
Development of Beta-Amyloid-Specific CAR-Tregs for the Treatment of Alzheimer’s Disease

 ,
,  , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
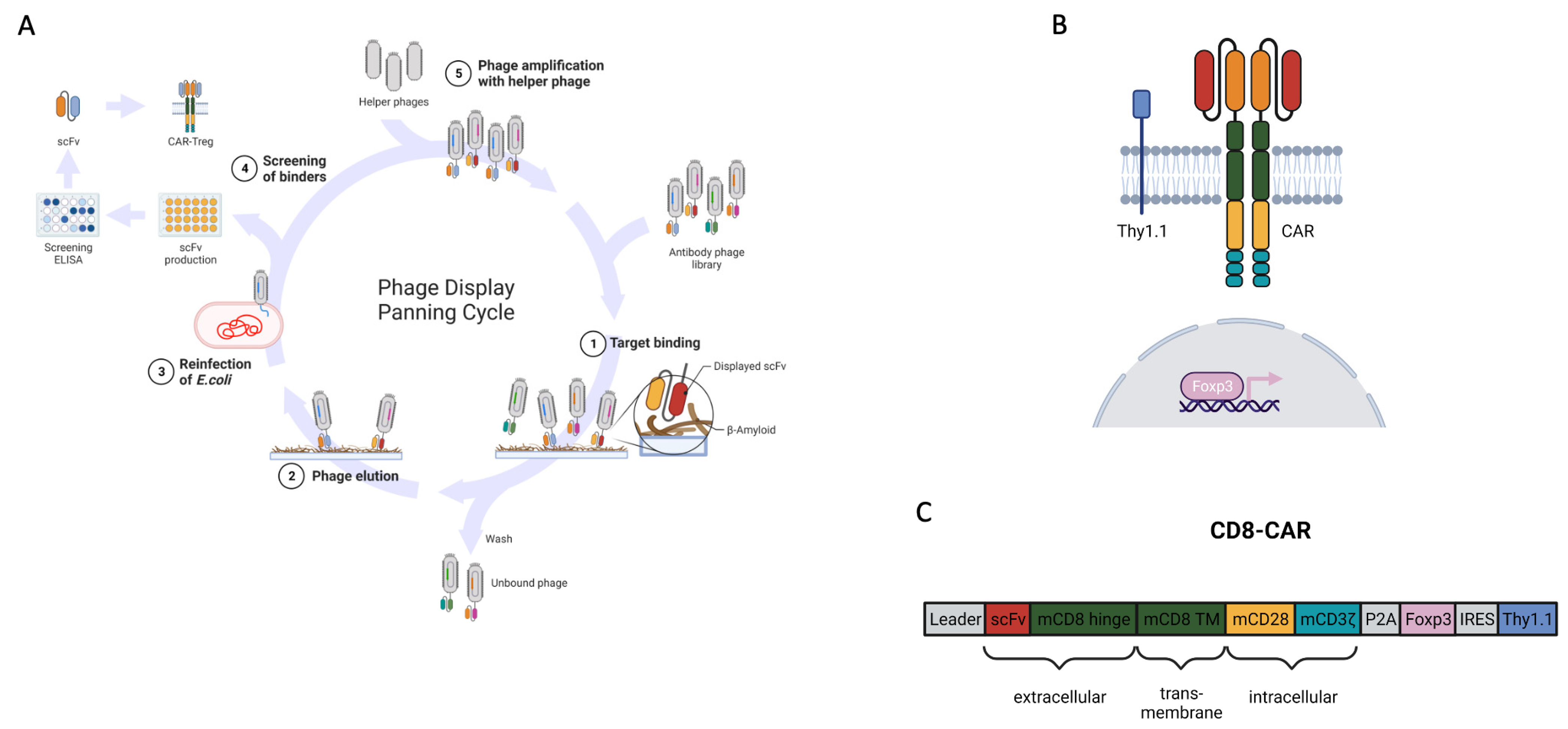
2.1. Generation of the CAR
2.2. Brain Lysates
2.3. Production of Retroviral Particles
2.4. Flow Cytometry Staining
2.5. NFAT-GFP Assay
2.6. nTreg and CD8 T Effector Cell Isolation
2.7. Production of CAR-cTregs
2.8. Activation Assay
2.9. Suppression Assay
2.10. Statistical Analysis
2.11. Software
3. Results
3.1. Generation of the Beta-Amyloid-Specific CAR
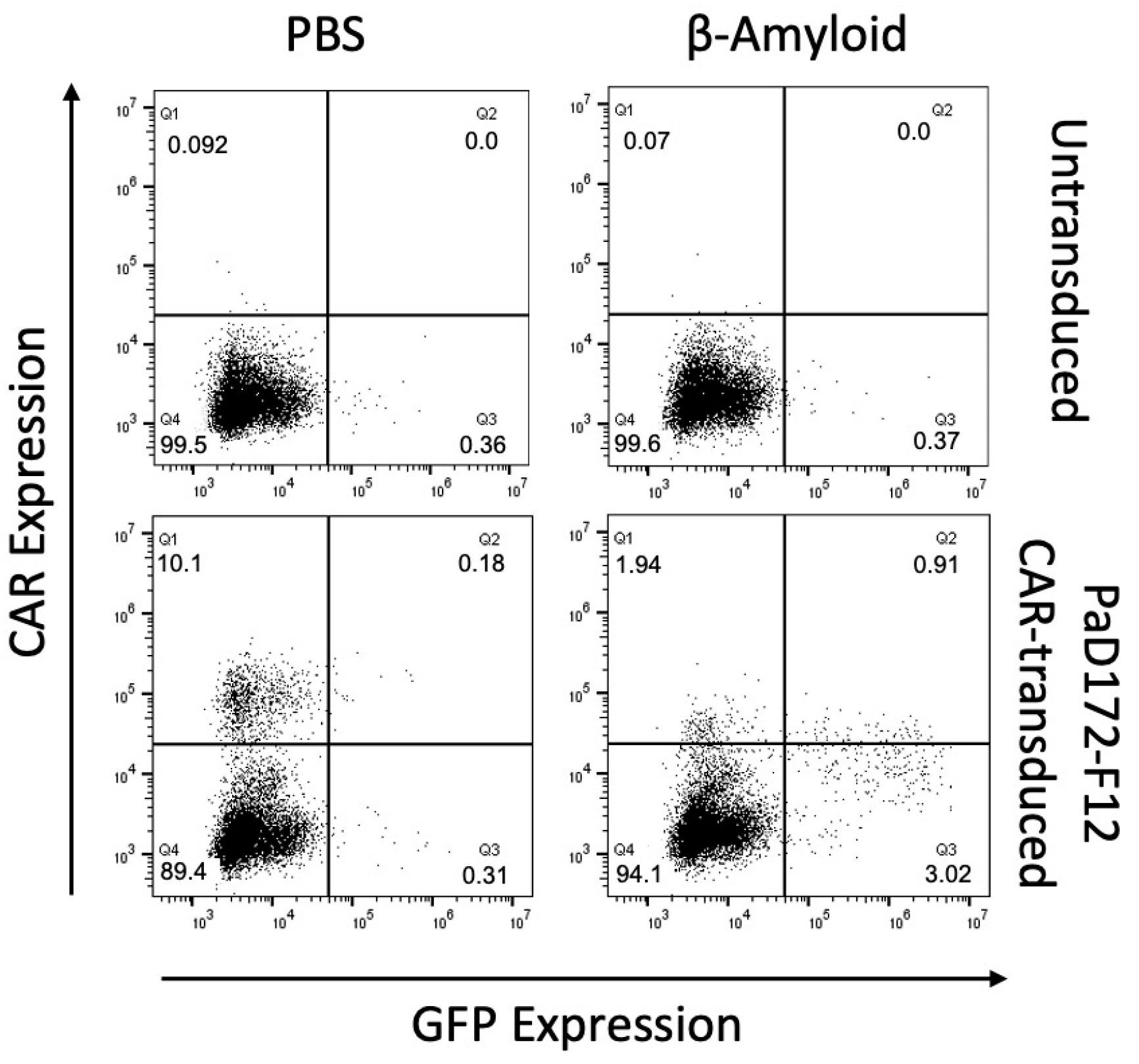
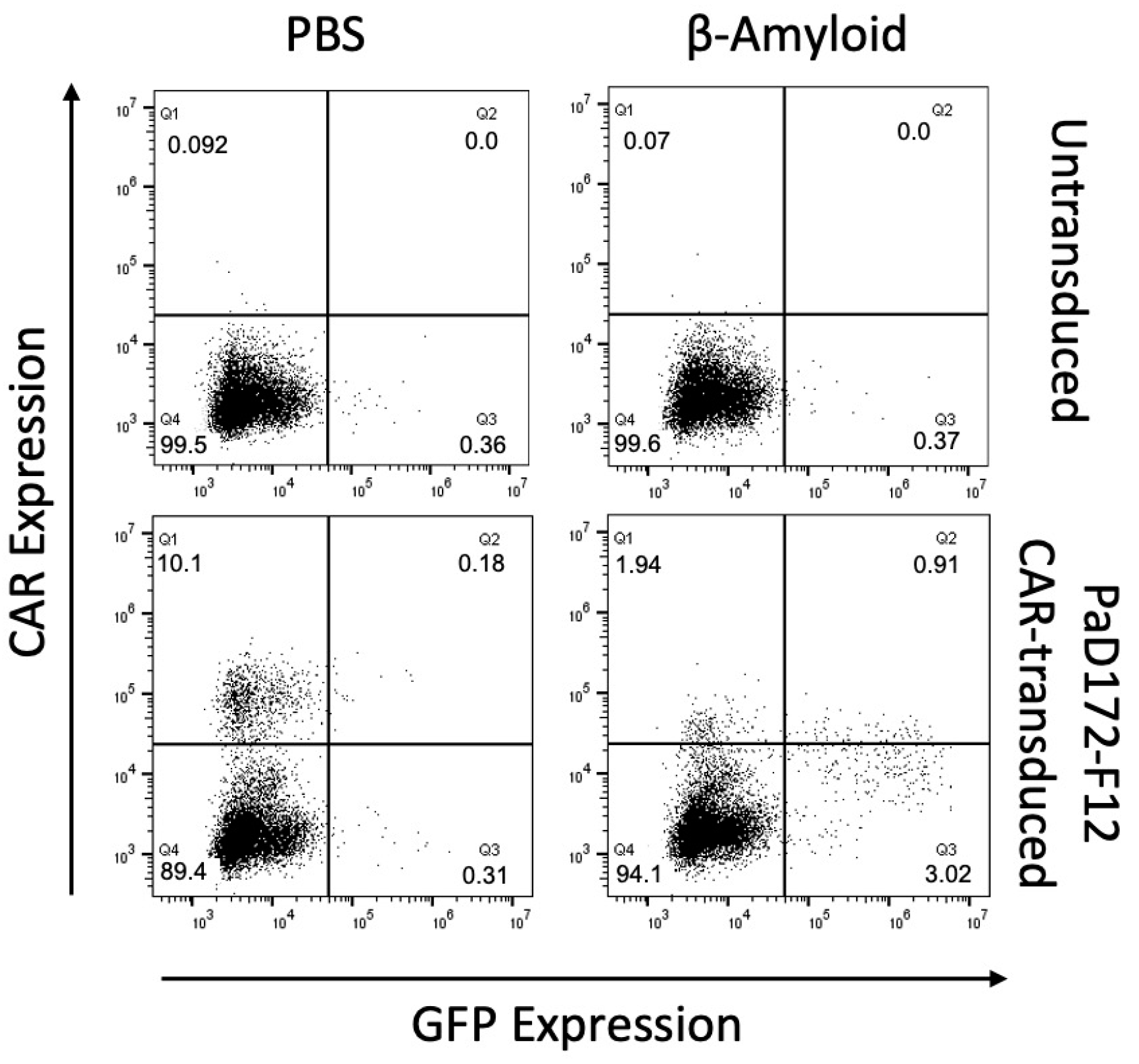
3.2. Beta-Amyloid-Specific CAR Can Be Activated by Human Peptide
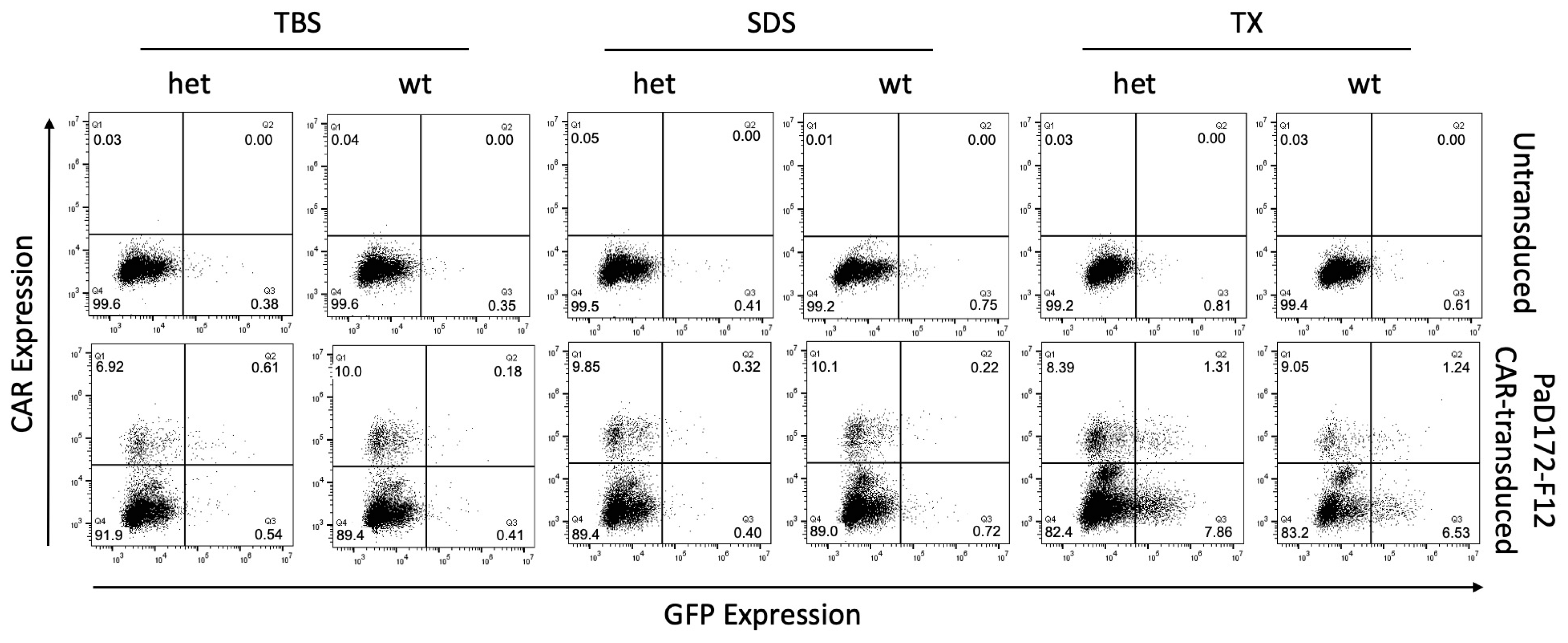
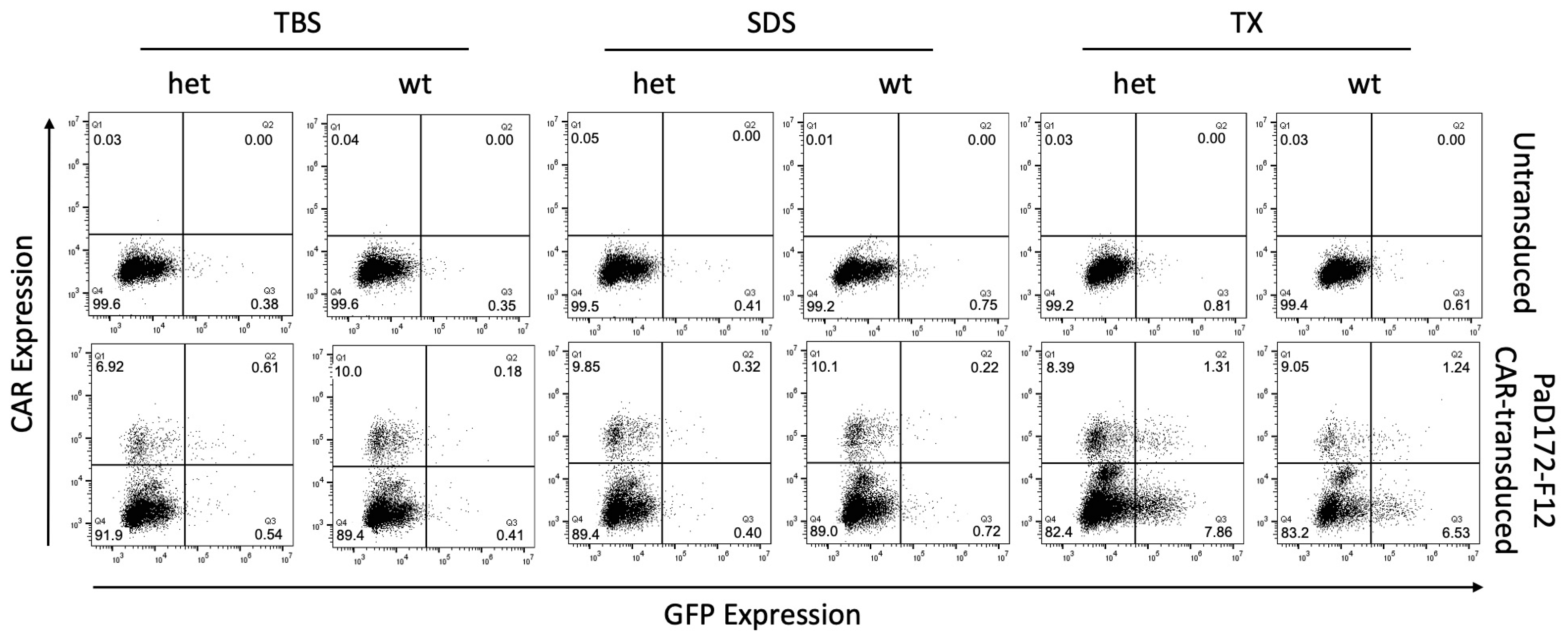
3.3. Murine Brain Lysates Contain Membrane-Bound Ab Plaques That Can Activate CARs
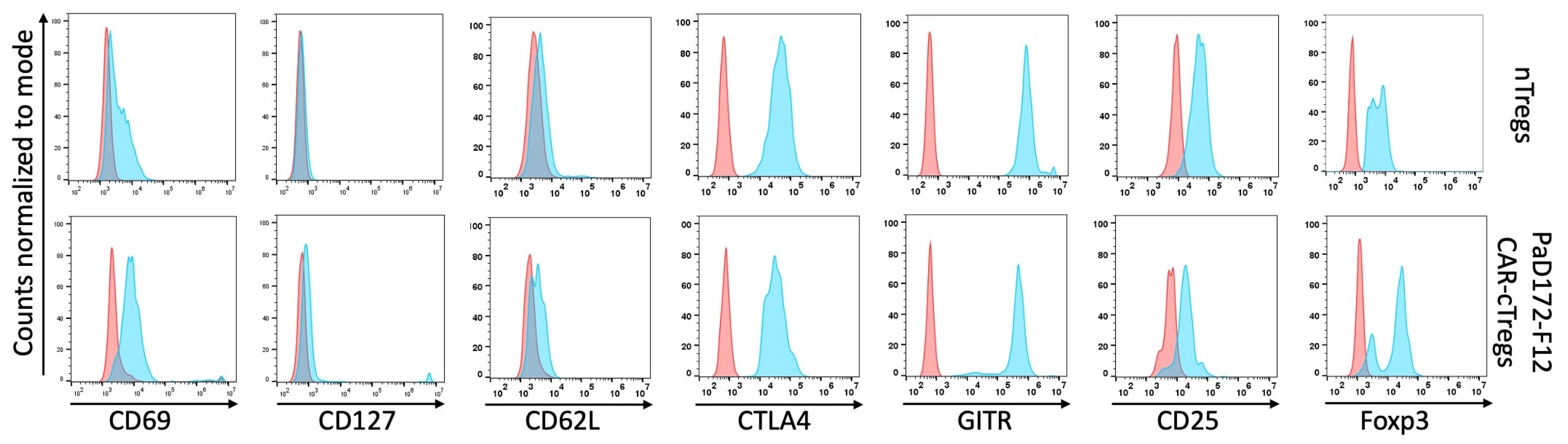
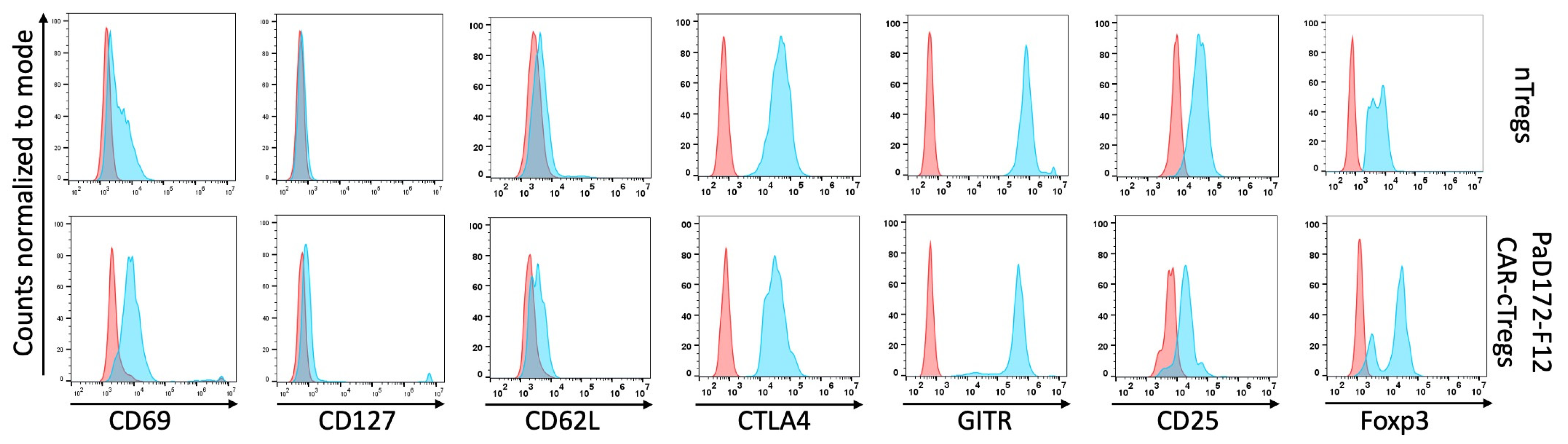
3.4. Aβ-Specific Converted CAR-Tregs Do Not Differ in Phenotype from Natural Tregs
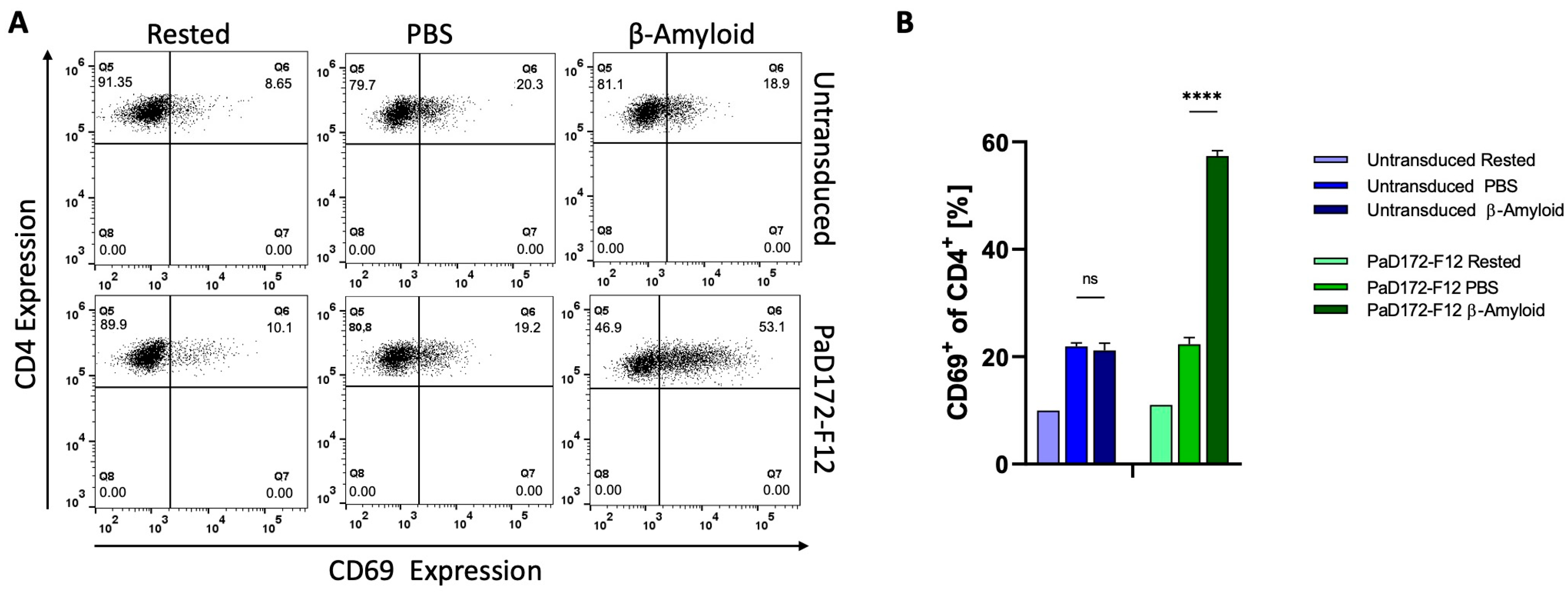
3.5. Primary Murine Aβ-Specific cTregs Can Be Activated by Aβ-Peptide
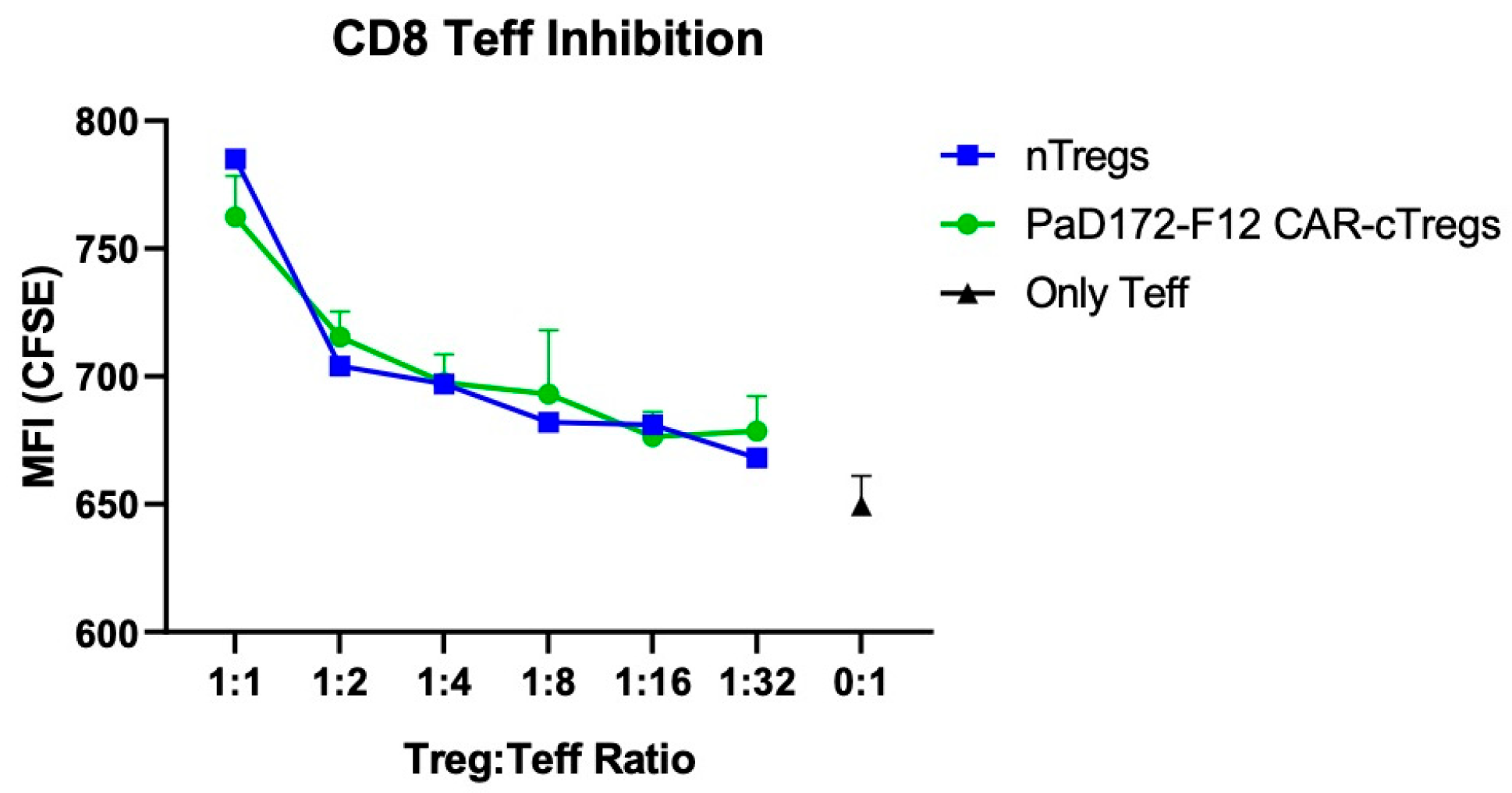
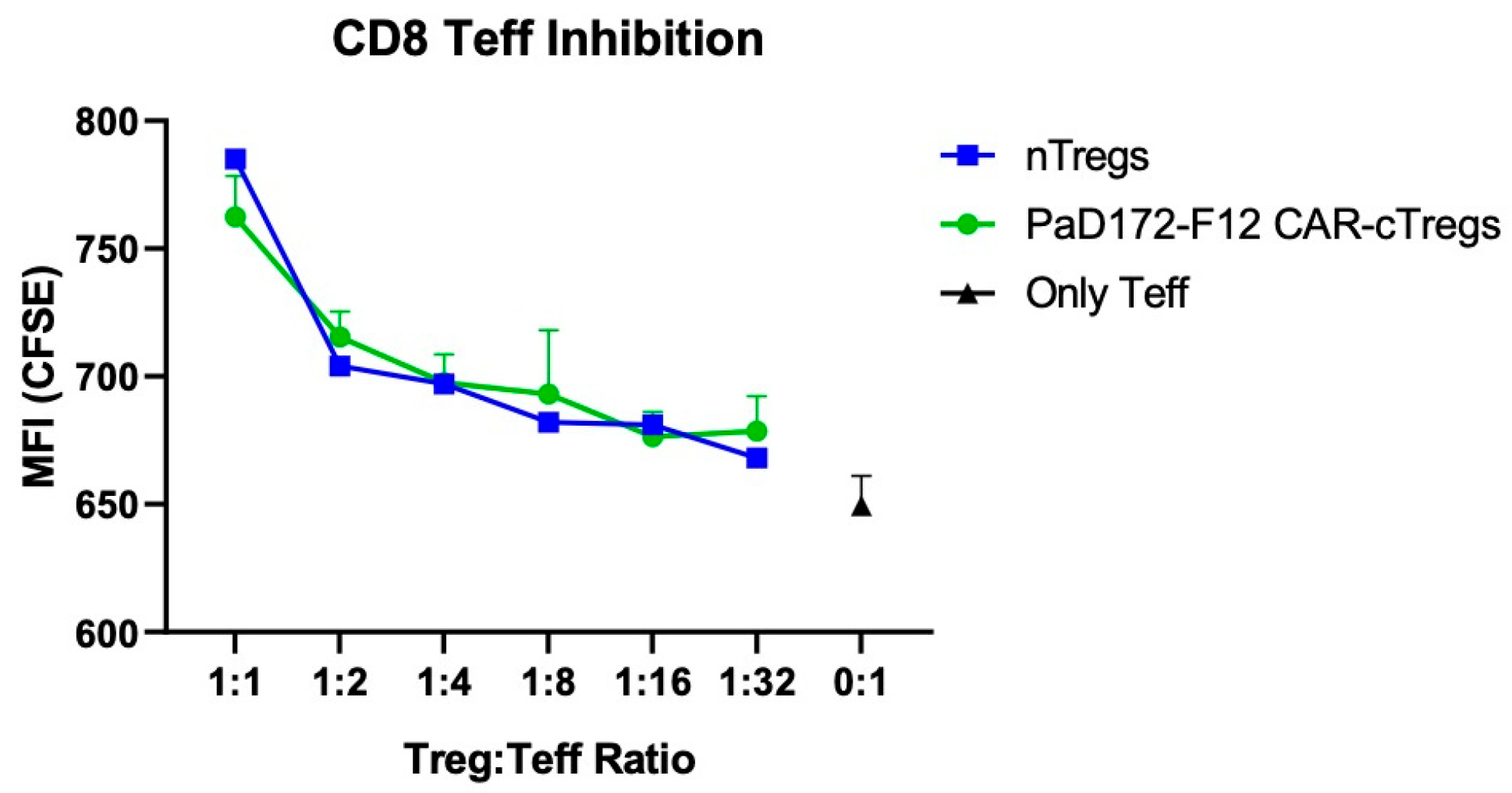
3.6. Aβ-Specific CAR-Tregs Can Inhibit Activated CD8+ T Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pampuscenko, K.; Morkuniene, R.; Sneideris, T.; Smirnovas, V.; Budvytyte, R.; Valincius, G.; Brown, G.C.; Borutaite, V. Extracellular tau induces microglial phagocytosis of living neurons in cell cultures. J. Neurochem. 