
Polycystin-1 Interacting Protein-1 (CU062) Interacts with the Ectodomain of Polycystin-1 (PC1)

 , , , , , , , , , and
, , , , , , , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Generation of Microhomology-Based Gene Disruptions with Cas12a
2.2. Transient Transfection of Plasmid DNA
2.3. Total Cellular Membrane Preparation
2.4. Immunoprecipitation with V5, OLLAS, and FLAG Antibodies
2.5. Western Analysis
2.6. Production of Monoclonal Antibodies
2.6.1. Verification
2.7. Real-Time Reverse Transcriptase Polymerase Chain Reaction (RT−PCR) for Mouse C21orf62
RT−PCR
2.8. Immunofluorescence (IF)
Microscopy
2.9. Statistics
2.10. Cell Lines
3. Institutional Animal Care and Use Committee (IACUC) (Animal Work)
4. Results
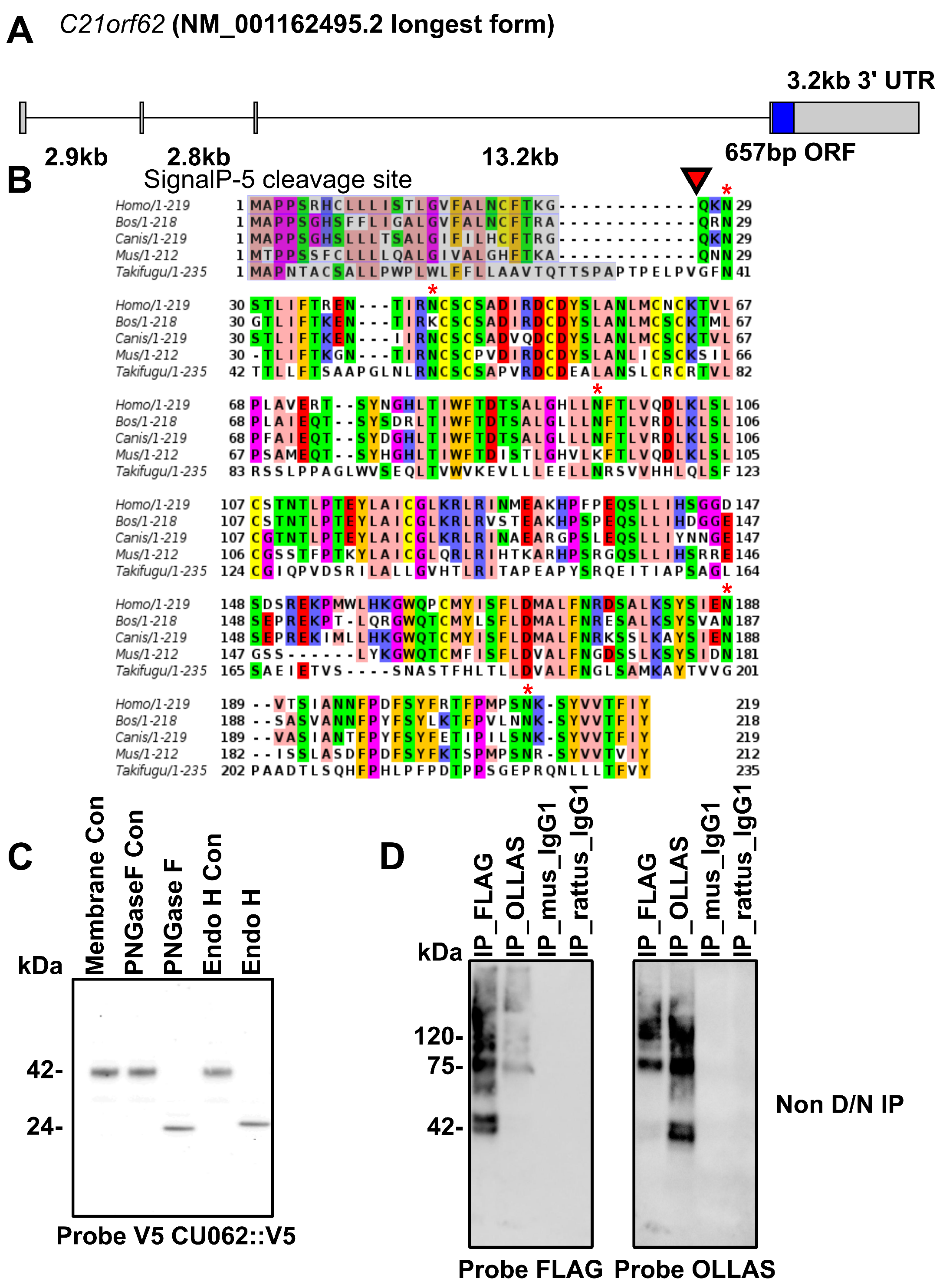
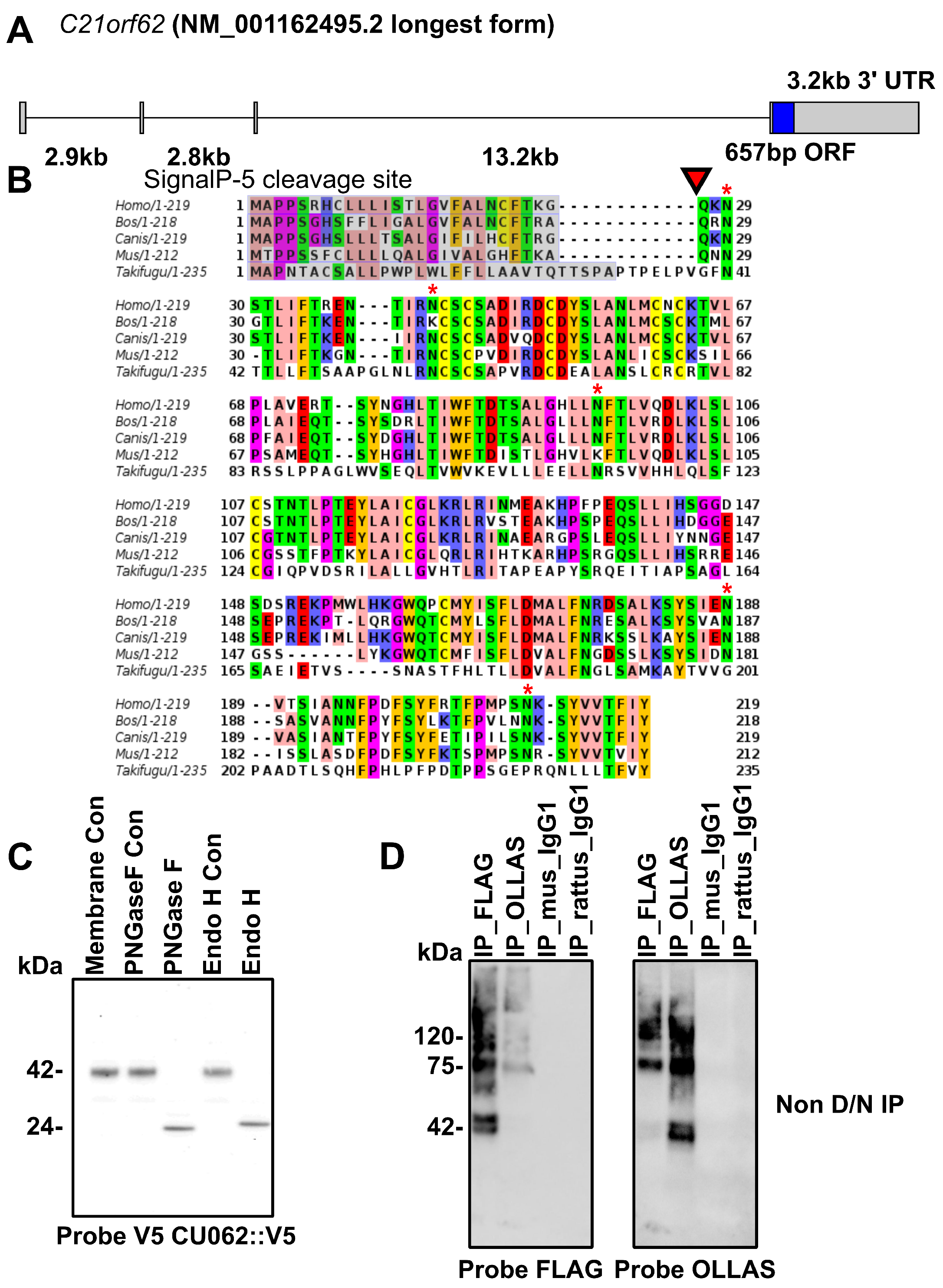
4.1. The CU062 Protein Is the Product of the C21orf62 Gene
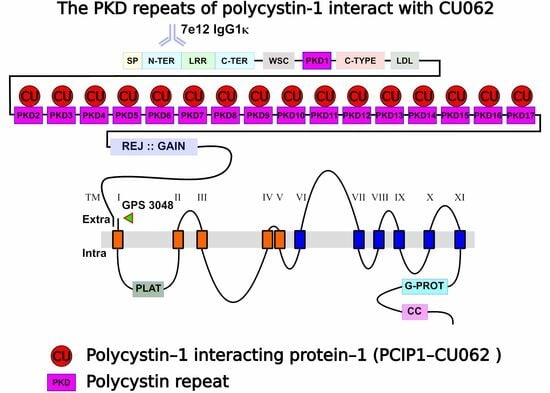
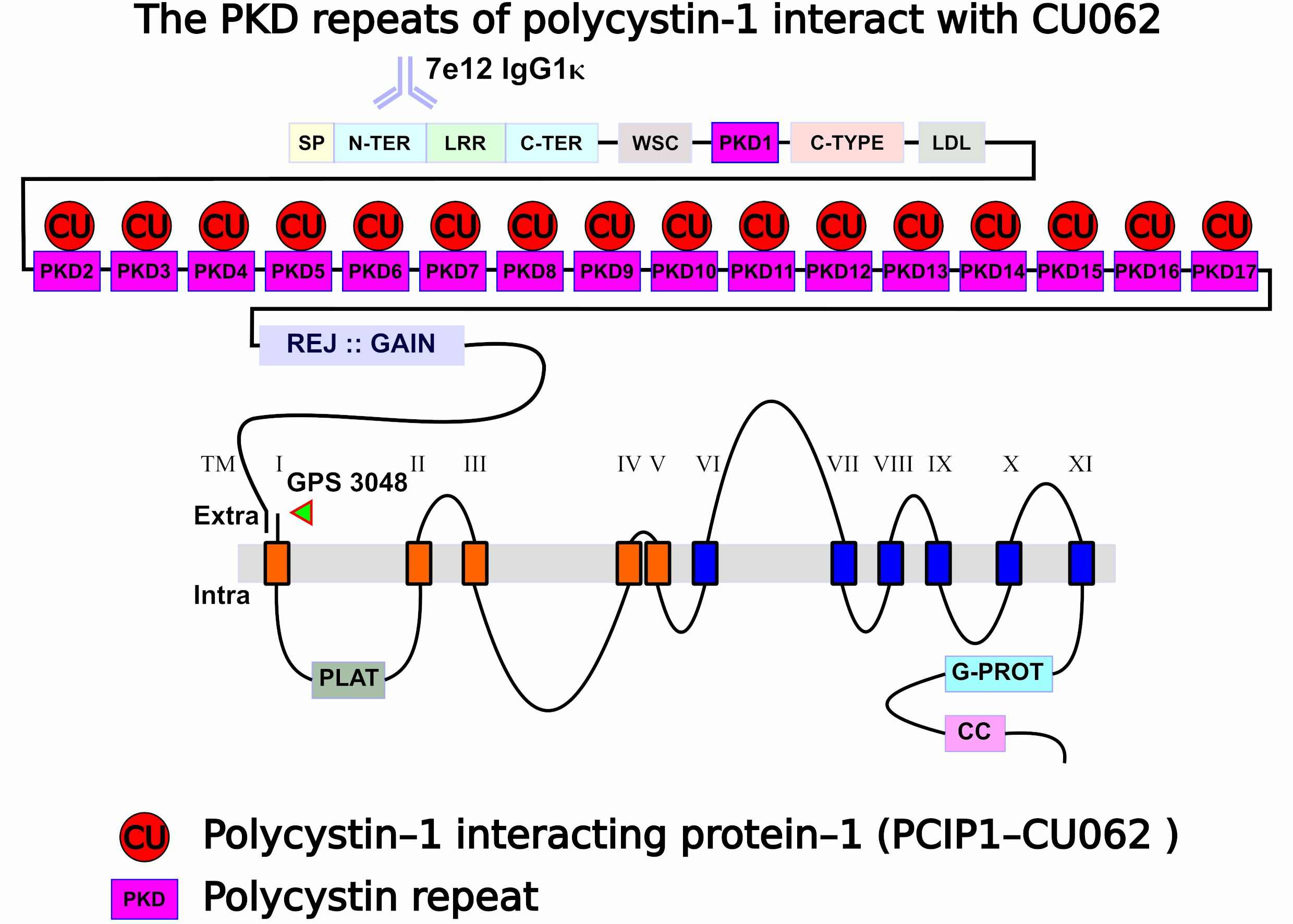
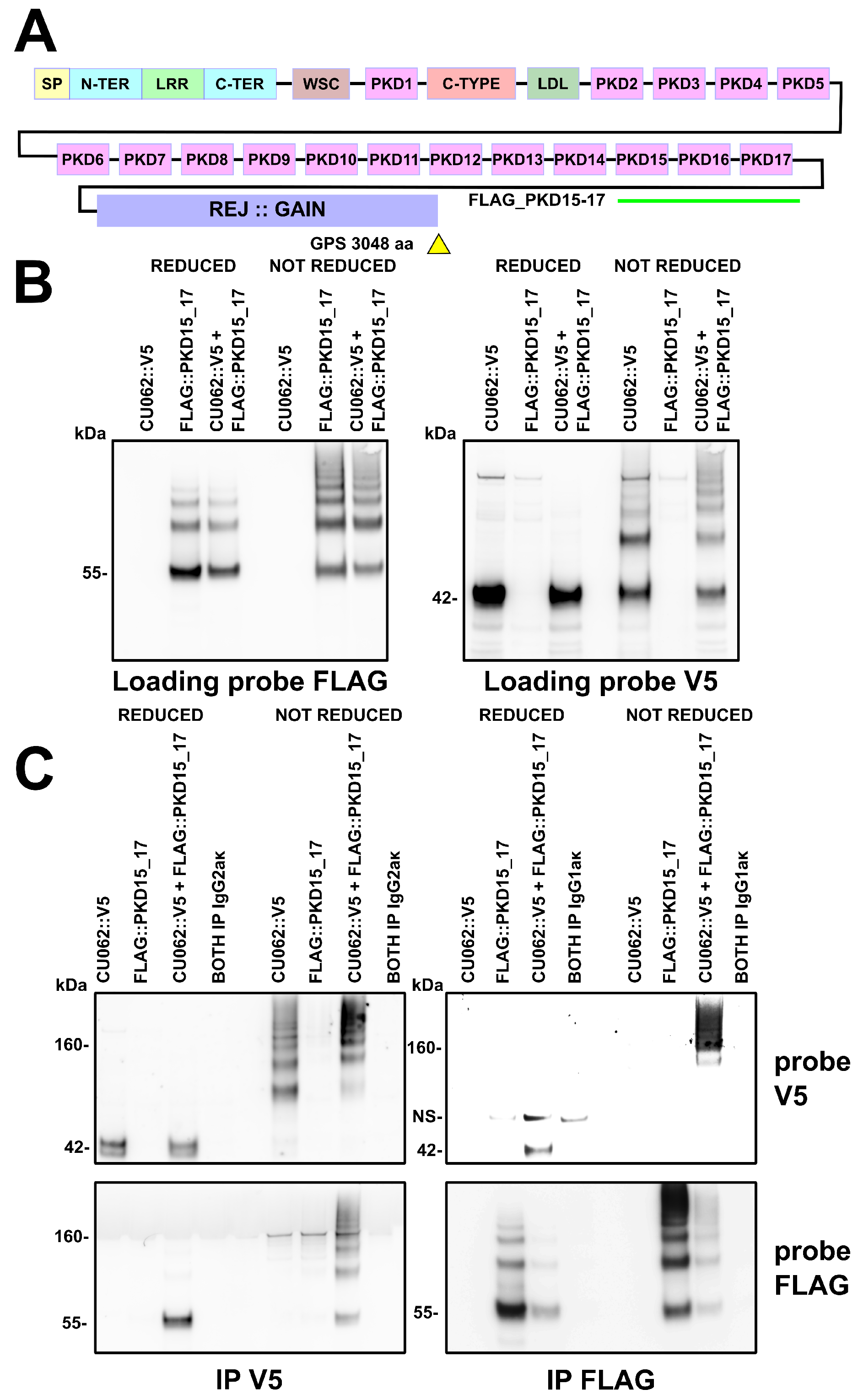
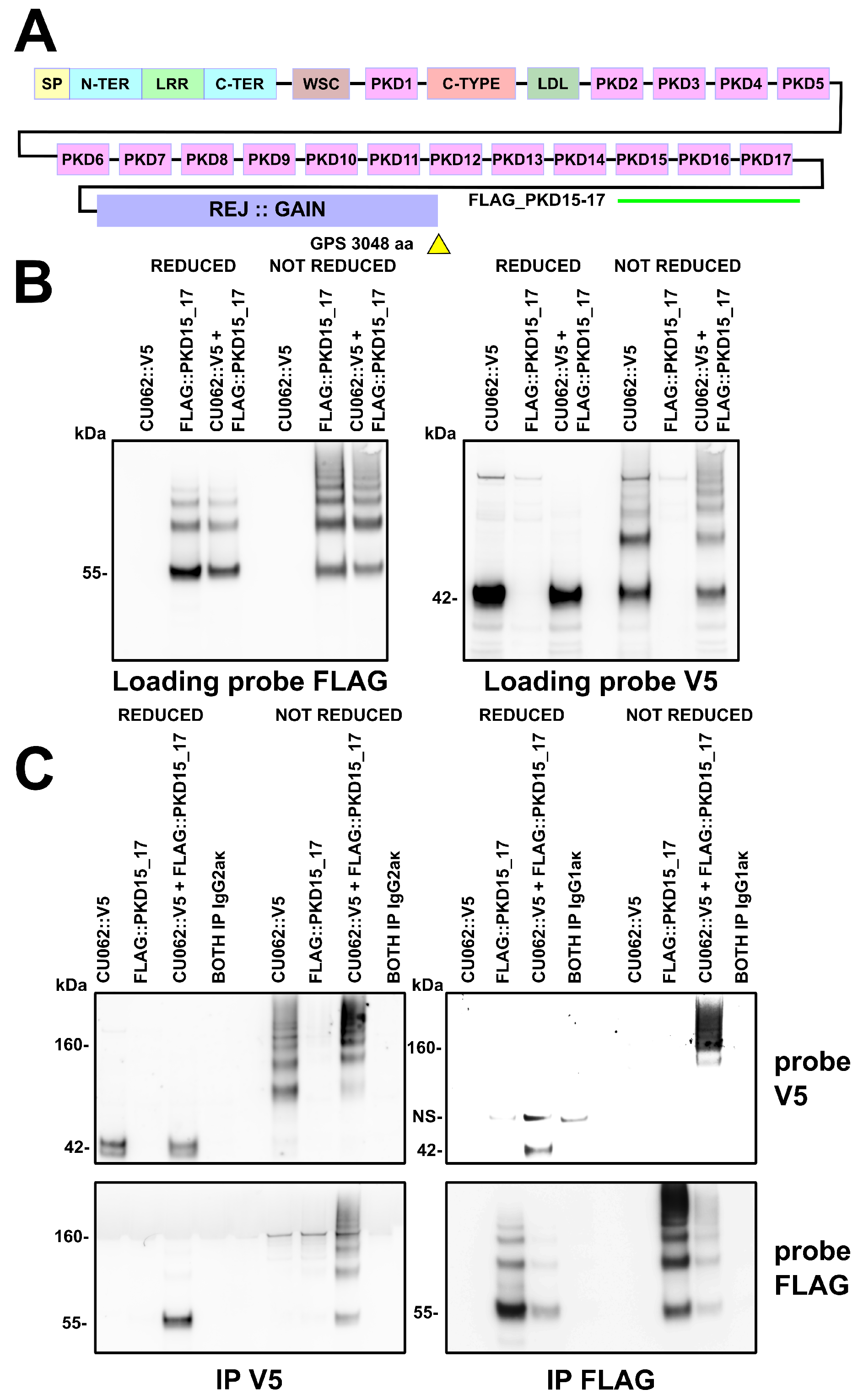
4.2. The CU062 Protein Interacts with PKD Domains 2–17 from PC1
4.2.1. The PKD/PKD, PKD/CU062 and the CU062/CU062 Interactions Are Not Carbohydrate-Dependent
4.2.2. The CU062 Protein Interacts Specifically with PC1 PKD Domains
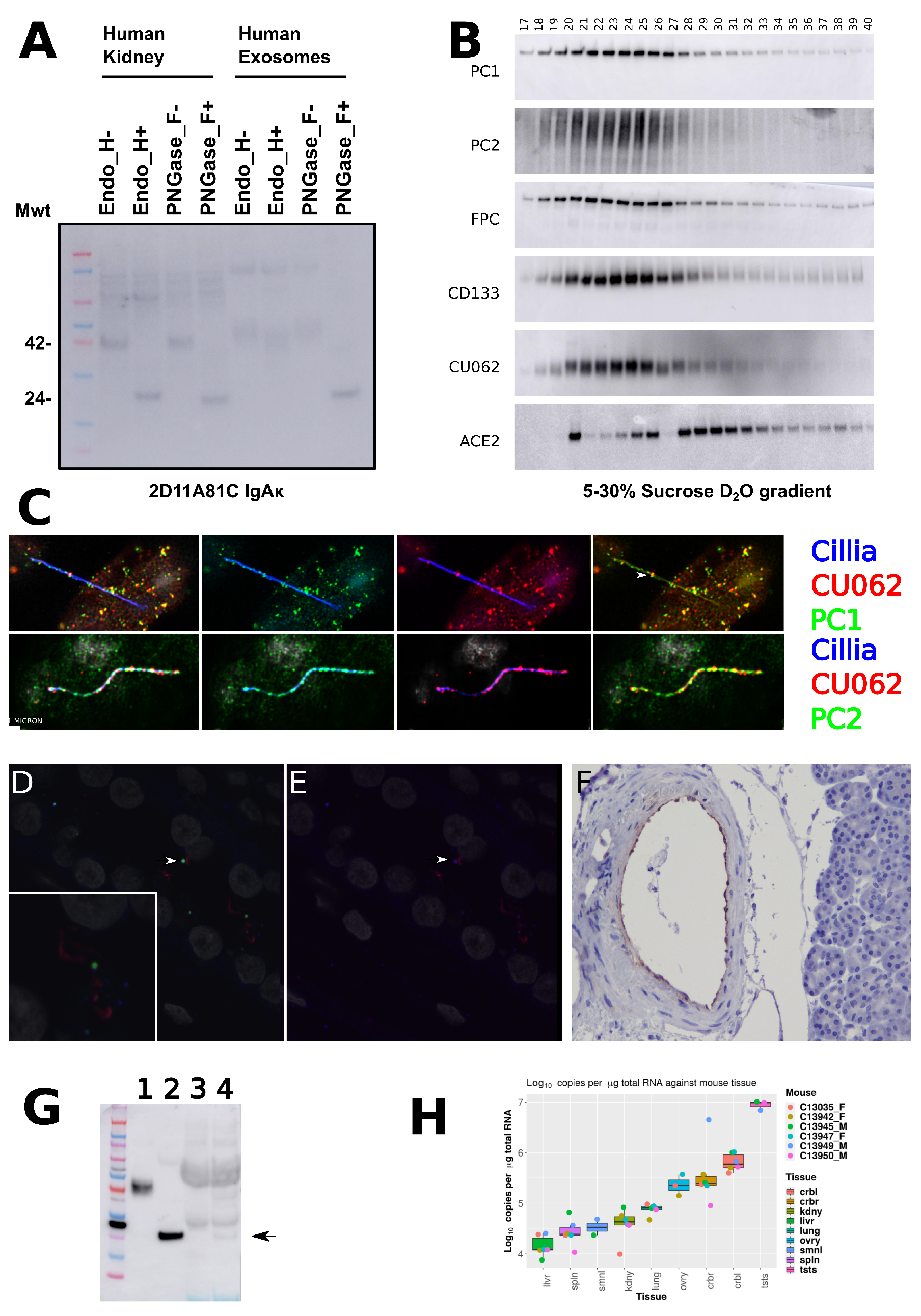
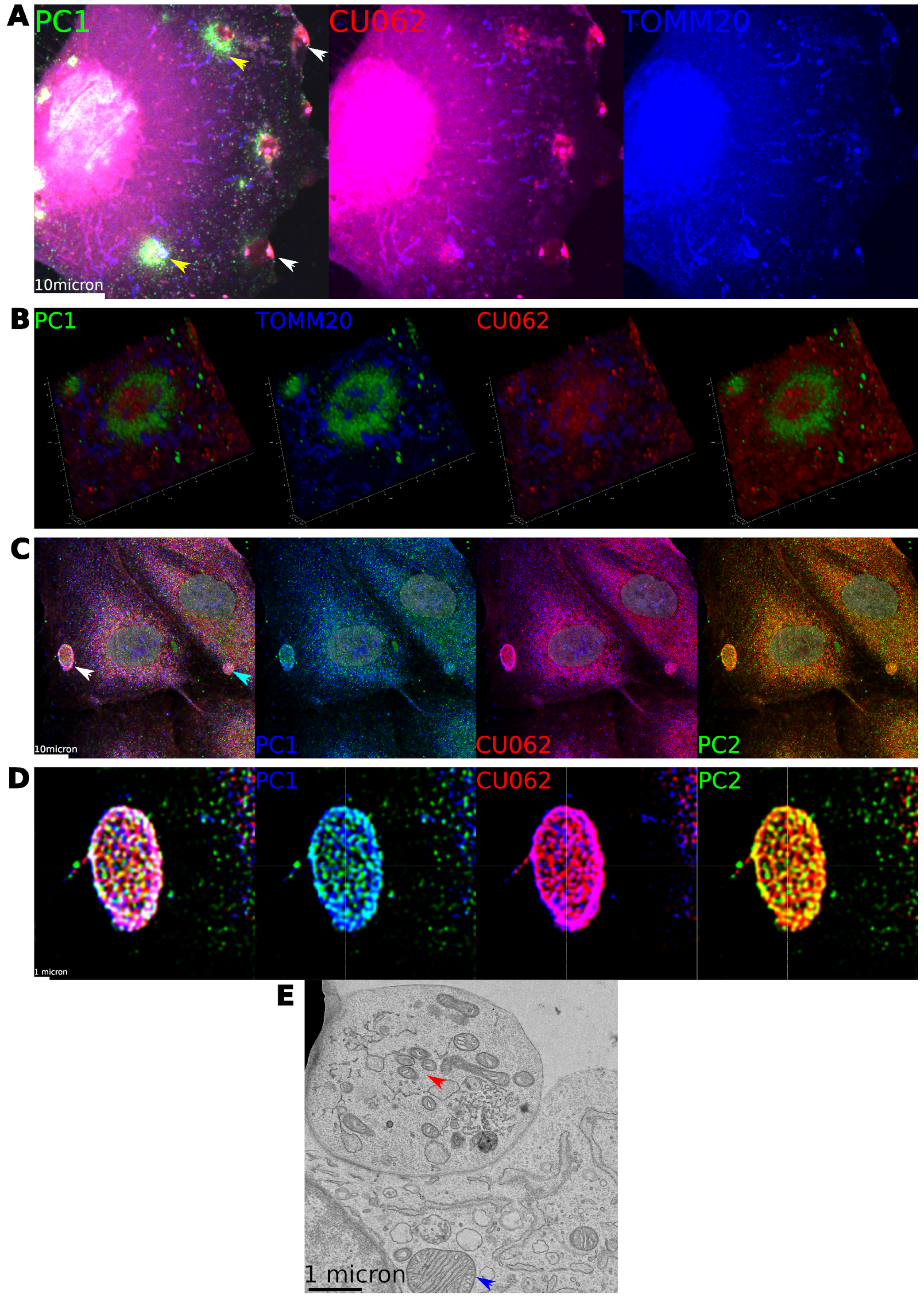
4.3. Localization of the CU062 Protein
4.4. Localization of PC1 and CU062 Depend on the Cell Type and Growth Status (Confluent vs. Nonconfluent)
4.4.1. Nonconfluent Migrating HPRE Cells
4.4.2. Confluent HPRE Cells
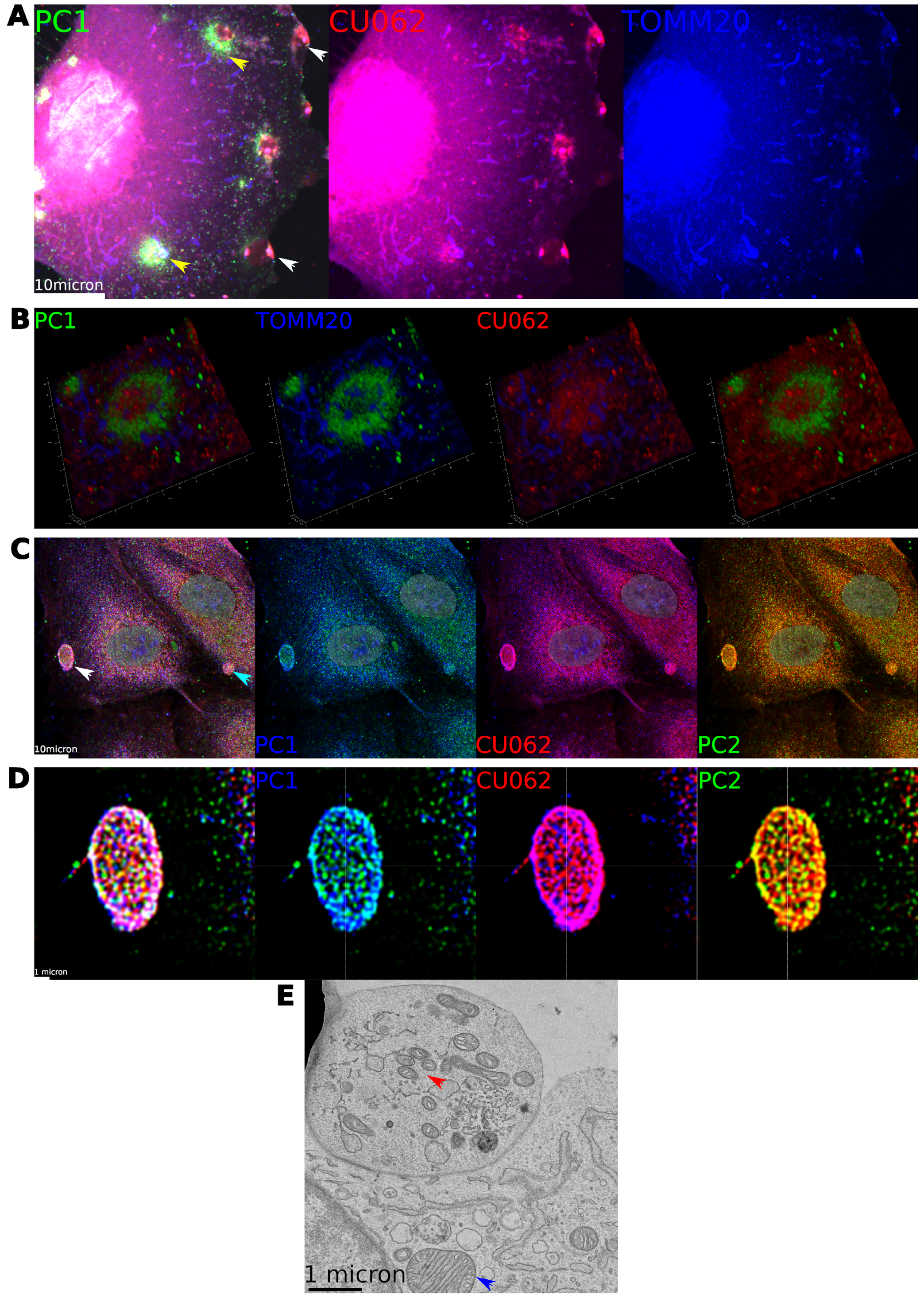
4.5. Localization of PC1 and CU062 with Mitochondria
5. Discussion
- 1.
- On the FAs of migrating PT cells.
- 2.
