
Mitochondrial Properties in Skeletal Muscle Fiber


Abstract
1. Introduction
2. Mitochondria in Different Types of Skeletal Muscle Fibers
2.1. Mitochondria Content and Oxidative Activity in Different Muscle Fiber Types
2.2. Mitochondria Oxidative Stress in Different Muscle Fiber Types
2.3. Mitochondria Calcium Uptake in Different Muscle Fiber Types
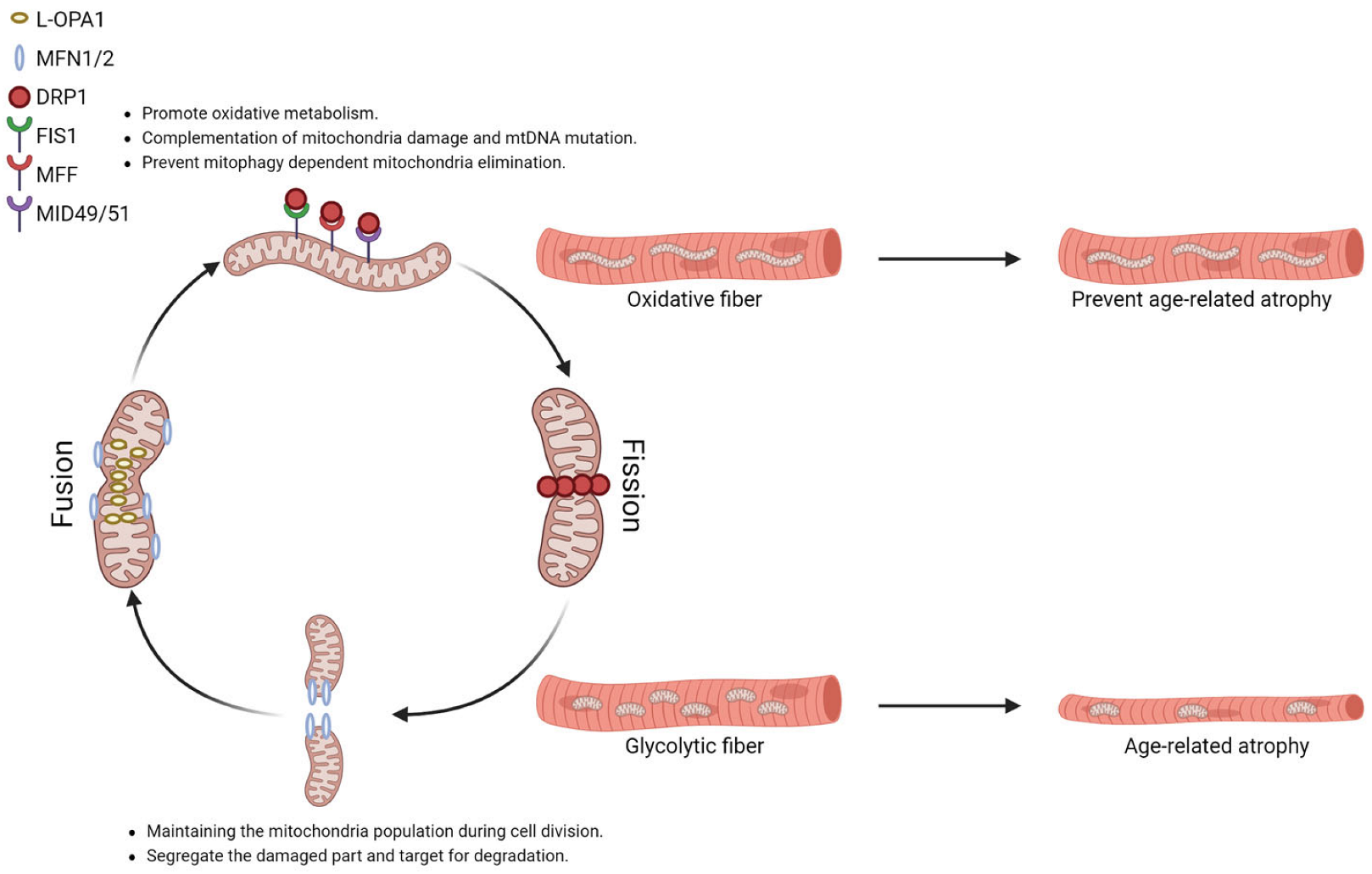
2.4. Mitochondrial Dynamics in Different Muscle Fiber Types
2.5. Mitochondrial Degradation in Different Muscle Fiber Types
3. Mitochondria in Human Myopathies
3.1. Primary Mitochondrial Myopathies
3.2. Other Myopathies with Mitochondria Symptoms
4. Mitochondria in Skeletal Muscle Aging
4.1. Effects of Aging on Mitochondria Content and Function
4.2. Effects of Aging on Mitochondria Quality Control
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.; Swann, K. Mitochondria and lipid metabolism in mammalian oocytes and early embryos. Int. J. Dev. Biol. 2019, 63, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, S.; Di Lisa, F.; Kaludercic, N. Mitochondrial reactive oxygen species in physiology and disease. Cell Calcium 2021, 94, 102344. [Google Scholar] [CrossRef] [PubMed]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef]
- Xiao, Y.; Karam, C.; Yi, J.; Zhang, L.; Li, X.; Yoon, D.; Wang, H.; Dhakal, K.; Ramlow, P.; Yu, T.; et al. ROS-related mitochondrial dysfunction in skeletal muscle of an ALS mouse model during the disease progression. Pharmacol. Res. 2018, 138, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Laverny, G.; Allenbach, Y.; Grelet, E.; Ueberschlag, V.; Echaniz-Laguna, A.; Lannes, B.; Alsaleh, G.; Charles, A.L.; Singh, F.; et al. IFN-beta-induced reactive oxygen species and mitochondrial damage contribute to muscle impairment and inflammation maintenance in dermatomyositis. Acta Neuropathol. 2017, 134, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Werth, J.L.; Thayer, S.A. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J. Neurosci. 1994, 14, 348–356. [Google Scholar] [CrossRef]
- Drago, I.; De Stefani, D.; Rizzuto, R.; Pozzan, T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 12986–12991. [Google Scholar] [CrossRef]
- Gherardi, G.; Nogara, L.; Ciciliot, S.; Fadini, G.P.; Blaauw, B.; Braghetta, P.; Bonaldo, P.; De Stefani, D.; Rizzuto, R.; Mammucari, C. Loss of mitochondrial calcium uniporter rewires skeletal muscle metabolism and substrate preference. Cell Death Differ. 2019, 26, 362–381. [Google Scholar] [CrossRef]
- Pérez-Schindler, J.; Handschin, C. Physiological Regulation of Skeletal Muscle Mass. In Nutrition and Skeletal Muscle; Academic Press: Cambridge, MA, USA, 2019; pp. 139–150. [Google Scholar]
- Holloszy, J.O. Biochemical Adaptations in Muscle. J. Biol. Chem. 1967, 242, 2278–2282. [Google Scholar] [CrossRef] [PubMed]
- Philp, A.M.; Saner, N.J.; Lazarou, M.; Ganley, I.G.; Philp, A. The influence of aerobic exercise on mitochondrial quality control in skeletal muscle. J. Physiol. 2021, 599, 3463–3476. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, A.B.; Gottlieb, R.A. Heart mitochondria: Gates of life and death. Cardiovasc. Res. 2008, 77, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Trigo, D.; Avelar, C.; Fernandes, M.; Sá, J.; da Cruz, E.S.O. Mitochondria, energy, and metabolism in neuronal health and disease. FEBS Lett. 2022, 596, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, H.L.; Hammers, D.W. Muscle Contraction. Cold Spring Harb. Perspect. Biol. 2018, 10, a023200. [Google Scholar] [CrossRef] [PubMed]
- Neunhauserer, D.; Zebedin, M.; Obermoser, M.; Moser, G.; Tauber, M.; Niebauer, J.; Resch, H.; Galler, S. Human skeletal muscle: Transition between fast and slow fibre types. Pflug. Arch. 2011, 461, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Bloemberg, D.; Quadrilatero, J. Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multicolor immunofluorescence analysis. PLoS ONE 2012, 7, e35273. [Google Scholar] [CrossRef]
- Ferraro, E.; Giammarioli, A.M.; Chiandotto, S.; Spoletini, I.; Rosano, G. Exercise-induced skeletal muscle remodeling and metabolic adaptation: Redox signaling and role of autophagy. Antioxid. Redox Signal. 2014, 21, 154–176. [Google Scholar] [CrossRef]
- Rodríguez Cruz, P.M.; Cossins, J.; Beeson, D.; Vincent, A. The Neuromuscular Junction in Health and Disease: Molecular Mechanisms Governing Synaptic Formation and Homeostasis. Front. Mol. Neurosci. 2020, 13, 610964. [Google Scholar] [CrossRef]
- Tintignac, L.A.; Brenner, H.R.; Rüegg, M.A. Mechanisms Regulating Neuromuscular Junction Development and Function and Causes of Muscle Wasting. Physiol. Rev. 2015, 95, 809–852. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.F.; Santos, G.; Schnyder, S.; Handschin, C. PGC-1alpha affects aging-related changes in muscle and motor function by modulating specific exercise-mediated changes in old mice. Aging Cell 2018, 17, e12697. [Google Scholar] [CrossRef] [PubMed]
- Garcia, S.; Nissanka, N.; Mareco, E.A.; Rossi, S.; Peralta, S.; Diaz, F.; Rotundo, R.L.; Carvalho, R.F.; Moraes, C.T. Overexpression of PGC-1alpha in aging muscle enhances a subset of young-like molecular patterns. Aging Cell 2018, 17, e12707. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Chin, S.; Li, P.; Liu, F.; Maratos-Flier, E.; Lebrasseur, N.K.; Yan, Z.; Spiegelman, B.M. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J. Biol. Chem. 2007, 282, 30014–30021. [Google Scholar] [CrossRef]
- Wattez, J.S.; Eury, E.; Hazen, B.C.; Wade, A.; Chau, S.; Ou, S.C.; Russell, A.P.; Cho, Y.; Kralli, A. Loss of skeletal muscle estrogen-related receptors leads to severe exercise intolerance. Mol. Metab. 2023, 68, 101670. [Google Scholar] [CrossRef]
- Sopariwala, D.H.; Rios, A.S.; Pei, G.; Roy, A.; Tomaz da Silva, M.; Thi Thu Nguyen, H.; Saley, A.; Van Drunen, R.; Kralli, A.; Mahan, K.; et al. Innately expressed estrogen-related receptors in the skeletal muscle are indispensable for exercise fitness. FASEB J. 2023, 37, e22727. [Google Scholar] [CrossRef]
- Rangwala, S.M.; Wang, X.; Calvo, J.A.; Lindsley, L.; Zhang, Y.; Deyneko, G.; Beaulieu, V.; Gao, J.; Turner, G.; Markovits, J. Estrogen-related receptor gamma is a key regulator of muscle mitochondrial activity and oxidative capacity. J. Biol. Chem. 2010, 285, 22619–22629. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; He, N.; Lin, C.S.; Wei, Z.; Hah, N.; Waizenegger, W.; He, M.X.; Liddle, C.; Yu, R.T.; Atkins, A.R.; et al. ERRγ Promotes Angiogenesis, Mitochondrial Biogenesis, and Oxidative Remodeling in PGC1α/β-Deficient Muscle. Cell Rep. 2018, 22, 2521–2529. [Google Scholar] [CrossRef] [PubMed]
- Matsakas, A.; Yadav, V.; Lorca, S.; Evans, R.M.; Narkar, V.A. Revascularization of ischemic skeletal muscle by estrogen-related receptor-γ. Circ. Res. 2012, 110, 1087–1096. [Google Scholar] [CrossRef]
- Sopariwala, D.H.; Rios, A.S.; Park, M.K.; Song, M.S.; Kumar, A.; Narkar, V.A. Estrogen-related receptor alpha is an AMPK-regulated factor that promotes ischemic muscle revascularization and recovery in diet-induced obese mice. FASEB BioAdv. 2022, 4, 602–618. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Kumar, V.; Vellichirammal, N.N.; Park, S.Y.; Rudebush, T.L.; Yu, L.; Son, W.M.; Pekas, E.J.; Wafi, A.M.; Hong, J.; et al. Functional, proteomic and bioinformatic analyses of Nrf2- and Keap1-null skeletal muscle. J. Physiol. 2020, 598, 5427–5451. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.; Widjaja, K.; Nguy, B.; Lushaj, E.B.; Moore, T.M.; Hevener, A.L.; McKenzie, D.; Aiken, J.M.; Wanagat, J. Digital PCR Quantitation of Muscle Mitochondrial DNA: Age, Fiber Type, and Mutation-Induced Changes. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Frankish, B.P.; Najdovska, P.; Xu, H.; Wette, S.G.; Murphy, R.M. Effects of voluntary wheel running on mitochondrial content and dynamics in rat skeletal muscle. J. Muscle Res. Cell Motil. 2021, 42, 67–76. [Google Scholar] [CrossRef]
