
JNK Signalling Regulates Self-Renewal of Proliferative Urine-Derived Renal Progenitor Cells via Inhibition of Ferroptosis

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. siRNA Transfection
2.3. Proliferation Assay
2.4. Flow Cytometry-Based Measurement of Cell Death
2.5. Immunofluorescence
2.6. Western Blotting
2.7. Flow Cytometry-Based Measurement of Lipid Peroxidation
2.8. Cell Survival Assay
2.9. GSH Assay
2.10. Seahorse XF Cell Mito Stress Test
2.11. TMRM Staining
2.12. Quantitative RT-PCR
2.13. Gene Expression Analysis
2.14. Gene Ontology (GO) and Pathway Analysis
2.15. Metascape Analysis
3. Results
3.1. High Concentrations of JNK Inhibitor Induce Cell Death
3.2. JNK Inhibition Leads to Decreased Proliferation and Loss of Progenitor State
3.3. Cell Cycle-Related Processes Are Regulated by JNK Inhibition
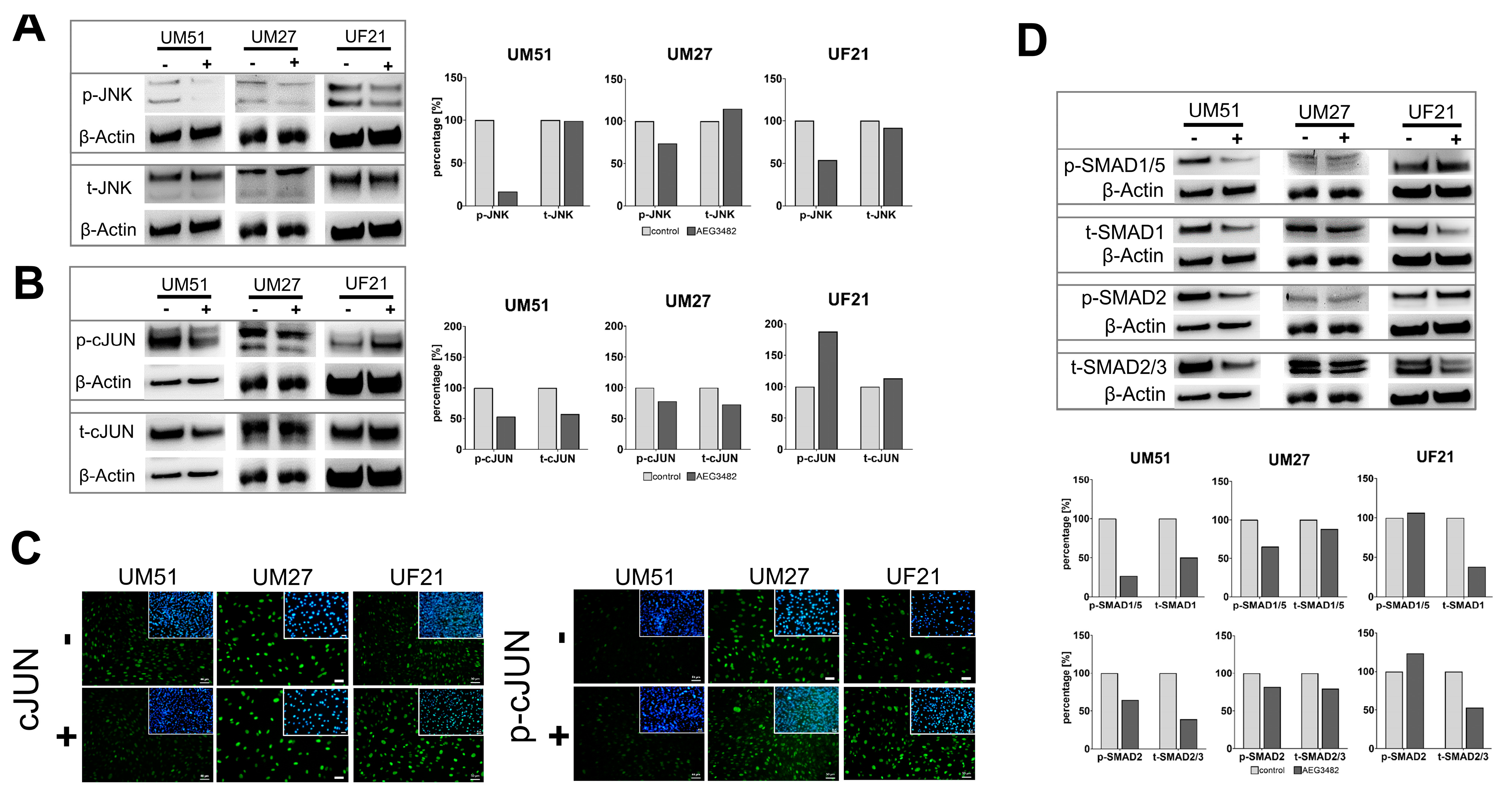
3.4. The Downstream Target Phospho-cJUN Is Downregulated by JNK Inhibition
3.5. UdRPCs Are More Susceptible to Ferroptosis upon JNK Inhibition
3.6. JNK Inhibition Partially Disrupts the Mitochondrial Membrane Potential and Reduces Respiration
4. Discussion
4.1. JNK Inhibition Decreases the Proliferation and Leads to Loss of Progenitor Character
4.2. Active JNK Signalling Protects Urine-Derived Renal Progenitor Cells against Ferroptosis
4.3. JNK Signalling Is Involved in the Metabolic Activity Associated with the Maintenance of Self-Renewal in UdRPCs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oliveira Arcolino, F.; Tort Piella, A.; Papadimitriou, E.; Bussolati, B.; Antonie, D.J.; Murray, P.; Van Den Heuvel, L.; Levtchenko, E. Human urine as a noninvasive source of kidney cells. Stem Cells Int. 2015, 2015, 362562. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.S.; Wruck, W.; Spitzhorn, L.S.; Nguyen, L.; Bohndorf, M.; Martins, S.; Asar, F.; Ncube, A.; Erichsen, L.; Graffmann, N.; et al. The FGF, TGFβ and WNT axis Modulate Self-renewal of Human SIX2+ Urine Derived Renal Progenitor Cells. Sci. Rep. 2020, 10, 739. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Takizawa, H.; Nakamura, A.; Turner, B.J.; Shabanpoor, F.; Aoki, Y. Application of Urine-Derived Stem Cells to Cellular Modeling in Neuromuscular and Neurodegenerative Diseases. Front. Mol. Neurosci. 2019, 12, 297. [Google Scholar] [CrossRef] [PubMed]
- Pavathuparambil Abdul Manaph, N.; Al-Hawaas, M.; Bobrovskaya, L.; Coates, P.T.; Zhou, X.F. Urine-derived cells for human cell therapy. Stem Cell Res. Ther. 2018, 9, 189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wei, G.; Li, P.; Zhou, X.; Zhang, Y. Urine-derived stem cells: A novel and versatile progenitor source for cell-based therapy and regenerative medicine. Genes Dis. 2014, 1, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; Liu, G.; Shi, Y.; Wu, R.; Yang, B.; He, T.; Fan, Y.; Lu, X.; Zhou, X.; Liu, H.; et al. Multipotential differentiation of human urine-derived stem cells: Potential for therapeutic applications in urology. Stem Cells 2013, 31, 1840–1856. [Google Scholar] [CrossRef]