- On the RFs, especially the migrasomes where CU062 forms a shell around the vesicle.
- 3.
- In novel structures, FRs, induced by CCCP.
- 4.
- As extracellular vesicles attached to the primary cilia of CD cells where they may have signaling roles.
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Iglesias, C.G.; Torres, V.E.; Offord, K.P.; Holley, K.E.; Beard, C.M.; Kurland, L.T. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935–1980. Am. J. Kidney Dis. 1983, 2, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.E.P.K.D. The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell 1994, 77, 881–894. [Google Scholar]
- Cai, Y.; Maeda, Y.; Cedzich, A.; Torres, V.E.; Wu, G.; Hayashi, T.; Mochizuki, T.; Park, J.H.; Witzgall, R.; Somlo, S. Identification and characterization of polycystin-2, the PKD2 gene product. J. Biol. Chem. 1999, 274, 28557–28565. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, S.; Consugar, M.B.; Chapman, A.B.; Torres, V.E.; Guay-Woodford, L.M.; Grantham, J.J.; Bennett, W.M.; Meyers, C.M.; Walker, D.L.; Bae, K.; et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 2143–2160. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Boletta, A.; Bhunia, A.K.; Xu, H.; Liu, L.; Ahrabi, A.K.; Watnick, T.J.; Zhou, F.; Germino, G.G. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc. Natl. Acad. Sci. USA 2002, 99, 16981–76986. [Google Scholar] [CrossRef]
- Hughes, J.; Ward, C.J.; Peral, B.; Aspinwall, R.; Clark, K.; Millan, J.L.S.; Gamble, V.; Harris, P.C. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat. Genet. 