- Glancy, B.; Balaban, R.S. Protein composition and function of red and white skeletal muscle mitochondria. Am. J. Physiol. Cell Physiol. 2011, 300, C1280–C1290. [Google Scholar] [CrossRef]
- Mogensen, M.; Sahlin, K. Mitochondrial efficiency in rat skeletal muscle: Influence of respiration rate, substrate and muscle type. Acta Physiol. Scand. 2005, 185, 229–236. [Google Scholar] [CrossRef]
- Schwerzmann, K.; Hoppeler, H.; Kayar, S.R.; Weibel, E.R. Oxidative capacity of muscle and mitochondria: Correlation of physiological, biochemical, and morphometric characteristics. Proc. Natl. Acad. Sci. USA 1989, 86, 1583–1587. [Google Scholar] [CrossRef]
- Murgia, M.; Nagaraj, N.; Deshmukh, A.S.; Zeiler, M.; Cancellara, P.; Moretti, I.; Reggiani, C.; Schiaffino, S.; Mann, M. Single muscle fiber proteomics reveals unexpected mitochondrial specialization. EMBO Rep. 2015, 16, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.; Nogara, L.; Baraldo, M.; Reggiani, C.; Mann, M.; Schiaffino, S. Protein profile of fiber types in human skeletal muscle: A single-fiber proteomics study. Skelet. Muscle 2021, 11, 24. [Google Scholar] [CrossRef]
- Hoeks, J.; Arany, Z.; Phielix, E.; Moonen-Kornips, E.; Hesselink, M.K.; Schrauwen, P. Enhanced lipid-but not carbohydrate-supported mitochondrial respiration in skeletal muscle of PGC-1alpha overexpressing mice. J. Cell. Physiol. 2012, 227, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.S.; Befroy, D.E.; Codella, R.; Kim, S.; Reznick, R.M.; Hwang, Y.J.; Liu, Z.X.; Lee, H.Y.; Distefano, A.; Samuel, V.T.; et al. Paradoxical effects of increased expression of PGC-1alpha on muscle mitochondrial function and insulin-stimulated muscle glucose metabolism. Proc. Natl. Acad. Sci. USA 2008, 105, 19926–19931. [Google Scholar] [CrossRef] [PubMed]
- Narkar, V.A.; Downes, M.; Yu, R.T.; Embler, E.; Wang, Y.X.; Banayo, E.; Mihaylova, M.M.; Nelson, M.C.; Zou, Y.; Juguilon, H.; et al. AMPK and PPARdelta agonists are exercise mimetics. Cell 2008, 134, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Zhang, C.L.; Yu, R.T.; Cho, H.K.; Nelson, M.C.; Bayuga-Ocampo, C.R.; Ham, J.; Kang, H.; Evans, R.M. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004, 2, e294. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Ali, F.; Chambon, C.; Duteil, D.; Bornert, J.M.; Tardivel, A.; Desvergne, B.; Wahli, W.; Chambon, P.; Metzger, D. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006, 4, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Waizenegger, W.; Lin, C.S.; Sorrentino, V.; He, M.X.; Wall, C.E.; Li, H.; Liddle, C.; Yu, R.T.; Atkins, A.R.; et al. PPARdelta Promotes Running Endurance by Preserving Glucose. Cell Metab. 2017, 25, 1186–1193.e4. [Google Scholar] [CrossRef]
- Jansson, E.; Sjödin, B.; Tesch, P. Changes in muscle fibre type distribution in man after physical training. A sign of fibre type transformation? Acta Physiol. Scand. 1978, 104, 235–237. [Google Scholar] [CrossRef]
- Howald, H.; Hoppeler, H.; Claassen, H.; Mathieu, O.; Straub, R. Influences of endurance training on the ultrastructural composition of the different muscle fiber types in humans. Pflug. Arch. 1985, 403, 369–376. [Google Scholar] [CrossRef]
- Trappe, S.; Harber, M.; Creer, A.; Gallagher, P.; Slivka, D.; Minchev, K.; Whitsett, D. Single muscle fiber adaptations with marathon training. J. Appl. Physiol. 2006, 101, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Little, J.P.; Safdar, A.; Cermak, N.; Tarnopolsky, M.A.; Gibala, M.J. Acute endurance exercise increases the nuclear abundance of PGC-1alpha in trained human skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R912–R917. [Google Scholar] [CrossRef] [PubMed]
- Chilibeck, P.D.; Syrotuik, D.G.; Bell, G.J. The effect of strength training on estimates of mitochondrial density and distribution throughout muscle fibres. Eur. J. Appl. Physiol. Occup. Physiol. 1999, 80, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Green, H.; Goreham, C.; Ouyang, J.; Ball-Burnett, M.; Ranney, D. Regulation of fiber size, oxidative potential, and capillarization in human muscle by resistance exercise. Am. J. Physiol. 1999, 276, R591–R596. [Google Scholar] [CrossRef] [PubMed]
- Haun, C.T.; Vann, C.G.; Osburn, S.C.; Mumford, P.W.; Roberson, P.A.; Romero, M.A.; Fox, C.D.; Johnson, C.A.; Parry, H.A.; Kavazis, A.N.; et al. Muscle fiber hypertrophy in response to 6 weeks of high-volume resistance training in trained young men is largely attributed to sarcoplasmic hypertrophy. PLoS ONE 2019, 14, e0215267. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.D.; Romero, M.A.; Mobley, C.B.; Mumford, P.W.; Roberson, P.A.; Haun, C.T.; Vann, C.G.; Osburn, S.C.; Holmes, H.H.; Greer, R.A.; et al. Skeletal muscle mitochondrial volume and myozenin-1 protein differences exist between high versus low anabolic responders to resistance training. PeerJ 2018, 6, e5338. [Google Scholar] [CrossRef] [PubMed]
- Tesch, P.A.; Komi, P.V.; Häkkinen, K. Enzymatic adaptations consequent to long-term strength training. Int. J. Sports Med. 1987, 8 (Suppl. S1), 66–69. [Google Scholar] [CrossRef]
- Tesch, P.A.; Thorsson, A.; Colliander, E.B. Effects of eccentric and concentric resistance training on skeletal muscle substrates, enzyme activities and capillary supply. Acta Physiol. Scand. 1990, 140, 575–580. [Google Scholar] [CrossRef]
- Wang, N.; Hikida, R.S.; Staron, R.S.; Simoneau, J.A. Muscle fiber types of women after resistance training—Quantitative ultrastructure and enzyme activity. Pflug. Arch. 1993, 424, 494–502. [Google Scholar] [CrossRef]
- Colliander, E.B.; Tesch, P.A. Effects of eccentric and concentric muscle actions in resistance training. Acta Physiol. Scand. 1990, 140, 31–39. [Google Scholar] [CrossRef]
- Salvadego, D.; Domenis, R.; Lazzer, S.; Porcelli, S.; Rittweger, J.; Rizzo, G.; Mavelli, I.; Simunic, B.; Pisot, R.; Grassi, B. Skeletal muscle oxidative function in vivo and ex vivo in athletes with marked hypertrophy from resistance training. J. Appl. Physiol. 2013, 114, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Porter, C.; Reidy, P.T.; Bhattarai, N.; Sidossis, L.S.; Rasmussen, B.B. Resistance Exercise Training Alters Mitochondrial Function in Human Skeletal Muscle. Med. Sci. Sports Exerc. 2015, 47, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Borras, C.; Pallardo, F.V.; Sastre, J.; Ji, L.L.; Vina, J. Decreasing xanthine oxidase-mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J. Physiol. 2005, 567 Pt 1, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; O’Moore, K.M.; Dickman, J.R.; Ji, L.L. Exercise activation of muscle peroxisome proliferator-activated receptor-gamma coactivator-1alpha signaling is redox sensitive. Free Radic. Biol. Med. 2009, 47, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, M.E.; Zhang, S.J.; Bruton, J.; Silva, J.P.; Reid, M.B.; Westerblad, H.; Katz, A. Role of reactive oxygen species in contraction-mediated glucose transport in mouse skeletal muscle. J. Physiol. 2006, 575 Pt 1, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Chambers, M.A.; Moylan, J.S.; Smith, J.D.; Goodyear, L.J.; Reid, M.B. Stretch-stimulated glucose uptake in skeletal muscle is mediated by reactive oxygen species and p38 MAP-kinase. J. Physiol. 2009, 587 Pt 13, 3363–3373. [Google Scholar] [CrossRef] [PubMed]
- Merry, T.L.; Steinberg, G.R.; Lynch, G.S.; McConell, G.K. Skeletal muscle glucose uptake during contraction is regulated by nitric oxide and ROS independently of AMPK. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E577–E585. [Google Scholar] [CrossRef]