- Erichsen, L.; Thimm, C.; Bohndorf, M.; Rahman, M.S.; Wruck, W.; Adjaye, J. Activation of the Renin–Angiotensin System Disrupts the Cytoskeletal Architecture of Human Urine-Derived Podocytes. Cells 2022, 11, 1095. [Google Scholar] [CrossRef]
- Javelaud, D.; Mauviel, A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-β: Implications for carcinogenesis. Oncogene 2005, 24, 5742–5750. [Google Scholar] [CrossRef]
- Awazu, M. Mitogen-activated Protein Kinases in the Development of Normal and Diseased Kidneys. Child Kidney Dis 2017, 21, 1–7. [Google Scholar] [CrossRef]
- Engel, M.E.; McDonnell, M.A.; Law, B.K.; Moses, H.L. Interdependent SMAD and JNK Signaling in Transforming Growth Factor-β-mediated Transcription. J. Biol. Chem. 1999, 274, 37413–37420. [Google Scholar] [CrossRef]
- Tournier, C.; Hess, P.; Yang, D.D.; Xu, J.; Turner, T.K.; Nimnual, A.; Bar-Sagi, D.; Jones, S.N.; Flavell, R.A.; Davis, R.J. Requirement of JNK for stress-induced activation of the cytochrome c- mediated death pathway. Science 2000, 288, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.O.; Jonassen, J.A.; Preval, K.M.; Davis, R.J.; Pazour, G.J. RESEARCH ARTICLE c-Jun N-terminal kinase (JNK) signaling contributes to cystic burden in polycystic kidney disease. PLoS Genet. 2021, 17, e1009711. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Xiao, X. Ferroptosis and kidney diseases. Int. Urol. Nephrol. 2020, 52, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, H.; Yang, S.; Wu, X.; He, D.; Cao, K.; Zhang, W. Emerging Role of Ferroptosis in Acute Kidney Injury. Oxid. Med. Cell. Longev. 2019, 2019, 8010614. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Conrad, M.; Lorenz, S.M.; Proneth, B. Targeting Ferroptosis: New Hope for As-Yet-Incurable Diseases. Trends Mol. Med. 2021, 27, 113–122. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed]
- Tonnus, W.; Meyer, C.; Steinebach, C.; Belavgeni, A.; von Mässenhausen, A.; Gonzalez, N.Z.; Maremonti, F.; Gembardt, F.; Himmerkus, N.; Latk, M.; et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat. Commun. 2021, 12, 4402. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Warnes, G.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Mächler, M.; Magnusson, A.; Möller, S. gplots: Various R Programming Tools for Plotting Data; R Core Team: Vienna, Austria, 2005; Volume 2. [Google Scholar]
- Nguyen, L.; Wruck, W.; Erichsen, L.; Graffmann, N.; Adjaye, J. The Nephrotoxin Puromycin Aminonucleoside Induces Injury in Kidney Organoids Differentiated from Induced Pluripotent Stem Cells. Cells 2022, 11, 635. [Google Scholar] [CrossRef]