1995, 10, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Weston, B.S.; Malhas, A.N.; Price, R.G. Structure-function relationships of the extracellular domain of the autosomal dominant polycystic kidney disease-associated protein, polycystin-1. FEBS Lett. 2003, 538, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Schröder, S.; Fraternali, F.; Quan, X.; Scott, D.; Qian, F.; Pfuhl, M. When a module is not a domain: The case of the REJ module and the redefinition of the architecture of polycystin-1. Biochem. J. 2011, 435, 651–660. [Google Scholar] [CrossRef]
- Ibraghimov-Beskrovnaya, O.; Bukanov, N.O.; Donohue, L.C.; Dackowski, W.R.; Klinger, K.W.; Landes, G.M. Strong homophilic interactions of the Ig-like domains of polycystin-1, the protein product of an autosomal dominant polycystic kidney disease gene, PKD1. Hum. Mol. Genet. 2000, 9, 1641–1649. [Google Scholar] [CrossRef]
- Bycroft, M.; Bateman, A.; Clarke, J.; Hamill, S.J.; Sandford, R.; Thomas, R.L.; Chothia, C. The structure of a PKD domain from polycystin-1: Implications for polycystic kidney disease. EMBO J. 1999, 18, 297–305. [Google Scholar] [CrossRef]
- Ward, C.J.; Hogan, M.C.; Rossetti, S.; Walker, D.; Sneddon, T.; Wang, X.; Kubly, V.; Cunningham, J.M.; Bacallao, R.; Ishibashi, M.; et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat. Genet. 2002, 30, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Onuchic, L.F.; Furu, L.; Nagasawa, Y.; Hou, X.; Eggermann, T.; Ren, Z.; Bergmann, C.; Senderek, J.; Esquivel, E.; Zeltner, R.; et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am. J. Hum. Genet. 2002, 70, 1305–1317. [Google Scholar] [CrossRef] [PubMed]
- Yoder, B.K.; Hou, X.; Guay-Woodford, L.M. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 2002, 13, 2508–2516. [Google Scholar] [CrossRef] [PubMed]