2020, 154, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Faridar, A.; Thome, A.D.; Zhao, W.; Thonhoff, J.R.; Beers, D.R.; Pascual, B.; Masdeu, J.C.; Appel, S.H. Restoring regulatory T-cell dys-function in Alzheimer’s disease through ex vivo expansion. Brain Commun. 2020, 2, fcaa112. [Google Scholar] [CrossRef] [PubMed]
- Singh-Bains, M.K.; Linke, V.; Austria, M.D.; Tan, A.Y.; Scotter, E.L.; Mehrabi, N.F.; Faull, R.L.; Dragunow, M. Altered microglia and neurovasculature in the Alzheimer’s disease cerebellum. Neurobiol. Dis. 2019, 132, 104589. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.; Apostolova, L.G.; Atri, A.; Salloway, S.; Weiner, M. Aducanumab: Appropriate Use Recommendations. J. Prev. Alzheimer’s Dis. 2021, 8, 398–410. [Google Scholar] [CrossRef]
- Yuan, J.; Maserejian, N.; Liu, Y.; Devine, S.; Gillis, C.; Massaro, J.; Au, R. Severity Distribution of Alzheimer’s Disease Dementia and Mild Cognitive Impairment in the Framingham Heart Study. J. Alzheimer’s Dis. 2021, 79, 807–817. [Google Scholar] [CrossRef]
- Xie, L.; Choudhury, G.R.; Winters, A.; Yang, S.H.; Jin, K. Cerebral regulatory T cells restrain microglia/macrophage-mediated in-flammatory responses via IL-10. Eur. J. Immunol. 2015, 45, 180–191. [Google Scholar] [CrossRef]
- Dansokho, C.; Ahmed, D.A.; Aid, S.; Toly-Ndour, C.; Chaigneau, T.; Calle, V.; Cagnard, N.; Holzenberger, M.; Piaggio, E.; Aucouturier, P.; et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain 2016, 139, 1237–1251. [Google Scholar] [CrossRef]