- Falcon, S.; Gentleman, R. Using GOstats to test gene lists for GO term association. Bioinformatics 2007, 23, 257–258. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Lindström, N.O.; De Sena Brandine, G.; Tran, T.; Ransick, A.; Suh, G.; Guo, J.; Kim, A.D.; Parvez, R.K.; Ruffins, S.W.; Rutledge, E.A.; et al. Progressive Recruitment of Mesenchymal Progenitors Reveals a Time-Dependent Process of Cell Fate Acquisition in Mouse and Human Nephrogenesis. Dev. Cell 2018, 45, 651–660. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Barnett, L.M.A.; Cummings, B.S. Nephrotoxicity and Renal Pathophysiology: A Contemporary Perspective. Toxicol. Sci. 2018, 164, 379–390. [Google Scholar] [CrossRef]
- Lima-Posada, I.; Portas-Cortés, C.; Pérez-Villalva, R.; Fontana, F.; Rodríguez-Romo, R.; Prieto, R.; Sánchez-Navarro, A.; Rodríguez-González, G.L.; Gamba, G.; Zambrano, E.; et al. Gender Differences in the Acute Kidney Injury to Chronic Kidney Disease Transition. Sci. Rep. 2017, 7, 12270. [Google Scholar] [CrossRef] [PubMed]
- Blank, U.; Brown, A.; Adams, D.C.; Karolak, M.J.; Oxburgh, L. BMP7 promotes proliferation of nephron progenitor cells via a JNK-dependent mechanism. Development 2009, 136, 3557–3566. [Google Scholar] [CrossRef] [PubMed]
- Muthukrishnan, S.D.; Yang, X.; Friesel, R.; Oxburgh, L. Concurrent BMP7 and FGF9 signalling governs AP-1 function to promote self-renewal of nephron progenitor cells. Nat. Commun. 2015, 6, 10027. [Google Scholar] [CrossRef]
- Liu, J.; Edgington-Giordano, F.; Dugas, C.; Abrams, A.; Katakam, P.; Satou, R.; Saifudeen, Z. Regulation of Nephron Progenitor Cell Self-Renewal by Intermediary Metabolism. J. Am. Soc. Nephrol. 2017, 28, 3323–3335. [Google Scholar] [CrossRef] [PubMed]
- Oxburgh, L.; Rosen, C.J. New Insights into Fuel Choices of Nephron Progenitor Cells. J. Am. Soc. Nephrol. 2017, 28, 3133–3135. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Tomita, M.; Asada, M.; Asada, N.; Nakamura, J.; Oguchi, A.; Higashi, A.Y.; Endo, S.; Robertson, E.; Kimura, T.; Kita, T.; et al. Bmp7 Maintains Undifferentiated Kidney Progenitor Population and Determines Nephron Numbers at Birth. PLoS ONE 2013, 8, e73554. [Google Scholar] [CrossRef]
- Barak, H.; Huh, S.-H.; Chen, S.; Jeanpierre, C.; Martinovic, J.; Parisot, M.; Bole-Feysot, C.; Nitschké, P.; Salomon, R.; Antignac, C.; et al. FGF9 and FGF20 Maintain the Stemness of Nephron Progenitors in Mice and Man. Dev. Cell 2012, 22, 1191–1207. [Google Scholar] [CrossRef]
- Wu, A.; Feng, B.; Yu, J.; Yan, L.; Che, L.; Zhuo, Y.; Luo, Y.; Yu, B.; Wu, D.; Chen, D. Fibroblast growth factor 21 attenuates iron overload-induced liver injury and fibrosis by inhibiting ferroptosis. Redox Biol. 2021, 46, 102131. [Google Scholar] [CrossRef]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423.e7. [Google Scholar] [CrossRef] [PubMed]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta-Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [PubMed]