- Bouviere, J.; Fortunato, R.S.; Dupuy, C.; Werneck-de-Castro, J.P.; Carvalho, D.P.; Louzada, R.A. Exercise-Stimulated ROS Sensitive Signaling Pathways in Skeletal Muscle. Antioxidants 2021, 10, 537. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863. [Google Scholar] [CrossRef]
- Anderson, E.J.; Neufer, P.D. Type II skeletal myofibers possess unique properties that potentiate mitochondrial H2O2 generation. Am. J. Physiol. Cell Physiol. 2006, 290, C844–C851. [Google Scholar] [CrossRef]
- Drechsel, D.A.; Patel, M. Respiration-dependent H2O2 removal in brain mitochondria via the thioredoxin/peroxiredoxin system. J. Biol. Chem. 2010, 285, 27850–27858. [Google Scholar] [CrossRef] [PubMed]
- Zoccarato, F.; Cavallini, L.; Alexandre, A. Respiration-dependent removal of exogenous H2O2 in brain mitochondria: Inhibition by Ca2+. J. Biol. Chem. 2004, 279, 4166–4174. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A.; Andreyev, A.Y.; Zhang, S.F.; Starkova, N.N.; Korneeva, M.; Syromyatnikov, M.; Popov, V.N. Scavenging of H2O2 by mouse brain mitochondria. J. Bioenerg. Biomembr. 2014, 46, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Banh, S.; Treberg, J.R. The pH sensitivity of H2O2 metabolism in skeletal muscle mitochondria. FEBS Lett. 2013, 587, 1799–1804. [Google Scholar] [CrossRef] [PubMed]
- Treberg, J.R.; Munro, D.; Banh, S.; Zacharias, P.; Sotiri, E. Differentiating between apparent and actual rates of H2O2 metabolism by isolated rat muscle mitochondria to test a simple model of mitochondria as regulators of H2O2 concentration. Redox Biol. 2015, 5, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Boss, O.; Samec, S.; Paoloni-Giacobino, A.; Rossier, C.; Dulloo, A.; Seydoux, J.; Muzzin, P.; Giacobino, J.P. Uncoupling protein-3: A new member of the mitochondrial carrier family with tissue-specific expression. FEBS Lett. 1997, 408, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Puig, A.; Solanes, G.; Grujic, D.; Flier, J.S.; Lowell, B.B. UCP3: An uncoupling protein homologue expressed preferentially and abundantly in skeletal muscle and brown adipose tissue. Biochem. Biophys. Res. Commun. 1997, 235, 79–82. [Google Scholar] [CrossRef]
- Vidal-Puig, A.J.; Grujic, D.; Zhang, C.Y.; Hagen, T.; Boss, O.; Ido, Y.; Szczepanik, A.; Wade, J.; Mootha, V.; Cortright, R.; et al. Energy metabolism in uncoupling protein 3 gene knockout mice. J. Biol. Chem. 2000, 275, 16258–16266. [Google Scholar] [CrossRef]
- Brand, M.D.; Pamplona, R.; Portero-Otin, M.; Requena, J.R.; Roebuck, S.J.; Buckingham, J.A.; Clapham, J.C.; Cadenas, S. Oxidative damage and phospholipid fatty acyl composition in skeletal muscle mitochondria from mice underexpressing or overexpressing uncoupling protein 3. Biochem. J. 2002, 368 Pt 2, 597–603. [Google Scholar] [CrossRef]
- Hesselink, M.K.; Keizer, H.A.; Borghouts, L.B.; Schaart, G.; Kornips, C.F.; Slieker, L.J.; Sloop, K.W.; Saris, W.H.; Schrauwen, P. Protein expression of UCP3 differs between human type 1, type 2a, and type 2b fibers. FASEB J. 2001, 15, 1071–1073. [Google Scholar] [CrossRef][Green Version]
- Russell, A.P.; Somm, E.; Praz, M.; Crettenand, A.; Hartley, O.; Melotti, A.; Giacobino, J.P.; Muzzin, P.; Gobelet, C.; Dériaz, O. UCP3 protein regulation in human skeletal muscle fibre types I, IIa and IIx is dependent on exercise intensity. J. Physiol. 2003, 550 Pt 3, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, P.; Troost, F.J.; Xia, J.; Ravussin, E.; Saris, W.H. Skeletal muscle UCP2 and UCP3 expression in trained and untrained male subjects. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 966–972. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Russell, A.; Wadley, G.; Snow, R.; Giacobino, J.P.; Muzzin, P.; Garnham, A.; Cameron-Smith, D. Slow component of [V]O2 kinetics: The effect of training status, fibre type, UCP3 mRNA and citrate synthase activity. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 157–164. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Russell, A.P.; Wadley, G.; Hesselink, M.K.; Schaart, G.; Lo, S.; Leger, B.; Garnham, A.; Kornips, E.; Cameron-Smith, D.; Giacobino, J.P.; et al. UCP3 protein expression is lower in type I, IIa and IIx muscle fiber types of endurance-trained compared to untrained subjects. Pflug. Arch. 2003, 445, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, M.; Bagger, M.; Pedersen, P.K.; Fernstrom, M.; Sahlin, K. Cycling efficiency in humans is related to low UCP3 content and to type I fibres but not to mitochondrial efficiency. J. Physiol. 2006, 571 Pt 3, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, G.; Cumming, K.T.; Holden, G.; Hallen, J.; Ronnestad, B.R.; Sveen, O.; Skaug, A.; Paur, I.; Bastani, N.E.; Ostgaard, H.N.; et al. Vitamin C and E supplementation hampers cellular adaptation to endurance training in humans: A double-blind, randomised, controlled trial. J. Physiol. 2014, 592, 1887–1901. [Google Scholar] [CrossRef]
- Paulsen, G.; Hamarsland, H.; Cumming, K.T.; Johansen, R.E.; Hulmi, J.J.; Borsheim, E.; Wiig, H.; Garthe, I.; Raastad, T. Vitamin C and E supplementation alters protein signalling after a strength training session, but not muscle growth during 10 weeks of training. J. Physiol. 2014, 592, 5391–5408. [Google Scholar] [CrossRef]
- Deluca, H.F.; Engstrom, G.W. Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. USA 1961, 47, 1744–1750. [Google Scholar] [CrossRef]
- Vasington, F.D.; Murphy, J.V. Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J. Biol. Chem. 1962, 237, 2670–2677. [Google Scholar] [CrossRef]
- Boncompagni, S.; Rossi, A.E.; Micaroni, M.; Beznoussenko, G.V.; Polishchuk, R.S.; Dirksen, R.T.; Protasi, F. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol. Biol. Cell 2009, 20, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Hansford, R.G.; Chappell, J.B. The effect of Ca2+ on the oxidation of glycerol phosphate by blowfly flight-muscle mitochondria. Biochem. Biophys. Res. Commun. 1967, 27, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M.; McCormack, J.G. The calcium sensitive dehydrogenases of vertebrate mitochondria. Cell Calcium 1986, 7, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Vecellio Reane, D.; Vallese, F.; Checchetto, V.; Acquasaliente, L.; Butera, G.; De Filippis, V.; Szabo, I.; Zanotti, G.; Rizzuto, R.; Raffaello, A. A MICU1 Splice Variant Confers High Sensitivity to the Mitochondrial Ca2+ Uptake Machinery of Skeletal Muscle. Mol. Cell 2016, 64, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.V.; Szabadkai, G.; Sharpe, J.A.; Parry, D.A.; Torelli, S.; Childs, A.M.; Kriek, M.; Phadke, R.; Johnson, C.A.; Roberts, N.Y.; et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014, 46, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Smith, D.; Kamer, K.J.; Griffin, H.; Childs, A.M.; Pysden, K.; Titov, D.; Duff, J.; Pyle, A.; Taylor, R.W.; Yu-Wai-Man, P.; et al. Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurol. Genet. 2016, 2, e59. [Google Scholar] [CrossRef]
- Kwong, J.Q.; Huo, J.; Bround, M.J.; Boyer, J.G.; Schwanekamp, J.A.; Ghazal, N.; Maxwell, J.T.; Jang, Y.C.; Khuchua, Z.; Shi, K.; et al. The mitochondrial calcium uniporter underlies metabolic fuel preference in skeletal muscle. J. Clin. Investig. 2018, 3, e121689. [Google Scholar] [CrossRef]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef]
- Debattisti, V.; Horn, A.; Singh, R.; Seifert, E.L.; Hogarth, M.W.; Mazala, D.A.; Huang, K.T.; Horvath, R.; Jaiswal, J.K.; Hajnoczky, G. Dysregulation of Mitochondrial Ca2+ Uptake and Sarcolemma Repair Underlie Muscle Weakness and Wasting in Patients and Mice Lacking MICU1. Cell Rep. 2019, 29, 1274–1286.e6. [Google Scholar] [CrossRef]