- Baek, H.; Ye, M.; Kang, G.H.; Lee, C.; Lee, G.; Choi, D.B.; Jung, J.; Kim, H.; Lee, S.; Kim, J.S.; et al. Neuroprotective effects of CD4+CD25+Foxp3+ regulatory T cells in a 3xTg-AD Alzheimer’s disease model. Oncotarget 2016, 7, 69347–69357. [Google Scholar] [CrossRef] [PubMed]
- Theil, A.; Tuve, S.; Oelschlägel, U.; Maiwald, A.; Döhler, D.; Oßmann, D.; Zenkel, A.; Wilhelm, C.; Middeke, J.M.; Shayegi, N.; et al. Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease. Cytotherapy 2015, 17, 473–486. [Google Scholar] [CrossRef]
- Hull, C.M.; Nickolay, L.E.; Estorninho, M.; Richardson, M.W.; Riley, J.L.; Peakman, M.; Maher, J.; Tree, T.I. Generation of human is-let-specific regulatory T cells by TCR gene transfer. J. Autoimmun. 2017, 79, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Zhang, A.H.; Yoon, J.; Culp, W.E.; Lees, J.R.; Wucherpfennig, K.W.; Scott, D.W. Engineered MBP-specific human Tregs ameliorate MOG-induced EAE through IL-2-triggered inhibition of effector T cells. J. Autoimmun. 2018, 92, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Noyan, F.; Zimmermann, K.; Hardtke-Wolenski, M.; Knoefel, A.; Schulde, E.; Geffers, R.; Hust, M.; Huehn, J.; Galla, M.; Morgan, M.; et al. Prevention of Allograft Rejection by Use of Regulatory T Cells With an MHC-Specific Chimeric Antigen Receptor. Am. J. Transplant. 2016, 17, 917–930. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Henriksen, K.J.; Bi, M.; Finger, E.B.; Szot, G.; Ye, J.; Masteller, E.L.; McDevitt, H.; Bonyhadi, M.; Bluestone, J.A. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J. Exp. Med. 2004, 199, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Műzes, G.; Sipos, F. CAR-Based Therapy for autoimmune diseases: A novel powerful option. Cells 2023, 12, 1534. [Google Scholar] [CrossRef] [PubMed]
- Droste, P.; Frenzel, A.; Steinwand, M.; Pelat, T.; Thullier, P.; Hust, M.; Lashuel, H.; Dübel, S. Structural differences of amyloid-beta fibrils revealed by antibodies from phage display. BMC. Biotechnol. 2015, 15, 57. [Google Scholar] [CrossRef]
- Tenspolde, M.; Zimmermann, K.; Weber, L.C.; Hapke, M.; Lieber, M.; Dywicki, J.; Frenzel, A.; Hust, M.; Galla, M.; Buitrago-Molina, L.E.; et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J. Autoimmun. 2019, 103, 102289. [Google Scholar] [CrossRef]
- Henschel, P.; Landwehr-Kenzel, S.; Engels, N.; Schienke, A.; Kremer, J.; Riet, T.; Redel, N.; Iordanidis, K.; Saetzler, V.; John, K.; et al. Supraphysiological FOXP3 expression in human CAR-Tregs results in improved stability, efficacy, and safety of CAR-Treg products for clinical application. J. Autoimmun. 2023, 138, 103057. [Google Scholar] [CrossRef]
- Pieper, T.; Roth, K.D.R.; Glaser, V.; Riet, T.; Buitrago-Molina, L.E.; Hagedorn, M.; Lieber, M.; Hust, M.; Noyan, F.; Jaeckel, E.; et al. Generation of Chimeric Antigen Receptors against Tetraspanin 7. Cells 2023, 12, 1453. [Google Scholar] [CrossRef]