- Nauli, S.M.; Alenghat, F.J.; Luo, Y.; Williams, E.; Vassilev, P.; Li, X.; Elia, A.E.H.; Lu, W.; Brown, E.M.; Quinn, S.J.; et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003, 33, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.D.; Geng, L.; Li, X.; Burrow, C.R. The PKD1 gene product, “polycystin-1”, is a tyrosine-phosphorylated protein that colocalizes with alpha2beta1-integrin in focal clusters in adherent renal epithelia. Lab. Investig. 1999, 79, 1311–1323. [Google Scholar] [PubMed]
- Scheffers, M.S.; van der Bent, P.; Prins, F.; Spruit, L.; Breuning, M.H.; Litvinov, S.V.; de Heer, E.; Peters, D.J. Polycystin-1, the product of the polycystic kidney disease 1 gene, co-localizes with desmosomes in MDCK cells. Hum. Mol. Genet. 2000, 9, 2743–2750. [Google Scholar] [CrossRef] [PubMed]
- Hogan, M.C.; Manganelli, L.; Woollard, J.R.; Masyuk, A.I.; Masyuk, T.V.; Tammachote, R.; Huang, B.Q.; Leontovich, A.A.; Beito, T.G.; Madden, B.J.; et al. Characterization of PKD Protein-Positive Exosome-Like Vesicles. J. Am. Soc. Nephrol. 2009. [Google Scholar] [CrossRef]
- Lea, W.A.; McGreal, K.; Sharma, M.; Parnell, S.C.; Zelenchuk, L.; Charlesworth, M.C.; Madden, B.J.; Johnson, K.L.; McCormick, D.J.; Hogan, M.C.; et al. Analysis of the polycystin complex (PCC) in human urinary exosome-like vesicles (ELVs). Sci. Rep. 2020, 10, 1500. [Google Scholar] [CrossRef]
- Hogan, M.C.; Bakeberg, J.L.; Gainullin, V.G.; Irazabal, M.V.; Harmon, A.J.; Lieske, J.C.; Charlesworth, M.C.; Johnson, K.L.; Madden, B.J.; Zenka, R.M.; et al. Identification of biomarkers for PKD1 using urinary exosomes. J. Am. Soc. Nephrol. 2014, 26, 1661. [Google Scholar] [CrossRef]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef]