- Adedoyin, O.; Boddu, R.; Traylor, A.; Lever, J.M.; Bolisetty, S.; George, J.F.; Agarwal, A. Heme oxygenase-1 mitigates Ferroptosis in renal proximal tubule cells. Am. J. Physiol.-Ren. Physiol. 2018, 314, F702–F714. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, H.; Yamanaka, M.; Igarashi, K. Ferroptosis: Regulation by competition between NRF2 and BACH1 and propagation of the death signal. FEBS J. 2022, 290, 1688–1704. [Google Scholar] [CrossRef]
- Sharma, A.; Jeet, S.; Flora, S. Review Article Positive and Negative Regulation of Ferroptosis and Its Role in Maintaining Metabolic and Redox Homeostasis. Oxidative Med. Cell. Longev. 2021, 2021, 9074206. [Google Scholar] [CrossRef]
- Berndt, C.; Lillig, C.H. Glutathione, Glutaredoxins, and Iron. Antioxidants Redox Signal. 2017, 27, 1235–1251. [Google Scholar] [CrossRef]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial membrane potential probes and the proton gradient: A practical usage guide. Biotechniques 2011, 50, 98–115. [Google Scholar] [CrossRef]
- van der Stel, W.; Yang, H.; le Dévédec, S.E.; van de Water, B.; Beltman, J.B.; Danen, E.H.J. High-content high-throughput imaging reveals distinct connections between mitochondrial morphology and functionality for OXPHOS complex I, III, and V inhibitors. Cell Biol. Toxicol. 2022, 39, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Sun, Z.; Xiao, X.; Liu, D.; Qi, H.; Tian, G.; Chen, M.; Chen, L.; Su, X. Transient Inhibition of the JNK Pathway Promotes Human Hematopoietic Stem Cell Quiescence and Engraftment. Stem Cells Transl. Med. 2022, 11, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Chambers, J.W.; LoGrasso, P.V. Mitochondrial c-Jun N-terminal Kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J. Biol. Chem. 2011, 286, 16052–16062. [Google Scholar] [CrossRef] [PubMed]
- Niizuma, K.; Endo, H.; Chan, P.H. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J. Neurochem. 2010, 109, 133–138. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, L.; Thewes, L.; Westerhoff, M.; Wruck, W.; Reichert, A.S.; Berndt, C.; Adjaye, J. JNK Signalling Regulates Self-Renewal of Proliferative Urine-Derived Renal Progenitor Cells via Inhibition of Ferroptosis. Cells 2023, 12, 2197. https://doi.org/10.3390/cells12172197
Nguyen L, Thewes L, Westerhoff M, Wruck W, Reichert AS, Berndt C, Adjaye J. JNK Signalling Regulates Self-Renewal of Proliferative Urine-Derived Renal Progenitor Cells via Inhibition of Ferroptosis. Cells. 2023; 12(17):2197. https://doi.org/10.3390/cells12172197
Chicago/Turabian StyleNguyen, Lisa, Leonie Thewes, Michelle Westerhoff, Wasco Wruck, Andreas S. Reichert, Carsten Berndt, and James Adjaye. 2023. "JNK Signalling Regulates Self-Renewal of Proliferative Urine-Derived Renal Progenitor Cells via Inhibition of Ferroptosis" Cells 12, no. 17: 2197. https://doi.org/10.3390/cells12172197
APA StyleNguyen, L., Thewes, L., Westerhoff, M., Wruck, W., Reichert, A. S., Berndt, C., & Adjaye, J. (2023). JNK Signalling Regulates Self-Renewal of Proliferative Urine-Derived Renal Progenitor Cells via Inhibition of Ferroptosis. Cells, 12(17), 2197. https://doi.org/10.3390/cells12172197