; Kamei, N.; Yamaguchi, S.; Shibata, T.; Murakoshi, M.; Horikoshi, S.; Tomino, Y.; Ohsawa, I.; Gotoh, H.; et al. Circulating TNF Receptors 1 and 2 Predict Mortality in Patients with End-Stage Renal Disease Undergoing Dialysis. Sci. Rep. 2017, 7, 43520. [Google Scholar] [CrossRef]
- Bae, E.; Cha, R.-H.; Kim, Y.C.; An, J.N.; Kim, D.K.; Yoo, K.D.; Lee, S.M.; Kim, M.-H.; Park, J.T.; Kang, S.-W.; et al. Circulating TNF Receptors Predict Cardiovascular Disease in Patients with Chronic Kidney Disease. Medicine 2017, 96, e6666. [Google Scholar] [CrossRef] [PubMed]
- Saulnier, P.-J.; Gand, E.; Ragot, S.; Ducrocq, G.; Halimi, J.-M.; Hulin-Delmotte, C.; Llaty, P.; Montaigne, D.; Rigalleau, V.; Roussel, R.; et al. Association of Serum Concentration of TNFR1 with All-Cause Mortality in Patients with Type 2 Diabetes and Chronic Kidney Disease: Follow-up of the SURDIAGENE Cohort. Diabetes Care 2014, 37, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- McCoy, I.E.; Hsu, J.Y.; Bonventre, J.V.; Parikh, C.R.; Go, A.S.; Liu, K.D.; Ricardo, A.C.; Srivastava, A.; Cohen, D.L.; He, J.; et al. Acute Kidney Injury Associates with Long-Term Increases in Plasma TNFR1, TNFR2, and KIM-1: Findings from the CRIC Study. J. Am. Soc. Nephrol. 2022, 33, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Ferrando, E.S.; Hanslin, K.; Hultström, M.; Larsson, A.; Frithiof, R.; Lipcsey, M.; The Uppsala Intensive Care COVID-19 Research Group. Soluble TNF Receptors Predict Acute Kidney Injury and Mortality in Critically Ill COVID-19 Patients: A Prospective Observational Study. Cytokine 2022, 149, 155727. [Google Scholar] [CrossRef]
- Bergquist, M.; Samuelsson, L.; Larsson, A.; Tydén, J.; Johansson, J.; Lipcsey, M. TNFR1, TNFR2, Neutrophil Gelatinase-Associated Lipocalin and Heparin Binding Protein in Identifying Sepsis and Predicting Outcome in an Intensive Care Cohort. Sci. Rep. 2020, 10, 15350. [Google Scholar] [CrossRef]
- Menez, S.; Moledina, D.G.; Garg, A.X.; Thiessen-Philbrook, H.; McArthur, E.; Jia, Y.; Liu, C.; Obeid, W.; Mansour, S.G.; Koyner, J.L.; et al. Results from the TRIBE-AKI Study Found Associations between Post-Operative Blood Biomarkers and Risk of Chronic Kidney Disease after Cardiac Surgery. Kidney Int. 2021, 99, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G.; Vasquez-Rios, G.; Mansour, S.G.; Moledina, D.G.; Thiessen-Philbrook, H.; Wurfel, M.M.; Bhatraju, P.; Himmelfarb, J.; Siew, E.; Garg, A.X.; et al. Plasma Soluble Tumor Necrosis Factor Receptor Concentrations and Clinical Events After Hospitalization: Findings From the ASSESS-AKI and ARID Studies. Am. J. Kidney Dis. 2023, 81, 190–200. [Google Scholar] [CrossRef]
- Wen, Y.; Xu, L.; Melchinger, I.; Thiessen-Philbrook, H.; Moledina, D.G.; Coca, S.G.; Hsu, C.; Go, A.S.; Liu, K.D.; Siew, E.D.; et al. Longitudinal Biomarkers and Kidney Disease Progression after Acute Kidney Injury. JCI Insight 2023, 8, e167731. [Google Scholar] [CrossRef]
- Lin, J.; Hu, F.B.; Rimm, E.B.; Rifai, N.; Curhan, G.C. The Association of Serum Lipids and Inflammatory Biomarkers with Renal Function in Men with Type II Diabetes Mellitus. Kidney Int. 2006, 69, 336–342. [Google Scholar] [CrossRef]
- Lin, J.; Hu, F.B.; Mantzoros, C.; Curhan, G.C. Lipid and Inflammatory Biomarkers and Kidney Function Decline in Type 2 Diabetes. Diabetologia 2009, 53, 263. [Google Scholar] [CrossRef]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial Cell Cycle Arrest in G2/M Mediates Kidney Fibrosis after Injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef]
- Kefaloyianni, E.; Muthu, M.L.; Kaeppler, J.; Sun, X.; Sabbisetti, V.; Chalaris, A.; Rose-John, S.; Wong, E.; Sagi, I.; Waikar, S.S.; et al. ADAM17 Substrate Release in Proximal Tubule Drives Kidney Fibrosis. JCI Insight 2016, 1, e87023. [Google Scholar] [CrossRef] [PubMed]