- Menezes, L.F.; Lin, C.C.; Zhou, F.; Germino, G.G. Fatty acid oxidation is impaired in an orthologous mouse model of autosomal dominant polycystic kidney disease. EBioMedicine 2016, 5, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Podrini, C.; Rowe, I.; Pagliarini, R.; Costa, A.S.; Chiaravalli, M.; Di Meo, I.; Kim, H.; Distefano, G.; Tiranti, V.; Qian, F.; et al. Dissection of metabolic reprogramming in polycystic kidney disease reveals coordinated rewiring of bioenergetic pathways. Commun. Biol. 2018, 1, 194. [Google Scholar] [CrossRef]
- Podrini, C.; Cassina, L.; Boletta, A. Metabolic reprogramming and the role of mitochondria in polycystic kidney disease. Cell. Signal. 2020, 67, 109495. [Google Scholar] [CrossRef] [PubMed]
- Tavano, S.; Heisenberg, C.P. Migrasomes take center stage. Nat. Cell Biol. 2019, 21, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Jiang, D.; Hu, X.; Du, W.; Ji, L.; Yang, Y.; Li, X.; Sho, T.; Wang, X.; Li, Y.; et al. Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell 2021, 184, 2896–2910. [Google Scholar] [CrossRef]
- Loghman-Adham, M.; Nauli, S.M.; Soto, C.E.; Kariuki, B.; Zhou, J. Immortalized epithelial cells from human autosomal dominant polycystic kidney cysts. Am. J. Physiol.-Ren. Physiol. 2003, 285, F397–F412. [Google Scholar] [CrossRef]
- Herbert, B.S.; Grimes, B.R.; Xu, W.M.; Werner, M.; Ward, C.; Rossetti, S.; Harris, P.; Bello-Reuss, E.; Ward, H.H.; Miller, C.; et al. A telomerase immortalized human proximal tubule cell line with a truncation mutation (Q4004X) in polycystin-1. PLoS ONE 2013, 8, e55191. [Google Scholar] [CrossRef]
- Ong, A.C.; Harris, P.C.; Biddolph, S.; Bowker, C.; Ward, C.J. Characterisation and expression of the PKD-1 protein, polycystin, in renal and extrarenal tissues. Kidney Int. 1999, 55, 2091–2116. [Google Scholar]
- de StGroth, S.F.; Scheidegger, D. Production of monoclonal antibodies: Strategy and tactics. J. Immunol. Methods 1980, 35, 1–21. [Google Scholar] [CrossRef]