- Sembrowich, W.L.; Quintinskie, J.J.; Li, G. Calcium uptake in mitochondria from different skeletal muscle types. J. Appl. Physiol. 1985, 59, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Mammucari, C.; Romanello, V.; Barberi, L.; Pietrangelo, L.; Fusella, A.; Mosole, S.; Gherardi, G.; Hofer, C.; Lofler, S.; et al. Physical exercise in aging human skeletal muscle increases mitochondrial calcium uniporter expression levels and affects mitochondria dynamics. Physiol. Rep. 2016, 4, e13005. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Hartnell, L.M.; Malide, D.; Yu, Z.X.; Combs, C.A.; Connelly, P.S.; Subramaniam, S.; Balaban, R.S. Mitochondrial reticulum for cellular energy distribution in muscle. Nature 2015, 523, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Nakada, K.; Inoue, K.; Ono, T.; Isobe, K.; Ogura, A.; Goto, Y.I.; Nonaka, I.; Hayashi, J.I. Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA. Nat. Med. 2001, 7, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Weaver, D.; Shirihai, O.; Hajnoczky, G. Mitochondrial ‘kiss-and-run’: Interplay between mitochondrial motility and fusion-fission dynamics. EMBO J. 2009, 28, 3074–3089. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef]
- Song, Z.; Ghochani, M.; McCaffery, J.M.; Frey, T.G.; Chan, D.C. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 2009, 20, 3525–3532. [Google Scholar] [CrossRef]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef]
- Noone, J.; O’Gorman, D.J.; Kenny, H.C. OPA1 regulation of mitochondrial dynamics in skeletal and cardiac muscle. Trends Endocrinol. Metab. 2022, 33, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370 Pt 3, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell 1990, 61, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Al Ojaimi, M.; Salah, A.; El-Hattab, A.W. Mitochondrial Fission and Fusion: Molecular Mechanisms, Biological Functions, and Related Disorders. Membranes 2022, 12, 893. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Favaro, G.; Romanello, V.; Varanita, T.; Andrea Desbats, M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef] [PubMed]
- Dulac, M.; Leduc-Gaudet, J.P.; Reynaud, O.; Ayoub, M.B.; Guerin, A.; Finkelchtein, M.; Hussain, S.N.; Gouspillou, G. Drp1 knockdown induces severe muscle atrophy and remodelling, mitochondrial dysfunction, autophagy impairment and denervation. J. Physiol. 2020, 598, 3691–3710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sliter, D.A.; Bleck, C.K.E.; Ding, S. Fis1 deficiencies differentially affect mitochondrial quality in skeletal muscle. Mitochondrion 2019, 49, 217–226. [Google Scholar] [CrossRef]
- Brown, A.D.; Fogarty, M.J.; Sieck, G.C. Mitochondrial morphology and function varies across diaphragm muscle fiber types. Respir. Physiol. Neurobiol. 2022, 295, 103780. [Google Scholar] [CrossRef]
- Mishra, P.; Varuzhanyan, G.; Pham, A.H.; Chan, D.C. Mitochondrial Dynamics is a Distinguishing Feature of Skeletal Muscle Fiber Types and Regulates Organellar Compartmentalization. Cell Metab. 2015, 22, 1033–1044. [Google Scholar] [CrossRef]
- Cartoni, R.; Léger, B.; Hock, M.B.; Praz, M.; Crettenand, A.; Pich, S.; Ziltener, J.L.; Luthi, F.; Dériaz, O.; Zorzano, A.; et al. Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J. Physiol. 2005, 567 Pt 1, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Fealy, C.E.; Mulya, A.; Lai, N.; Kirwan, J.P. Exercise training decreases activation of the mitochondrial fission protein dynamin-related protein-1 in insulin-resistant human skeletal muscle. J. Appl. Physiol. 2014, 117, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Alavi, M.V.; Bette, S.; Schimpf, S.; Schuettauf, F.; Schraermeyer, U.; Wehrl, H.F.; Ruttiger, L.; Beck, S.C.; Tonagel, F.; Pichler, B.J.; et al. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain 2007, 130 Pt 4, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.B.; Bush, Z.; McGinnis, G.R.; Rowe, G.C. Adult skeletal muscle deletion of Mitofusin 1 and 2 impedes exercise performance and training capacity. J. Appl. Physiol. 2019, 126, 341–353. [Google Scholar] [CrossRef]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017, 25, 1374–1389.e6. [Google Scholar] [CrossRef]
- White, R.B.; Bierinx, A.S.; Gnocchi, V.F.; Zammit, P.S. Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev. Biol. 2010, 10, 21. [Google Scholar] [CrossRef]
- Ontell, M.; Feng, K.C.; Klueber, K.; Dunn, R.F.; Taylor, F. Myosatellite cells, growth, and regeneration in murine dystrophic muscle: A quantitative study. Anat. Rec. 1984, 208, 159–174. [Google Scholar] [CrossRef]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabo, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Civiletto, G.; Varanita, T.; Cerutti, R.; Gorletta, T.; Barbaro, S.; Marchet, S.; Lamperti, C.; Viscomi, C.; Scorrano, L.; Zeviani, M. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab. 2015, 21, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Touvier, T.; De Palma, C.; Rigamonti, E.; Scagliola, A.; Incerti, E.; Mazelin, L.; Thomas, J.L.; D’Antonio, M.; Politi, L.; Schaeffer, L.; et al. Muscle-specific Drp1 overexpression impairs skeletal muscle growth via translational attenuation. Cell Death Dis. 2015, 6, e1663. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ren, S.; Clish, C.; Jain, M.; Mootha, V.; McCaffery, J.M.; Chan, D.C. Titration of mitochondrial fusion rescues Mff-deficient cardiomyopathy. J. Cell Biol. 2015, 211, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Lo Verso, F.; Carnio, S.; Vainshtein, A.; Sandri, M. Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy 2014, 10, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Laker, R.C.; Drake, J.C.; Wilson, R.J.; Lira, V.A.; Lewellen, B.M.; Ryall, K.A.; Fisher, C.C.; Zhang, M.; Saucerman, J.J.; Goodyear, L.J.; et al. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat. Commun. 2017, 8, 548. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.W.; Erlich, A.T.; Crilly, M.J.; Hood, D.A. Parkin is required for exercise-induced mitophagy in muscle: Impact of aging. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E404–E415. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Lira, V.A.; Okutsu, M.; Zhang, M.; Greene, N.P.; Laker, R.C.; Breen, D.S.; Hoehn, K.L.; Yan, Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J. 2013, 27, 4184–4193. [Google Scholar] [CrossRef]
- Ju, J.S.; Jeon, S.I.; Park, J.Y.; Lee, J.Y.; Lee, S.C.; Cho, K.J.; Jeong, J.M. Autophagy plays a role in skeletal muscle mitochondrial biogenesis in an endurance exercise-trained condition. J. Physiol. Sci. 2016, 66, 417–430. [Google Scholar] [CrossRef]
- Chen, C.C.W.; Erlich, A.T.; Hood, D.A. Role of Parkin and endurance training on mitochondrial turnover in skeletal muscle. Skelet. Muscle 2018, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H.; et al. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Quijano, C.; Chen, E.; Liu, H.; Cao, L.; Fergusson, M.M.; Rovira, I.I.; Gutkind, S.; Daniels, M.P.; Komatsu, M.; et al. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging 2009, 1, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Gouspillou, G.; Godin, R.; Piquereau, J.; Picard, M.; Mofarrahi, M.; Mathew, J.; Purves-Smith, F.M.; Sgarioto, N.; Hepple, R.T.; Burelle, Y.; et al. Protective role of Parkin in skeletal muscle contractile and mitochondrial function. J. Physiol. 2018, 596, 2565–2579. [Google Scholar] [CrossRef] [PubMed]
- Diwan, A.; Krenz, M.; Syed, F.M.; Wansapura, J.; Ren, X.; Koesters, A.G.; Li, H.; Kirshenbaum, L.A.; Hahn, H.S.; Robbins, J.; et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J. Clin. Investig. 2007, 117, 2825–2833. [Google Scholar] [CrossRef] [PubMed]