- Kremer, J.; Henschel, P.; Simon, D.; Riet, T.; Falk, C.; Hardtke-Wolenski, M.; Wedemeyer, H.; Noyan, F.; Jaeckel, E. Membrane-bound IL-2 improves the expansion, survival, and phenotype of CAR Tregs and confers resistance to calcineurin inhibitors. Front. Immunol. 2022, 13, 1005582. [Google Scholar] [CrossRef] [PubMed]
- Jaeckel, E.; Lipes, M.A.; Boehmer, V.H. Recessive tolerance to preproinsulin 2 reduces but does not abolish type 1 diabetes. Nat. Immunol. 2004, 5, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef]
- Trzonkowski, P.; Bieniaszewska, M.; Juścińska, J.; Dobyszuk, A.; Krzystyniak, A.; Marek, N.; Myśliwska, J.; Hellmann, A. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127− T regulatory cells. Clin. Immunol. 2009, 133, 22–26. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Zhao, W.; Wang, J.; Zhang, X.; Wen, S.; Neal, D.; Thonhoff, J.R.; Alsuliman, A.S.; Shpall, E.J.; Rezvani, K.; et al. ALS patients’ regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight. 2017, 2, e89530. [Google Scholar] [CrossRef]
- Thonhoff, J.R.; Beers, D.R.; Zhao, W.; Pleitez, M.; Simpson, E.P.; Berry, J.D.; Cudkowicz, M.E.; Appel, S.H. Expanded autologous regu-latory T-lymphocyte infusions in ALS: A phase I, first-in-human study. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e465. [Google Scholar] [CrossRef]
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V resions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968. [Google Scholar] [CrossRef]
- Toleikis, L.; Frenzel, A. Cloning Single-Chain Antibody Fragments (ScFv) from Hyrbidoma. Cells 2012, 907, 59–71. [Google Scholar] [CrossRef]