- Nauli, S.M.; Rossetti, S.; Kolb, R.J.; Alenghat, F.J.; Consugar, M.B.; Harris, P.C.; Ingber, D.E.; Loghman-Adham, M.; Zhou, J. Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J. Am. Soc. Nephrol. 2006, 17, 1015–1025. [Google Scholar] [CrossRef] [PubMed]
- Slavov, D.; Hattori, M.; Sakaki, Y.; Rosenthal, A.; Shimizu, N.; Minoshima, S.; Kudoh, J.; Yaspo, M.L.; Ramser, J.; Reinhardt, R.; et al. Criteria for gene identification and features of genome organization: Analysis of 6.5 Mb of DNA sequence from human chromosome 21. Gene 2000, 247, 215–232. [Google Scholar] [CrossRef] [PubMed]
- Armenteros, J.J.A.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Crispin, M.; Sonnen, A.P.; Harvey, D.J.; Chang, V.T.; Evans, E.J.; Scanlan, C.N.; Stuart, D.I.; Gilbert, R.J.; Davis, S.J. Use of the α-mannosidase I inhibitor kifunensine allows the crystallization of apo CTLA-4 homodimer produced in long-term cultures of Chinese hamster ovary cells. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 785–789. [Google Scholar] [CrossRef]
- Pisitkun, T.; Shen, R.F.; Knepper, M.A. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. USA 2004, 101, 13368–13373. [Google Scholar] [CrossRef]
- Bakeberg, J.L.; Tammachote, R.; Woollard, J.R.; Hogan, M.C.; Tuan, H.F.; Li, M.; van Deursen, J.M.; Wu, Y.; Huang, B.Q.; Torres, V.E.; et al. Epitope-tagged Pkhd1 tracks the processing, secretion, and localization of fibrocystin. J. Am. Soc. Nephrol. 2011, 22, 2266–2277. [Google Scholar] [CrossRef]