- Diwan, A.; Koesters, A.G.; Odley, A.M.; Pushkaran, S.; Baines, C.P.; Spike, B.T.; Daria, D.; Jegga, A.G.; Geiger, H.; Aronow, B.J.; et al. Unrestrained erythroblast development in Nix−/− mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc. Natl. Acad. Sci. USA 2007, 104, 6794–6799. [Google Scholar] [CrossRef]
- Zhang, W.; Ren, H.; Xu, C.; Zhu, C.; Wu, H.; Liu, D.; Wang, J.; Liu, L.; Li, W.; Ma, Q.; et al. Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. Elife 2016, 5, e21407. [Google Scholar] [CrossRef]
- Zhang, W.; Ma, Q.; Siraj, S.; Ney, P.A.; Liu, J.; Liao, X.; Yuan, Y.; Li, W.; Liu, L.; Chen, Q. Nix-mediated mitophagy regulates platelet activation and life span. Blood Adv. 2019, 3, 2342–2354. [Google Scholar] [CrossRef]
- Sandoval, H.; Thiagarajan, P.; Dasgupta, S.K.; Schumacher, A.; Prchal, J.T.; Chen, M.; Wang, J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008, 454, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Xu, Z.; Liu, L.; Guo, Q.; Wu, H.; Liang, X.; Zhou, D.; Xiao, L.; Liu, L.; Liu, Y.; et al. Mitophagy Directs Muscle-Adipose Crosstalk to Alleviate Dietary Obesity. Cell Rep. 2018, 23, 1357–1372. [Google Scholar] [CrossRef] [PubMed]
- D’Acunzo, P.; Perez-Gonzalez, R.; Kim, Y.; Hargash, T.; Miller, C.; Alldred, M.J.; Erdjument-Bromage, H.; Penikalapati, S.C.; Pawlik, M.; Saito, M.; et al. Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in Down syndrome. Sci. Adv. 2021, 7, eabe5085. [Google Scholar] [CrossRef] [PubMed]
- Todkar, K.; Chikhi, L.; Desjardins, V.; El-Mortada, F.; Pepin, G.; Germain, M. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 2021, 12, 1971. [Google Scholar] [CrossRef] [PubMed]
- Phinney, D.G.; Di Giuseppe, M.; Njah, J.; Sala, E.; Shiva, S.; St Croix, C.M.; Stolz, D.B.; Watkins, S.C.; Di, Y.P.; Leikauf, G.D.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nat. Commun. 2015, 6, 8472. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.W.S.; Lu, G.; Dong, H.; Cho, Y.L.; Natalia, A.; Wang, L.; Chan, C.; Kappei, D.; Taneja, R.; Ling, S.C.; et al. A degradative to secretory autophagy switch mediates mitochondria clearance in the absence of the mATG8-conjugation machinery. Nat. Commun. 2022, 13, 3720. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Buajitti, E.; Rosella, L.C.; Zabzuni, E.; Young, L.T.; Andreazza, A.C. Prevalence and health care costs of mitochondrial disease in Ontario, Canada: A population-based cohort study. PLoS ONE 2022, 17, e0265744. [Google Scholar] [CrossRef]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.Q.; Tarnopolsky, M.A. Mitochondrial neuropathy and neurogenic features in mitochondrial myopathy. Mitochondrion 2021, 56, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.J.; Shen, L.; Gonzalez, M.; Leipzig, J.; Lott, M.T.; Stassen, A.P.; Diroma, M.A.; Navarro-Gomez, D.; Yeske, P.; Bai, R.; et al. Mitochondrial Disease Sequence Data Resource (MSeqDR): A global grass-roots consortium to facilitate deposition, curation, annotation, and integrated analysis of genomic data for the mitochondrial disease clinical and research communities. Mol. Genet. Metab. 2015, 114, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.J.; Sondheimer, N. Mitochondrial genetic diseases. Curr. Opin. Pediatr. 2010, 22, 711–716. [Google Scholar] [CrossRef] [PubMed]
- McCormick, E.M.; Zolkipli-Cunningham, Z.; Falk, M.J. Mitochondrial disease genetics update: Recent insights into the molecular diagnosis and expanding phenotype of primary mitochondrial disease. Curr. Opin. Pediatr. 2018, 30, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Filosto, M.; Tonin, P.; Vattemi, G.; Spagnolo, M.; Rizzuto, N.; Tomelleri, G. Antioxidant agents have a different expression pattern in muscle fibers of patients with mitochondrial diseases. Acta Neuropathol. 2002, 103, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Felczak, P.; Lewandowska, E.; Stepniak, I.; Oldak, M.; Pollak, A.; Lechowicz, U.; Pasennik, E.; Stepien, T.; Wierzba-Bobrowicz, T. Pathology of mitochondria in MELAS syndrome: An ultrastructural study. Pol. J. Pathol. 2017, 68, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Nelson, I.; Bonne, G.; Degoul, F.; Marsac, C.; Ponsot, G.; Lestienne, P. Kearns-Sayre syndrome with sideroblastic anemia: Molecular investigations. Neuropediatrics 1992, 23, 199–205. [Google Scholar] [CrossRef]
- Wiedemann, F.R.; Bartels, C.; Kirches, E.; Mawrin, C.; Wallesch, C.W. Unusual presentations of patients with the mitochondrial MERRF mutation A8344G. Clin. Neurol. Neurosurg. 2008, 110, 859–863. [Google Scholar] [CrossRef]
- Trevisson, E.; DiMauro, S.; Navas, P.; Salviati, L. Coenzyme Q deficiency in muscle. Curr. Opin. Neurol. 2011, 24, 449–456. [Google Scholar] [CrossRef]
- Ugalde, C.; Hinttala, R.; Timal, S.; Smeets, R.; Rodenburg, R.J.; Uusimaa, J.; van Heuvel, L.P.; Nijtmans, L.G.; Majamaa, K.; Smeitink, J.A. Mutated ND2 impairs mitochondrial complex I assembly and leads to Leigh syndrome. Mol. Genet. Metab. 2007, 90, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, A.S. Understanding mitochondrial myopathies: A review. PeerJ 2018, 6, e4790. [Google Scholar] [CrossRef] [PubMed]
- McClelland, C.; Manousakis, G.; Lee, M.S. Progressive External Ophthalmoplegia. Curr. Neurol. Neurosci. Rep. 2016, 16, 53. [Google Scholar] [CrossRef] [PubMed]
- Seneca, S.; Goemans, N.; Van Coster, R.; Givron, P.; Reybrouck, T.; Sciot, R.; Meulemans, A.; Smet, J.; Van Hove, J.L. A mitochondrial tRNA aspartate mutation causing isolated mitochondrial myopathy. Am. J. Med. Genet. A 2005, 137, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissière, A.; Campos, Y.; Rivera, H.; de la Aleja, J.G.; Carroccia, R.; et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 2008, 131 Pt 2, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Vilà, M.R.; Villarroya, J.; García-Arumí, E.; Castellote, A.; Meseguer, A.; Hirano, M.; Roig, M. Selective muscle fiber loss and molecular compensation in mitochondrial myopathy due to TK2 deficiency. J. Neurol. Sci. 2008, 267, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Maeda, K.; Iizuka, T.; Hayashi, M.; Hashizume, Y.; Sanada, M.; Kawai, H.; Kashiwagi, A. Accumulation of oxidative stress around the stroke-like lesions of MELAS patients. Mitochondrion 2009, 9, 306–313. [Google Scholar] [CrossRef]
- Chou, S.J.; Tseng, W.L.; Chen, C.T.; Lai, Y.F.; Chien, C.S.; Chang, Y.L.; Lee, H.C.; Wei, Y.H.; Chiou, S.H. Impaired ROS Scavenging System in Human Induced Pluripotent Stem Cells Generated from Patients with MERRF Syndrome. Sci. Rep. 2016, 6, 23661. [Google Scholar] [CrossRef]
- Brini, M.; Pinton, P.; King, M.P.; Davidson, M.; Schon, E.A.; Rizzuto, R. A calcium signaling defect in the pathogenesis of a mitochondrial DNA inherited oxidative phosphorylation deficiency. Nat. Med. 1999, 5, 951–954. [Google Scholar] [CrossRef]
- Joshi, P.R.; Hauburger, A.; Kley, R.; Claeys, K.G.; Schneider, I.; Kress, W.; Stoltenburg, G.; Weis, J.; Vorgerd, M.; Deschauer, M.; et al. Mitochondrial abnormalities in myofibrillar myopathies. Clin. Neuropathol. 2014, 33, 134–142. [Google Scholar] [CrossRef]
- Claeys, K.G.; Fardeau, M.; Schroder, R.; Suominen, T.; Tolksdorf, K.; Behin, A.; Dubourg, O.; Eymard, B.; Maisonobe, T.; Stojkovic, T.; et al. Electron microscopy in myofibrillar myopathies reveals clues to the mutated gene. Neuromuscul. Disord. 2008, 18, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Milner, D.J.; Weitzer, G.; Tran, D.; Bradley, A.; Capetanaki, Y. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J. Cell Biol. 1996, 134, 1255–1270. [Google Scholar] [CrossRef]
