
The Adiponectin Receptor Agonist, ALY688: A Promising Therapeutic for Fibrosis in the Dystrophic Muscle

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. In Vivo Studies of Muscle Function
2.3. Bright-Field Histochemistry
2.4. Immunofluorescence
2.5. Culture of Murine C2C12 Cell Lines
2.6. Culture of Human Myotubes
2.7. RNA Extraction and Real-Time Quantitative PCR
2.8. Protein Extraction and ELISAs
2.9. Statistical Analysis
3. Results
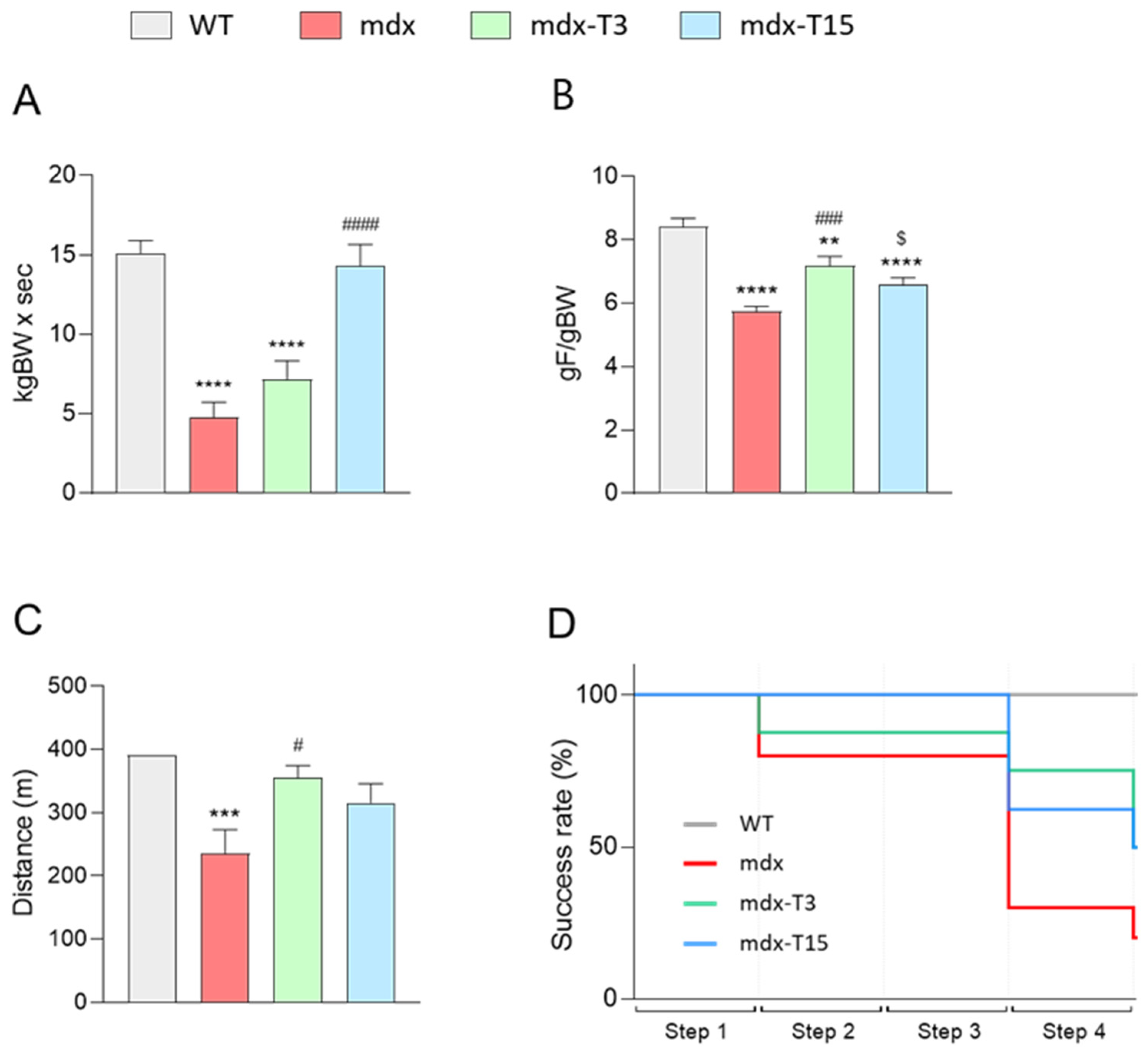
3.1. ALY688 Enhances Force and Endurance of Mdx Mice
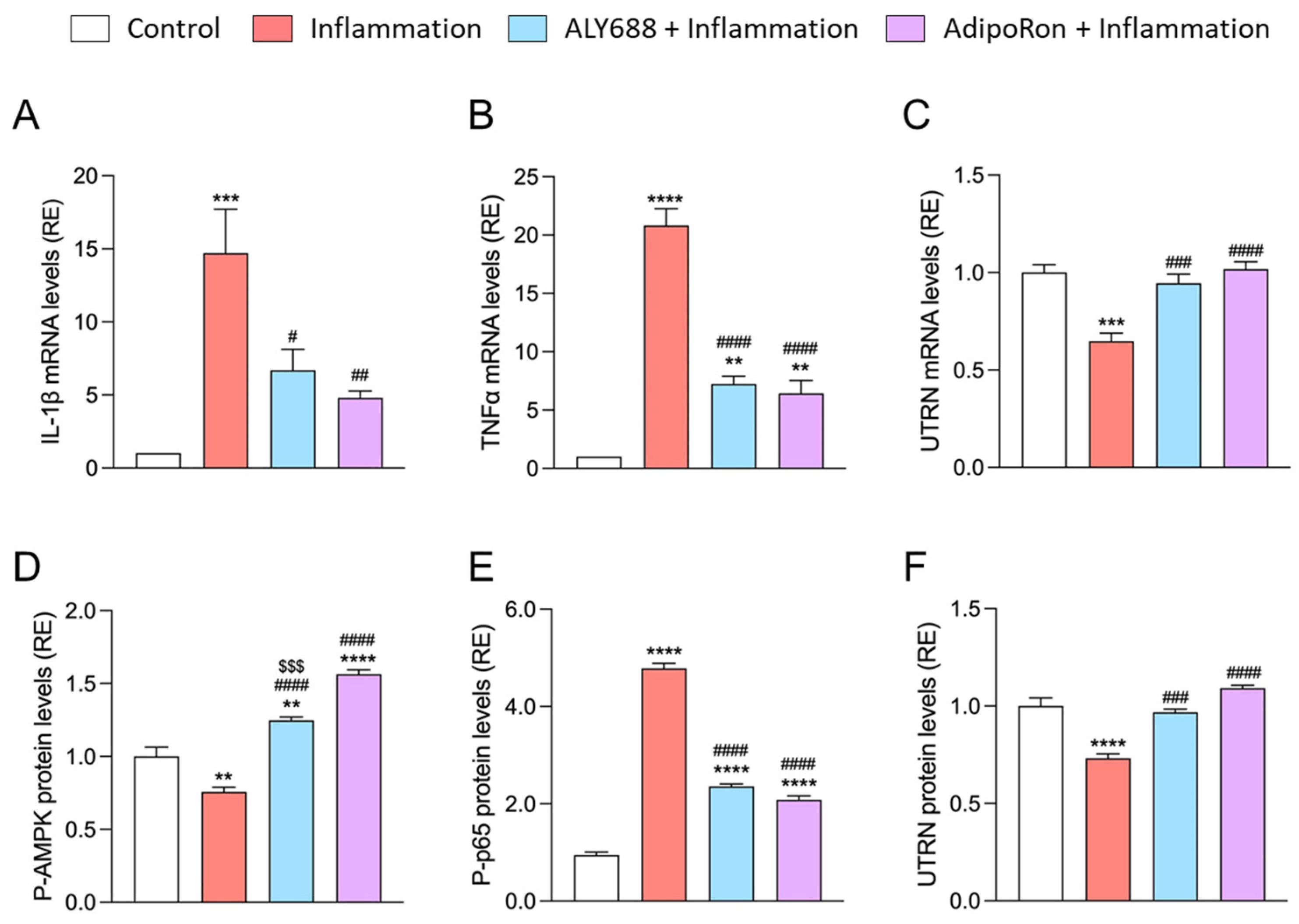
3.2. ALY688 Effectively Reduces Muscle Inflammation and Oxidative Stress
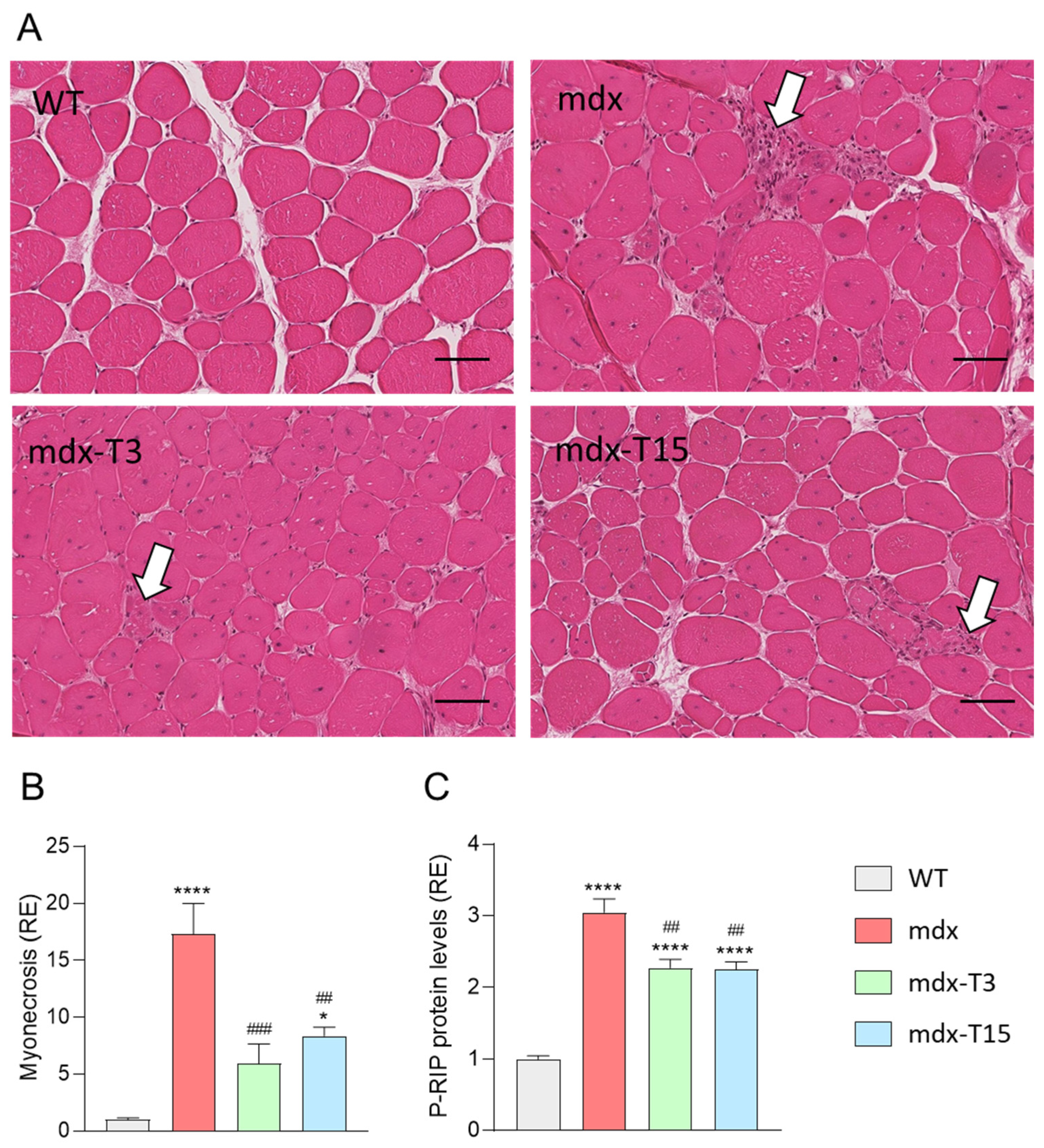
3.3. ALY688 Counteracts Myonecrosis in Mdx Mice
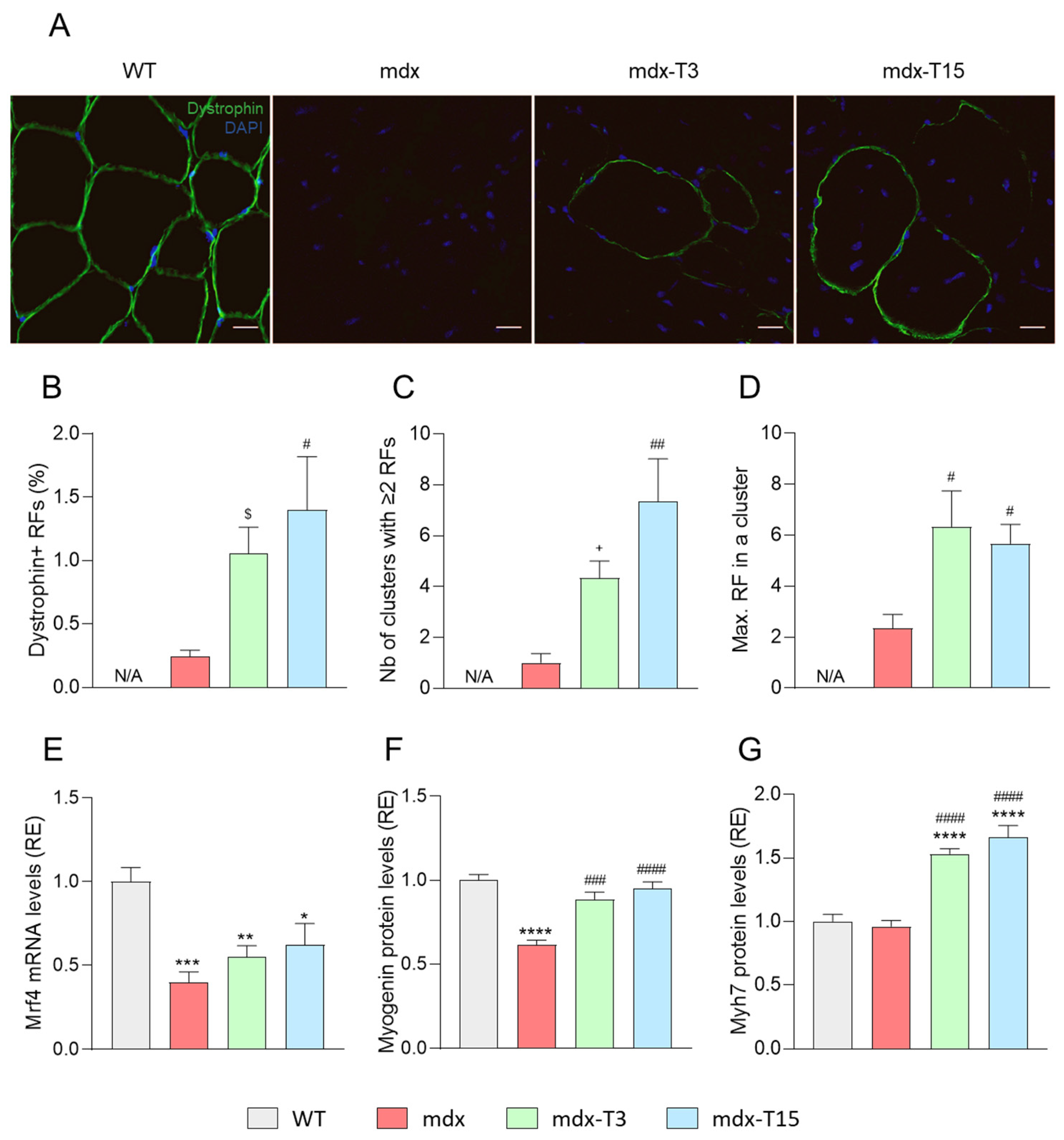
3.4. ALY688 Increases the Number of Revertant Myofibres and Enhances the Myogenic Program in Mdx Mice
3.5. ALY688 Attenuates Fibrosis in Mdx Mice
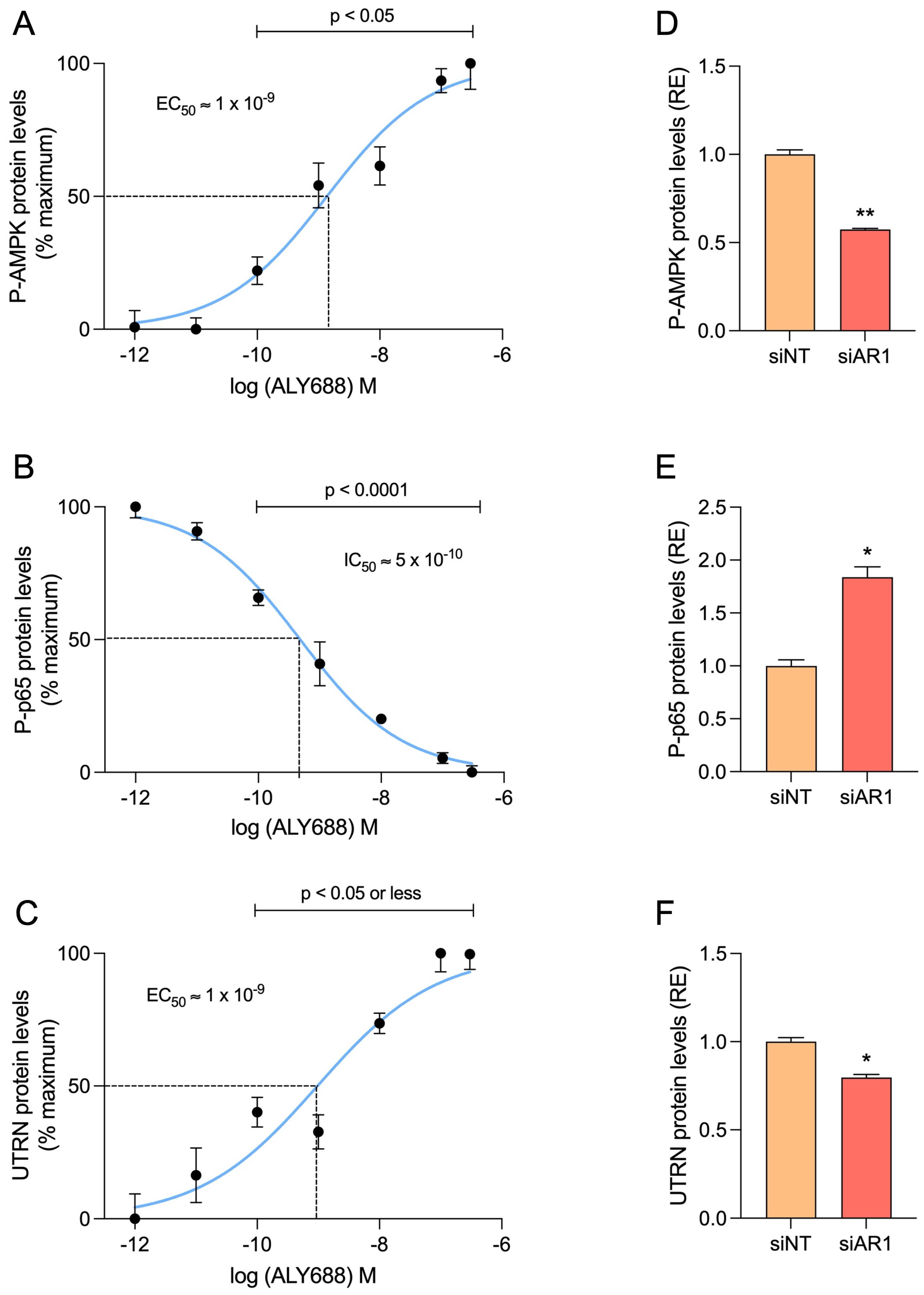
3.6. ALY688 Enhances Key Effectors of the AMPK Pathway in Mdx Mice