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. MAbs 2016, 8, 1177–1194. [Google Scholar] [CrossRef]
- Radichev, I.A.; Yoon, J.; Scott, D.W.; Griffin, K.; Savinov, A.Y. Towards antigen-specific Tregs for type 1 diabetes: Construction and functional assessment of pancreatic endocrine marker, HPi2-based chimeric antigen receptor. Cell. Immunol. 2020, 358, 104224. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Boroughs, A.C.; Larson, R.C.; Choi, B.D.; Bouffard, A.A.; Riley, L.S.; Schiferle, E.; Kulkarni, A.S.; Cetrulo, C.L.; Ting, D.; Blazar, B.R.; et al. Chimeric antigen receptor costimulation domains modulate human regulatory T cell function. J. Clin. Investig. 2019, 4, e126194. [Google Scholar] [CrossRef]
- Koristka, S.; Kegler, A.; Bergmann, R.; Arndt, C.; Feldmann, A.; Albert, S.; Cartellieri, M.; Ehninger, A.; Ehninger, G.; Middeke, J.M.; et al. Engrafting human regulatory T cells with a flexible modular chimeric antigen receptor technology. J. Autoimmun. 2018, 90, 116–131. [Google Scholar] [CrossRef] [PubMed]
- Bour-Jordan, H.; Bluestone, J.A. Regulating the regulators: Costimulatory signals control the homeostasis and function of regulatory T cells. Immunol. Rev. 2009, 229, 41–66. [Google Scholar] [CrossRef]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 712. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Rurik, J.G.; Tombácz, I.; Yadegari, A.; Fernández, P.O.M.; Shewale, S.V.; Li, L.; Kimura, T.; Soliman, O.Y.; Papp, T.E.; Tam, Y.K.; et al. CAR T cells produced in vivo to treat cardiac injury. Science 2022, 375, 91–96. [Google Scholar] [CrossRef]