- Milner, D.J.; Mavroidis, M.; Weisleder, N.; Capetanaki, Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J. Cell Biol. 2000, 150, 1283–1298. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Schaefer, J.; Meinhardt, M.; Reichmann, H. Mitochondrial abnormalities in the myofibrillar myopathies. Eur. J. Neurol. 2015, 22, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Filipe, A.; Chernorudskiy, A.; Arbogast, S.; Varone, E.; Villar-Quiles, R.N.; Pozzer, D.; Moulin, M.; Fumagalli, S.; Cabet, E.; Dudhal, S.; et al. Defective endoplasmic reticulum-mitochondria contacts and bioenergetics in SEPN1-related myopathy. Cell Death Differ. 2021, 28, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Boncompagni, S.; Rossi, A.E.; Micaroni, M.; Hamilton, S.L.; Dirksen, R.T.; Franzini-Armstrong, C.; Protasi, F. Characterization and temporal development of cores in a mouse model of malignant hyperthermia. Proc. Natl. Acad. Sci. USA 2009, 106, 21996–22001. [Google Scholar] [CrossRef] [PubMed]
- Miró, O.; Casademont, J.; Grau, J.M.; Jarreta, D.; Urbano-Márquez, A.; Cardellach, F. Histological and biochemical assessment of mitochondrial function in dermatomyositis. Br. J. Rheumatol. 1998, 37, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, L.E.; Drost, M.R.; Schaart, G.; de Laat, J.; van Doorn, P.A.; van der Ploeg, A.T.; Reuser, A.J. Muscle fiber-type distribution, fiber-type-specific damage, and the Pompe disease phenotype. J. Inherit. Metab. Dis. 2013, 36, 787–794. [Google Scholar] [CrossRef]
- Bartsakoulia, M.; Pyle, A.; Troncoso-Chandia, D.; Vial-Brizzi, J.; Paz-Fiblas, M.V.; Duff, J.; Griffin, H.; Boczonadi, V.; Lochmuller, H.; Kleinle, S.; et al. A novel mechanism causing imbalance of mitochondrial fusion and fission in human myopathies. Hum. Mol. Genet. 2018, 27, 1186–1195. [Google Scholar] [CrossRef]
- Liu, F.; Lou, J.; Zhao, D.; Li, W.; Zhao, Y.; Sun, X.; Yan, C. Dysferlinopathy: Mitochondrial abnormalities in human skeletal muscle. Int. J. Neurosci. 2016, 126, 499–509. [Google Scholar] [CrossRef]
- Lim, J.A.; Li, L.; Kakhlon, O.; Myerowitz, R.; Raben, N. Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy 2015, 11, 385–402. [Google Scholar] [CrossRef]
- Imoto, C.; Nonaka, I. The significance of type 1 fiber atrophy (hypotrophy) in childhood neuromuscular disorders. Brain Dev. 2001, 23, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Herasse, M.; Parain, K.; Marty, I.; Monnier, N.; Kaindl, A.M.; Leroy, J.P.; Richard, P.; Lunardi, J.; Romero, N.B.; Ferreiro, A. Abnormal distribution of calcium-handling proteins: A novel distinctive marker in core myopathies. J. Neuropathol. Exp. Neurol. 2007, 66, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Menshikova, E.V.; Ritov, V.B.; Fairfull, L.; Ferrell, R.E.; Kelley, D.E.; Goodpaster, B.H. Effects of exercise on mitochondrial content and function in aging human skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef]
- Gonzalez-Freire, M.; Scalzo, P.; D’Agostino, J.; Moore, Z.A.; Diaz-Ruiz, A.; Fabbri, E.; Zane, A.; Chen, B.; Becker, K.G.; Lehrmann, E.; et al. Skeletal muscle ex vivo mitochondrial respiration parallels decline in vivo oxidative capacity, cardiorespiratory fitness, and muscle strength: The Baltimore Longitudinal Study of Aging. Aging Cell 2018, 17, e12725. [Google Scholar] [CrossRef]
- Porter, C.; Hurren, N.M.; Cotter, M.V.; Bhattarai, N.; Reidy, P.T.; Dillon, E.L.; Durham, W.J.; Tuvdendorj, D.; Sheffield-Moore, M.; Volpi, E.; et al. Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E224–E232. [Google Scholar] [CrossRef]
- Crupi, A.N.; Nunnelee, J.S.; Taylor, D.J.; Thomas, A.; Vit, J.P.; Riera, C.E.; Gottlieb, R.A.; Goodridge, H.S. Oxidative muscles have better mitochondrial homeostasis than glycolytic muscles throughout life and maintain mitochondrial function during aging. Aging 2018, 10, 3327–3352. [Google Scholar] [CrossRef]
- Ferri, E.; Marzetti, E.; Calvani, R.; Picca, A.; Cesari, M.; Arosio, B. Role of Age-Related Mitochondrial Dysfunction in Sarcopenia. Int. J. Mol. Sci. 2020, 21, 5236. [Google Scholar] [CrossRef]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Capel, F.; Rimbert, V.; Lioger, D.; Diot, A.; Rousset, P.; Mirand, P.P.; Boirie, Y.; Morio, B.; Mosoni, L. Due to reverse electron transfer, mitochondrial H2O2 release increases with age in human vastus lateralis muscle although oxidative capacity is preserved. Mech. Ageing Dev. 2005, 126, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Lertwattanarak, R.; Lefort, N.; Molina-Carrion, M.; Joya-Galeana, J.; Bowen, B.P.; Garduno-Garcia Jde, J.; Abdul-Ghani, M.; Richardson, A.; DeFronzo, R.A.; et al. Reduction in reactive oxygen species production by mitochondria from elderly subjects with normal and impaired glucose tolerance. Diabetes 2011, 60, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Hey-Mogensen, M.; Gram, M.; Jensen, M.B.; Lund, M.T.; Hansen, C.N.; Scheibye-Knudsen, M.; Bohr, V.A.; Dela, F. A novel method for determining human ex vivo submaximal skeletal muscle mitochondrial function. J. Physiol. 2015, 593, 3991–4010. [Google Scholar] [CrossRef] [PubMed]
- Gram, M.; Vigelso, A.; Yokota, T.; Helge, J.W.; Dela, F.; Hey-Mogensen, M. Skeletal muscle mitochondrial H2O2 emission increases with immobilization and decreases after aerobic training in young and older men. J. Physiol. 2015, 593, 4011–4027. [Google Scholar] [CrossRef] [PubMed]
- Marzani, B.; Felzani, G.; Bellomo, R.G.; Vecchiet, J.; Marzatico, F. Human muscle aging: ROS-mediated alterations in rectus abdominis and vastus lateralis muscles. Exp. Gerontol. 2005, 40, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Gianni, P.; Jan, K.J.; Douglas, M.J.; Stuart, P.M.; Tarnopolsky, M.A. Oxidative stress and the mitochondrial theory of aging in human skeletal muscle. Exp. Gerontol. 2004, 39, 1391–1400. [Google Scholar] [CrossRef]
- Beltran Valls, M.R.; Wilkinson, D.J.; Narici, M.V.; Smith, K.; Phillips, B.E.; Caporossi, D.; Atherton, P.J. Protein carbonylation and heat shock proteins in human skeletal muscle: Relationships to age and sarcopenia. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 174–181. [Google Scholar] [CrossRef]
- Fano, G.; Mecocci, P.; Vecchiet, J.; Belia, S.; Fulle, S.; Polidori, M.C.; Felzani, G.; Senin, U.; Vecchiet, L.; Beal, M.F. Age and sex influence on oxidative damage and functional status in human skeletal muscle. J. Muscle Res. Cell Motil. 2001, 22, 345–351. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA 2007, 297, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Sesso, H.D.; Christen, W.G.; Bubes, V.; Smith, J.P.; MacFadyen, J.; Schvartz, M.; Manson, J.E.; Glynn, R.J.; Buring, J.E.; Gaziano, J.M. Multivitamins in the prevention of cardiovascular disease in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2012, 308, 1751–1760. [Google Scholar] [CrossRef] [PubMed]
- Lippman, S.M.; Klein, E.A.; Goodman, P.J.; Lucia, M.S.; Thompson, I.M.; Ford, L.G.; Parnes, H.L.; Minasian, L.M.; Gaziano, J.M.; Hartline, J.A.; et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009, 301, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Gaziano, J.M.; Glynn, R.J.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Sesso, H.D.; Buring, J.E. Vitamins E and C in the prevention of prostate and total cancer in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2009, 301, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Sesso, H.D.; Buring, J.E.; Christen, W.G.; Kurth, T.; Belanger, C.; MacFadyen, J.; Bubes, V.; Manson, J.E.; Glynn, R.J.; Gaziano, J.M. Vitamins E and C in the prevention of cardiovascular disease in men: The Physicians’ Health Study II randomized controlled trial. JAMA 2008, 300, 2123–2133. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [PubMed]