- Ma, L.; Li, Y.; Peng, J.; Wu, D.; Zhao, X.; Cui, Y.; Chen, L.; Yan, X.; Du, Y.; Yu, L. Discovery of the migrasome, an organelle mediating release of cytoplasmic contents during cell migration. Cell Res. 2015, 25, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zucker, B.; Zhang, S.; Elias, S.; Zhu, Y.; Chen, H.; Ding, T.; Li, Y.; Sun, Y.; Lou, J.; et al. Migrasome formation is mediated by assembly of micron-scale tetraspanin macrodomains. Nat. Cell Biol. 2019, 21, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Cassina, L.; Chiaravalli, M.; Boletta, A. Increased mitochondrial fragmentation in polycystic kidney disease acts as a modifier of disease progression. FASEB J. 2020, 34, 6493–6507. [Google Scholar] [CrossRef]
- Tanaka, Y.; Okada, Y.; Hirokawa, N. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature 2005, 435, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Charron, A.J.; Nakamura, S.; Bacallao, R.; Wandinger-Ness, A. Compromised cytoarchitecture and polarized trafficking in autosomal dominant polycystic kidney disease cells. J. Cell Biol. 2000, 149, 111–124. [Google Scholar] [CrossRef] [PubMed]
- AbouAlaiwi, W.A.; Ratnam, S.; Booth, R.L.; Shah, J.V.; Nauli, S.M. Endothelial cells from humans and mice with polycystic kidney disease are characterized by polyploidy and chromosome segregation defects through survivin down-regulation. Hum. Mol. Genet. 2011, 20, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Battini, L.; Macip, S.; Fedorova, E.; Dikman, S.; Somlo, S.; Montagna, C.; Gusella, G.L. Loss of polycystin-1 causes centrosome amplification and genomic instability. Hum. Mol. Genet. 2008, 17, 2819–2833. [Google Scholar] [CrossRef]
- Wilson, P.D.; Hreniuk, D.; Gabow, P.A. Abnormal extracellular matrix and excessive growth of human adult polycystic kidney disease epithelia. J. Cell. Physiol. 1992, 150, 360–369. [Google Scholar] [CrossRef]