- Jin, Z.; Maiti, S.; Huls, H.; Singh, H.; Olivares, S.; Mátés, L.; Izsvák, Z.; Ivics, Z.; Lee, A.D.; Champlin, R.E.; et al. The hyperactive Sleeping Beauty transposase SB100X improves the genetic modification of T cells to express a chimeric antigen receptor. Gene Ther. 2011, 18, 849–856. [Google Scholar] [CrossRef]
- Dimitri, A.; Herbst, F.; Fraietta, J.A. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol. Cancer 2022, 21, 78. [Google Scholar] [CrossRef]
- Morgan, M.A.; Galla, M.; Grez, M.; Fehse, B.; Schambach, A. Retroviral gene therapy in Germany with a view on previous experience and future perspectives. Gene Ther. 2021, 28, 494–512. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, O.; Wall, J.B.J.; Zheng, M.; Zhou, Y.; Wang, L.; Vaseghi, H.R.; Qian, L.; Liu, J. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017, 7, 2193. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Salas, E.; Francisco-Velilla, R.; Fernandez-Chamorro, J.; Embarek, A.M. Insights into Structural and Mechanistic Features of Viral IRES Elements. Front. Microbiol. 2018, 8, 2629. [Google Scholar] [CrossRef]
- Li, W.; Qiu, S.; Chen, J.; Jiang, S.; Chen, W.; Jiang, J.; Wang, F.; Si, W.; Shu, Y.; Wei, P.; et al. Chimeric Antigen Receptor Designed to Prevent Ubiquitination and Downregulation Showed Durable Antitumor Efficacy. Immunity 2020, 53, 456–470.e6. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef]