- Fiatarone, M.A.; O’Neill, E.F.; Ryan, N.D.; Clements, K.M.; Solares, G.R.; Nelson, M.E.; Roberts, S.B.; Kehayias, J.J.; Lipsitz, L.A.; Evans, W.J. Exercise training and nutritional supplementation for physical frailty in very elderly people. N. Engl. J. Med. 1994, 330, 1769–1775. [Google Scholar] [CrossRef]
- Bobeuf, F.; Labonte, M.; Dionne, I.J.; Khalil, A. Combined effect of antioxidant supplementation and resistance training on oxidative stress markers, muscle and body composition in an elderly population. J. Nutr. Health Aging 2011, 15, 883–889. [Google Scholar] [CrossRef]
- Bjornsen, T.; Salvesen, S.; Berntsen, S.; Hetlelid, K.J.; Stea, T.H.; Lohne-Seiler, H.; Rohde, G.; Haraldstad, K.; Raastad, T.; Kopp, U.; et al. Vitamin C and E supplementation blunts increases in total lean body mass in elderly men after strength training. Scand. J. Med. Sci. Sports 2016, 26, 755–763. [Google Scholar] [CrossRef]
- Balagopal, P.; Rooyackers, O.E.; Adey, D.B.; Ades, P.A.; Nair, K.S. Effects of aging on in vivo synthesis of skeletal muscle myosin heavy-chain and sarcoplasmic protein in humans. Am. J. Physiol. 1997, 273, E790–E800. [Google Scholar] [CrossRef]
- Kumar, V.; Selby, A.; Rankin, D.; Patel, R.; Atherton, P.; Hildebrandt, W.; Williams, J.; Smith, K.; Seynnes, O.; Hiscock, N.; et al. Age-related differences in the dose-response relationship of muscle protein synthesis to resistance exercise in young and old men. J. Physiol. 2009, 587, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Delbono, O.; O’Rourke, K.S.; Ettinger, W.H. Excitation-calcium release uncoupling in aged single human skeletal muscle fibers. J. Membr. Biol. 1995, 148, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Moreno, R.; Wang, Z.M.; Gerring, R.C.; Delbono, O. Sarcoplasmic reticulum Ca2+ release declines in muscle fibers from aging mice. Biophys. J. 2008, 94, 3178–3188. [Google Scholar] [CrossRef] [PubMed]
- Lamboley, C.R.; Wyckelsma, V.L.; Dutka, T.L.; McKenna, M.J.; Murphy, R.M.; Lamb, G.D. Contractile properties and sarcoplasmic reticulum calcium content in type I and type II skeletal muscle fibres in active aged humans. J. Physiol. 2015, 593, 2499–2514. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.F.; Yang, W.; Liao, Z.Y.; Wu, Y.X.; Fan, Z.; Guo, A.; Yu, J.; Chen, Q.N.; Wu, J.H.; Zhou, J.; et al. MICU3 regulates mitochondrial Ca2+-dependent antioxidant response in skeletal muscle aging. Cell Death Dis. 2021, 12, 1115. [Google Scholar] [CrossRef] [PubMed]
- Pietrangelo, L.; D’Incecco, A.; Ainbinder, A.; Michelucci, A.; Kern, H.; Dirksen, R.T.; Boncompagni, S.; Protasi, F. Age-dependent uncoupling of mitochondria from Ca2+ release units in skeletal muscle. Oncotarget 2015, 6, 35358–35371. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Pietrangelo, L.; Loefler, S.; Fruhmann, H.; Vogelauer, M.; Burggraf, S.; Pond, A.; Grim-Stieger, M.; Cvecka, J.; Sedliak, M.; et al. Lifelong physical exercise delays age-associated skeletal muscle decline. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 163–173. [Google Scholar] [CrossRef]
- Jin, J.Y.; Wei, X.X.; Zhi, X.L.; Wang, X.H.; Meng, D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol. Sin. 2021, 42, 655–664. [Google Scholar] [CrossRef]
- Archer, S.L. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. [Google Scholar] [CrossRef]
- Konopka, A.R.; Suer, M.K.; Wolff, C.A.; Harber, M.P. Markers of human skeletal muscle mitochondrial biogenesis and quality control: Effects of age and aerobic exercise training. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 371–378. [Google Scholar] [CrossRef]
- Balan, E.; Schwalm, C.; Naslain, D.; Nielens, H.; Francaux, M.; Deldicque, L. Regular Endurance Exercise Promotes Fission, Mitophagy, and Oxidative Phosphorylation in Human Skeletal Muscle Independently of Age. Front. Physiol. 2019, 10, 1088. [Google Scholar] [CrossRef] [PubMed]
- Distefano, G.; Standley, R.A.; Dubé, J.J.; Carnero, E.A.; Ritov, V.B.; Stefanovic-Racic, M.; Toledo, F.G.; Piva, S.R.; Goodpaster, B.H.; Coen, P.M. Chronological Age Does not Influence Ex-vivo Mitochondrial Respiration and Quality Control in Skeletal Muscle. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 535–542. [Google Scholar] [PubMed]
- Joseph, A.M.; Adhihetty, P.J.; Buford, T.W.; Wohlgemuth, S.E.; Lees, H.A.; Nguyen, L.M.; Aranda, J.M.; Sandesara, B.D.; Pahor, M.; Manini, T.M.; et al. The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 2012, 11, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Crane, J.D.; Devries, M.C.; Safdar, A.; Hamadeh, M.J.; Tarnopolsky, M.A. The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 119–128. [Google Scholar] [CrossRef]
- O’Leary, M.F.; Vainshtein, A.; Iqbal, S.; Ostojic, O.; Hood, D.A. Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am. J. Physiol. Cell Physiol. 2013, 304, C422–C430. [Google Scholar] [CrossRef]
- O’Connell, K.; Ohlendieck, K. Proteomic DIGE analysis of the mitochondria-enriched fraction from aged rat skeletal muscle. Proteomics 2009, 9, 5509–5524. [Google Scholar] [CrossRef]
- Iqbal, S.; Ostojic, O.; Singh, K.; Joseph, A.M.; Hood, D.A. Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle Nerve 2013, 48, 963–970. [Google Scholar] [CrossRef]
- Faitg, J.; Leduc-Gaudet, J.P.; Reynaud, O.; Ferland, G.; Gaudreau, P.; Gouspillou, G. Effects of Aging and Caloric Restriction on Fiber Type Composition, Mitochondrial Morphology and Dynamics in Rat Oxidative and Glycolytic Muscles. Front. Physiol. 2019, 10, 420. [Google Scholar] [CrossRef]
- Koltai, E.; Hart, N.; Taylor, A.W.; Goto, S.; Ngo, J.K.; Davies, K.J.; Radak, Z. Age-associated declines in mitochondrial biogenesis and protein quality control factors are minimized by exercise training. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R127–R134. [Google Scholar] [CrossRef]
- Capitanio, D.; Vasso, M.; De Palma, S.; Fania, C.; Torretta, E.; Cammarata, F.P.; Magnaghi, V.; Procacci, P.; Gelfi, C. Specific protein changes contribute to the differential muscle mass loss during ageing. Proteomics 2016, 16, 645–656. [Google Scholar] [CrossRef]
- Zhao, L.; Zou, X.; Feng, Z.; Luo, C.; Liu, J.; Li, H.; Chang, L.; Wang, H.; Li, Y.; Long, J.; et al. Evidence for association of mitochondrial metabolism alteration with lipid accumulation in aging rats. Exp. Gerontol. 2014, 56, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Joseph, A.M.; Adhihetty, P.J.; Wawrzyniak, N.R.; Wohlgemuth, S.E.; Picca, A.; Kujoth, G.C.; Prolla, T.A.; Leeuwenburgh, C. Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS ONE 2013, 8, e69327. [Google Scholar] [CrossRef] [PubMed]
- Leduc-Gaudet, J.P.; Picard, M.; St-Jean Pelletier, F.; Sgarioto, N.; Auger, M.J.; Vallée, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef] [PubMed]
- Yeo, D.; Kang, C.; Gomez-Cabrera, M.C.; Vina, J.; Ji, L.L. Intensified mitophagy in skeletal muscle with aging is downregulated by PGC-1alpha overexpression in vivo. Free Radic. Biol. Med. 2019, 130, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.D.; Davis, L.A.; Fogarty, M.J.; Sieck, G.C. Mitochondrial Fragmentation and Dysfunction in Type IIx/IIb Diaphragm Muscle Fibers in 24-Month Old Fischer 344 Rats. Front. Physiol. 2021, 12, 727585. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Irazoki, A.; Martinez-Vicente, M.; Aparicio, P.; Aris, C.; Alibakhshi, E.; Rubio-Valera, M.; Castellanos, J.; Lores, L.; Palacín, M.; Gumà, A.; et al. Coordination of mitochondrial and lysosomal homeostasis mitigates inflammation and muscle atrophy during aging. Aging Cell 2022, 21, e13583. [Google Scholar] [CrossRef]


{kind=link}