- Lian, X.; Wu, X.; Li, Z.; Zhang, Y.; Song, K.; Cai, G.; Li, Q.; Lin, S.; Chen, X.; Bai, X.Y. The combination of metformin and 2-deoxyglucose significantly inhibits cyst formation in miniature pigs with polycystic kidney disease. Br. J. Pharmacol. 2019, 176, 711–724. [Google Scholar] [CrossRef]
- Nigro, E.A.; Castelli, M.; Boletta, A. Role of the polycystins in cell migration, polarity, and tissue morphogenesis. Cells 2015, 4, 687–705. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lea, W.A.; Winklhofer, T.; Zelenchuk, L.; Sharma, M.; Rossol-Allison, J.; Fields, T.A.; Reif, G.; Calvet, J.P.; Bakeberg, J.L.; Wallace, D.P.; et al. Polycystin-1 Interacting Protein-1 (CU062) Interacts with the Ectodomain of Polycystin-1 (PC1). Cells 2023, 12, 2166. https://doi.org/10.3390/cells12172166
Lea WA, Winklhofer T, Zelenchuk L, Sharma M, Rossol-Allison J, Fields TA, Reif G, Calvet JP, Bakeberg JL, Wallace DP, et al. Polycystin-1 Interacting Protein-1 (CU062) Interacts with the Ectodomain of Polycystin-1 (PC1). Cells. 2023; 12(17):2166. https://doi.org/10.3390/cells12172166
Chicago/Turabian StyleLea, Wendy A., Thomas Winklhofer, Lesya Zelenchuk, Madhulika Sharma, Jessica Rossol-Allison, Timothy A. Fields, Gail Reif, James P. Calvet, Jason L. Bakeberg, Darren P. Wallace, and et al. 2023. "Polycystin-1 Interacting Protein-1 (CU062) Interacts with the Ectodomain of Polycystin-1 (PC1)" Cells 12, no. 17: 2166. https://doi.org/10.3390/cells12172166
APA StyleLea, W. A., Winklhofer, T., Zelenchuk, L., Sharma, M., Rossol-Allison, J., Fields, T. A., Reif, G., Calvet, J. P., Bakeberg, J. L., Wallace, D. P., & Ward, C. J. (2023). Polycystin-1 Interacting Protein-1 (CU062) Interacts with the Ectodomain of Polycystin-1 (PC1). Cells, 12(17), 2166. https://doi.org/10.3390/cells12172166