3.7. ALY688 Recapitulates Its Beneficial Effects on Human Myotubes Challenged by Pro-Inflammatory Cytokines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef]
- Ryder, S.; Leadley, R.M.; Armstrong, N.; Westwood, M.; de Kock, S.; Butt, T.; Jain, M.; Kleijnen, J. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: An evidence review. Orphanet J. Rare Dis. 2017, 12, 79. [Google Scholar] [CrossRef]
- Acharyya, S.; Villalta, S.A.; Bakkar, N.; Bupha-Intr, T.; Janssen, P.M.; Carathers, M.; Li, Z.W.; Beg, A.A.; Ghosh, S.; Sahenk, Z.; et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J. Clin. Investig. 2007, 117, 889–901. [Google Scholar] [CrossRef]
- Klingler, W.; Jurkat-Rott, K.; Lehmann-Horn, F.; Schleip, R. The role of fibrosis in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 184–195. [Google Scholar] [PubMed]
- Abou-Samra, M.; Selvais, C.M.; Dubuisson, N.; Brichard, S.M. Adiponectin and Its Mimics on Skeletal Muscle: Insulin Sensitizers, Fat Burners, Exercise Mimickers, Muscling Pills… or Everything Together? Int. J. Mol. Sci. 2020, 21, 2620. [Google Scholar] [CrossRef]