| Myopathy | Gene of Mutation | nDNA/mtDNA | Muscle and Mitochondria Phenotypes | Reference |
|---|---|---|---|---|
| CPEO | POLG1, POLG2, ATN1, C10ORF2, Opa1, TK2, and multiple mtDNA or mtRNA | nDNA/mtDNA | mitochondria with swollen cristae or paracrystalline inclusions. Reduce mitochondria respiration capacity. Increase ROS generation. Mitochondria fusion defect. | [167,174,176] |
| TK-2 DEFICIENCY | TK2 | nDNA | Selective loss of type II fiber. Increase the proportion of SDH staining positive fiber and Cox staining negative fiber. | [177] |
| KEARNS-SAYRE SYNDROME | Variable single mtDNA deletion | mtDNA | Reduce mitochondria respiration activity. | [169] |
| MELAS | MT-TL1, MT-TH, MT-TV, MT-ND5, and MT-ND5 | mtDNA | Enlarged mitochondria or slightly swollen small mitochondria. Reduce mitochondria respiration activity and increase ROS generation. | [168,178] |
| MERRF | MT-TK, MT-TL1, MT-TH, and MT-TS1 | mtDNA | Reduce mitochondria respiration activity, increase ROS generation, ROS clearance defect. | [170,178,179,180] |
| COQ10 DEFICIENCY | COQ2, COQ4, COQ6, COQ7, COQ8A, COQ8B, COQ9, PDSS1, and PDSS2 | nDNA | Reduce mitochondria respiration activity. | [171] |
| LEIGH SYNDROME | ND2, and SURF1 | nDNA | Reduce mitochondria respiration activity, reduce mitochondria complex I activity. | [172] |
| Myopathy | Gene of Mutation | nDNA/mtDNA | Muscle and Mitochondria Phenotypes |
|---|---|---|---|
| ab-crystallinopathy | CRYAB | nDNA | Increase the proportion of rubbed-out fibers (low complex II activity), Cox staining negative fibers (low complex IV activity), and paracrystalline inclusions. |
| BAG3 myopathy | BAG3 | nDNA | Increase the proportion of rubbed-out fibers (low complex II activity) |
| Desminopathy | DES | nDNA | Altered mitochondria distribution. Cox negative fibers (low complex IV activity). Enlarged, vacuolated mitochondria. Mitochondria with abnormal cristae. Mitochondria with paracrystalline inclusion. |
| DNAJB6 | DNAJB6 | nDNA | Increase proportion of rubbed-out fibers (low complex II activity), Cox staining negative fibers (low complex IV activity) |
| FHL1 | FHL1 | nDNA | Increase the proportion of rubbed-out fibers (low complex II activity), Cox staining negative fibers (low complex IV activity), and Ragged Red Fibers. |
| Filaminopathy | FLNC | nDNA | Increase the proportion of Cox staining negative fibers (low complex IV activity), Ragged Red Fibers. |
| Myotilinopathy | MYOT | nDNA | Increase proportion of rubbed-out fibers (low complex II activity), Cox staining negative fibers (low complex IV activity) |
| Plectinopathy | PLEC | nDNA | Abnormal mitochondria distribution, increased proportion of rubbed-out fibers (low complex II activity), Cox staining negative fibers (low complex IV activity), and paracrystalline inclusions |
| Titinopathy | TTN | nDNA | Focal areas of mitochondrial depletion, increased proportion of Cox staining negative fibers (low complex IV activity), and paracrystalline inclusions |
| ZASPopathy | ZASP | nDNA | Increase proportion of rubbed-out fibers (low complex II activity), Cox staining negative fibers (low complex IV activity) |
| Myopathy | Gene of Mutation | nDNA/mtDNA | Muscle and Mitochondria Phenotypes | Reference |
|---|---|---|---|---|
| dermatomyositis | Unknown | Increase the proportion of SDH staining positive fiber and Cox staining negative fiber. | [188] | |
| Pompe Disease | GAA | nDNA | Short and fragmented mitochondria reduce mitochondria respiration activity and ATP generation. Increase ROS generation. Mitochondria calcium overload. Increase expression of mitochondria dynamic-related protein but reduce mitophagy activity. | [189,192] |
| Metabolic myopathy | MIEF2 | nDNA | Elongated mitochondria, aberrant mitochondrial cristae organization. | [190] |
| Dysferlinopathy | DYSF | nDNA | Decrease complex I, III, and IV protein level and activity, decrease cell ATP level. | [191] |
| Species | Sex | Tissue | Model | Protein Fraction | Mitochondria Dynamic Proteins | Mitophagy/Autophagy Related Proteins | Reference |
|---|---|---|---|---|---|---|---|
| Humans | Male | Vastus Lateralis | Younger men (20 ± 1 years) vs. older men (74 ± 3 years) | Whole muscle lysate | Mfn1, Mfn2 and Fis1: NS | [231] | |
| Humans | Male | Vastus Lateralis | Younger men (22 ± 1 years) vs. older men (67 ± 2 years) | Whole muscle lysate | Opa1, Mfn2, Fis1: NS | [232] | |
| Humans | Male and female (combined) | Vastus Lateralis | Younger (24 ± 3 years) vs. older adults (78 ± 5 years) | Whole muscle lysate | Opa1, Mfn2, Fis1 and Drp1: NS | [233] | |
| Humans | Male and female (combined) | Vastus Lateralis | Younger (23 ± 1 years) vs. older adults (75 ± 1 years) | Whole muscle lysate | Mfn2, Fis1, Drp1: NS Opa1: ↓ | [234] | |
| Mice | Not specified | TA | Youths/Aged 6 vs. 18 months old | Whole muscle lysate | Opa1: ↓ | [129] | |
| Mice | Not specified | GAS | Youths/Aged 6 vs. 22 months old | Whole muscle lysate | Mfn1, Mfn2, Opa1 and Fis1: ↓ Drp1: NS | Lc3II, p62, Bnip3: ↑ | [246] |
| Mice (C57BL/6J) | Female | TA | Youths/Aged 2 vs. 24 months old | Whole muscle lysate | Fis1 and Mfn2: ↑ Drp1 and Opa1: NS | Mitochondria Pink1, Parkin: ↑ Lc3I, Lc3II, p62, Rheb, Beclin1: ↑ Bnip3: ↓ | [245] |
| Mice | Male | GAS | Youths/Aged 2–3 vs. 22–24 months old | Whole muscle lysate | Mfn2/Drp1 ratio: ↑ Opa1, Drp1, Mfn1 and Mfn2: NS | [244] | |
| Mice (C57BL/6J) | Male | TA and soleus | Youths/Aged 3 vs. 28–29 months old | Whole muscle lysate and mitochondria fraction | Mfn2 (TA and SOL): ↑ Long Opa1 (functional)/Short Opa1 (nonfunctional) ratio (SOL): trend for ↑ | Lc3II/Lc3I ratio (TA and SOL): NS Atg5 (TA and SOL): ↑ Mitochondria p62 (TA and SOL): ↑ | [201] |
| Mice | Male and female (combined) | QUAD | Youths/Middle-aged 3–6 vs. 8–15 months old | Whole muscle lysate | Mfn1 and Mfn2: ↑ Opa1 and Drp1: NS Fis1: ↓ | Beclin1: ↓ Ulk1: trend for ↓ p62: ↑ Lc3II, Atg5: NS | [243] |
| Rats (Fischer 344 Brown Norway) | Male | EDL | Youths/Aged 5 vs. 35 months old | Whole muscle lysate | Mfn2, Fis1 and Opa1: ↑ Drp1: NS | Ulk1, Beclin1, Atg7: ↑ Mitochondria Parkin, Lc3II: ↑ | [236] |
| Rats (Wistar) | Not specified | GAS | Youths/Aged 3 vs. 26 months old | Mitochondria fraction | Fis1: ↑ | [237] | |
| Rats (Fischer 344 Brown Norway) | Male | TA | Youths/Aged 5 vs. 35 months old | Mitochondria fraction | Fis1 and Drp1: ↑ Mfn2: ↓ Opa1: NS | [238] | |
| Rats (Sprague–Dawley) | Male | GAS and SOL | Youths/Aged 9 vs. 22 months old | Whole muscle lysate | Drp1 (SOL and GAS): ↑ Mfn2 and Fis1 (GAS): ↑ | [239] | |
| Rats (Wistar) | Male | GAS | Youths/Aged 3 vs. 26 months old | Whole muscle lysate | Fis1 and Mfn1: ↑ | [240] | |
| Rats (Sprague-Dawley) | Male | GAS and Triceps | Youths/Aged 3 vs. 22 months old | Whole muscle lysate | Opa1 and Mfn1 (GAS and TRI): ↑ Fis1 (GAS): ↓ Fis1 (TRI): ↑ | Beclin1, Bax, Lc3B (GAS): ↓ Pink1 (Triceps): ↓ | [241] |
| Rats (Sprague-Dawley) | Male | Muscles | Youths/Aged 5 vs. 25 months old | Whole muscle lysate | Drp1: ↑ Opa1: NS Mfn2 and Fis1: ↓ | p62: ↑ Lc3II: ↓ | [242] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, H.; Tsai, S.-Y. Mitochondrial Properties in Skeletal Muscle Fiber. Cells 2023, 12, 2183. https://doi.org/10.3390/cells12172183
Dong H, Tsai S-Y. Mitochondrial Properties in Skeletal Muscle Fiber. Cells. 2023; 12(17):2183. https://doi.org/10.3390/cells12172183
Chicago/Turabian StyleDong, Han, and Shih-Yin Tsai. 2023. "Mitochondrial Properties in Skeletal Muscle Fiber" Cells 12, no. 17: 2183. https://doi.org/10.3390/cells12172183
APA StyleDong, H., & Tsai, S.-Y. (2023). Mitochondrial Properties in Skeletal Muscle Fiber. Cells, 12(17), 2183. https://doi.org/10.3390/cells12172183