- Kefaloyianni, E.; Raja, M.R.K.; Schumacher, J.; Muthu, M.L.; Krishnadoss, V.; Waikar, S.S.; Herrlich, A. Proximal Tubule-Derived Amphiregulin Amplifies and Integrates Profibrotic EGF Receptor Signals in Kidney Fibrosis. J. Am. Soc. Nephrol. JASN 2019, 30, 2370–2383. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wang, C.; Nie, L.; Zhao, X.; Gu, J.; Guan, X.; Wang, S.; Xiao, T.; Xu, X.; He, T.; et al. Klotho Protects Against Indoxyl Sulphate-Induced Myocardial Hypertrophy. J. Am. Soc. Nephrol. 2015, 26, 2434–2446. [Google Scholar] [CrossRef]
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl Sulfate Induces Leukocyte-Endothelial Interactions through Up-Regulation of E-Selectin. J. Biol. Chem. 2010, 285, 38869–38875. [Google Scholar] [CrossRef] [PubMed]
- Abdelmageed, M.M.; Kefaloyianni, E.; Arthanarisami, A.; Komaru, Y.; Atkinson, J.J.; Herrlich, A. TNF or EGFR Inhibition Equally Block AKI-to-CKD Transition: Opportunities for Etanercept Treatment. Nephrol. Dial. Transpl. 2023, 38, 1139–1150. [Google Scholar] [CrossRef]
- Khamissi, F.Z.; Ning, L.; Kefaloyianni, E.; Dun, H.; Arthanarisami, A.; Keller, A.; Atkinson, J.J.; Li, W.; Wong, B.; Dietmann, S.; et al. Identification of Kidney Injury Released Circulating Osteopontin as Causal Agent of Respiratory Failure. Sci. Adv. 2022, 8, eabm5900. [Google Scholar] [CrossRef]
- Wu, H.; Villalobos, R.G.; Yao, X.; Reilly, D.; Chen, T.; Rankin, M.; Myshkin, E.; Breyer, M.D.; Humphreys, B.D. Mapping the Single-Cell Transcriptomic Response of Murine Diabetic Kidney Disease to Therapies. Cell Metab. 2022, 34, 1064–1078.e6. [Google Scholar] [CrossRef]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The Uremic Toxicity of Indoxyl Sulfate and P-Cresyl Sulfate: A Systematic Review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Yoo, T.-H.; Hwang, Y.; Lee, G.H.; Kim, B.; Jang, J.; Yu, H.T.; Kim, M.C.; Cho, J.-Y.; Lee, C.J.; et al. Indoxyl Sulfate (IS)-Mediated Immune Dysfunction Provokes Endothelial Damage in Patients with End-Stage Renal Disease (ESRD). Sci. Rep. 2017, 7, 3057. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kumar, S.; Dolzhenko, E.; Alvarado, G.F.; Guo, J.; Lu, C.; Chen, Y.; Li, M.; Dessing, M.C.; Parvez, R.K.; et al. Molecular Characterization of the Transition from Acute to Chronic Kidney Injury Following Ischemia/Reperfusion. JCI Insight 2017, 2, e94716. [Google Scholar] [CrossRef]
- Fu, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Dong, Z. Rodent Models of AKI-CKD Transition. Am. J. Physiol.-Ren. Physiol. 2018, 315, F1098–F1106. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arthanarisami, A.; Komaru, Y.; Katsouridi, C.; Schumacher, J.; Verges, D.K.; Ning, L.; Abdelmageed, M.M.; Herrlich, A.; Kefaloyianni, E. Acute Kidney Injury-Induced Circulating TNFR1/2 Elevations Correlate with Persistent Kidney Injury and Progression to Fibrosis. Cells 2023, 12, 2214. https://doi.org/10.3390/cells12182214
Arthanarisami A, Komaru Y, Katsouridi C, Schumacher J, Verges DK, Ning L, Abdelmageed MM, Herrlich A, Kefaloyianni E. Acute Kidney Injury-Induced Circulating TNFR1/2 Elevations Correlate with Persistent Kidney Injury and Progression to Fibrosis. Cells. 2023; 12(18):2214. https://doi.org/10.3390/cells12182214
Chicago/Turabian StyleArthanarisami, Akshayakeerthi, Yohei Komaru, Charikleia Katsouridi, Julian Schumacher, Deborah K. Verges, Liang Ning, Mai M. Abdelmageed, Andreas Herrlich, and Eirini Kefaloyianni. 2023. "Acute Kidney Injury-Induced Circulating TNFR1/2 Elevations Correlate with Persistent Kidney Injury and Progression to Fibrosis" Cells 12, no. 18: 2214. https://doi.org/10.3390/cells12182214
APA StyleArthanarisami, A., Komaru, Y., Katsouridi, C., Schumacher, J., Verges, D. K., Ning, L., Abdelmageed, M. M., Herrlich, A., & Kefaloyianni, E. (2023). Acute Kidney Injury-Induced Circulating TNFR1/2 Elevations Correlate with Persistent Kidney Injury and Progression to Fibrosis. Cells, 12(18), 2214. https://doi.org/10.3390/cells12182214