- Yamauchi, T.; Nio, Y.; Maki, T.; Kobayashi, M.; Takazawa, T.; Iwabu, M.; Okada-Iwabu, M.; Kawamoto, S.; Kubota, N.; Kubota, T.; et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 2007, 13, 332–339. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Ito, Y.; Tsuchida, A.; Yokomizo, T.; Kita, S.; Sugiyama, T.; Miyagishi, M.; Hara, K.; Tsunoda, M.; et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003, 423, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Abou-Samra, M.; Lecompte, S.; Schakman, O.; Noel, L.; Many, M.C.; Gailly, P.; Brichard, S.M. Involvement of adiponectin in the pathogenesis of dystrophinopathy. Skelet. Muscle 2015, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Abou-Samra, M.; Boursereau, R.; Lecompte, S.; Noel, L.; Brichard, S.M. Potential Therapeutic Action of Adiponectin in Duchenne Muscular Dystrophy. Am. J. Pathol. 2017, 187, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L., Jr.; Haspinger, E.; La Russa, F.; Maspero, F.; Graziano, P.; Kovalszky, I.; Lovas, S.; Nama, K.; Hoffmann, R.; Knappe, D.; et al. Design and development of a peptide-based adiponectin receptor agonist for cancer treatment. BMC Biotechnol. 2011, 11, 90. [Google Scholar] [CrossRef]
- Otvos, L., Jr. Potential Adiponectin Receptor Response Modifier Therapeutics. Front. Endocrinol. 2019, 10, 539. [Google Scholar] [CrossRef]
- Kumar, P.; Smith, T.; Rahman, K.; Thorn, N.E.; Anania, F.A. Adiponectin agonist ADP355 attenuates CCl4-induced liver fibrosis in mice. PLoS ONE 2014, 9, e110405. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Zhang, Z.; Huang, B.; Cheng, X.; Wang, D.; la Gahu, Z.; Xue, Z.; Da, Y.; Li, D.; et al. Adiponectin-derived active peptide ADP355 exerts anti-inflammatory and anti-fibrotic activities in thioacetamide-induced liver injury. Sci. Rep. 2016, 6, 19445. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Xue, C.; Li, J.; Feng, K.; Zeng, P.; Chen, Y.; Duan, Y.; Zhang, S.; Li, X.; Han, J.; et al. Adiponectin agonist ADP355 ameliorates doxorubicin-induced cardiotoxicity by decreasing cardiomyocyte apoptosis and oxidative stress. Biochem. Biophys. Res. Commun. 