- Davenport, A.J.; Jenkins, M.R.; Cross, R.S.; Yong, C.S.; Prince, H.M.; Ritchie, D.S.; Trapani, J.A.; Kershaw, M.H.; Darcy, P.K.; Neeson, P.J. CAR-T Cells Inflict Sequential Killing of Multiple Tumor Target Cells. Cancer Immunol. Res. 2015, 3, 483–494. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saetzler, V.; Riet, T.; Schienke, A.; Henschel, P.; Freitag, K.; Haake, A.; Heppner, F.L.; Buitrago-Molina, L.E.; Noyan, F.; Jaeckel, E.; et al. Development of Beta-Amyloid-Specific CAR-Tregs for the Treatment of Alzheimer’s Disease. Cells 2023, 12, 2115. https://doi.org/10.3390/cells12162115
Saetzler V, Riet T, Schienke A, Henschel P, Freitag K, Haake A, Heppner FL, Buitrago-Molina LE, Noyan F, Jaeckel E, et al. Development of Beta-Amyloid-Specific CAR-Tregs for the Treatment of Alzheimer’s Disease. Cells. 2023; 12(16):2115. https://doi.org/10.3390/cells12162115
Chicago/Turabian StyleSaetzler, Valerie, Tobias Riet, Andrea Schienke, Pierre Henschel, Kiara Freitag, Alexander Haake, Frank L. Heppner, Laura Elisa Buitrago-Molina, Fatih Noyan, Elmar Jaeckel, and et al. 2023. "Development of Beta-Amyloid-Specific CAR-Tregs for the Treatment of Alzheimer’s Disease" Cells 12, no. 16: 2115. https://doi.org/10.3390/cells12162115
APA StyleSaetzler, V., Riet, T., Schienke, A., Henschel, P., Freitag, K., Haake, A., Heppner, F. L., Buitrago-Molina, L. E., Noyan, F., Jaeckel, E., & Hardtke-Wolenski, M. (2023). Development of Beta-Amyloid-Specific CAR-Tregs for the Treatment of Alzheimer’s Disease. Cells, 12(16), 2115. https://doi.org/10.3390/cells12162115