2020, 533, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.K.; Mitchell, P.L.; Gross, S.; Marette, A.; Sweeney, G. ALY688 elicits adiponectin-mimetic signaling and improves insulin action in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2022, 322, C151–C163. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yang, X.; Li, Q.; Zeng, P.; Liu, Y.; Liu, L.; Chen, Y.; Yu, M.; Ma, C.; Li, X.; et al. Activation of Adiponectin Receptor Regulates Proprotein Convertase Subtilisin/Kexin Type 9 Expression and Inhibits Lesions in ApoE-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.D.; Khanna, S.; Kaminski, H.J.; Rao, J.S.; Merriam, A.P.; Richmonds, C.R.; Leahy, P.; Li, J.; Guo, W.; Andrade, F.H. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2002, 11, 263–272. [Google Scholar] [CrossRef]
- Kastenschmidt, J.M.; Ellefsen, K.L.; Mannaa, A.H.; Giebel, J.J.; Yahia, R.; Ayer, R.E.; Pham, P.; Rios, R.; Vetrone, S.A.; Mozaffar, T.; et al. QuantiMus: A Machine Learning-Based Approach for High Precision Analysis of Skeletal Muscle Morphology. Front. Physiol. 2019, 10, 1416. [Google Scholar] [CrossRef]
- Augusto, V.; Padovani, C.; Eduardo, G.; Campos, R. Skeletal muscle fiber types in C57BL6J mice. J. Morphol. Sci 2004, 21, 89–94. [Google Scholar]
- Abou-Samra, M.; Selvais, C.M.; Boursereau, R.; Lecompte, S.; Noel, L.; Brichard, S.M. AdipoRon, a new therapeutic prospect for Duchenne muscular dystrophy. J. Cachexia Sarcopenia Muscle 2020, 11, 518–533. [Google Scholar] [CrossRef]
- Dubuisson, N.; Davis-López de Carrizosa, M.A.; Versele, R.; Selvais, C.M.; Noel, L.; Van den Bergh, P.Y.D.; Brichard, S.M.; Abou-Samra, M. Inhibiting the inflammasome with MCC950 counteracts muscle pyroptosis and improves Duchenne muscular dystrophy. Front. Immunol. 2022, 13, 1049076. [Google Scholar] [CrossRef] [PubMed]
- van Putten, M. The Use of Hanging Wire Tests to Monitor Muscle Strength and Condition over Time. In DMDM.2.1.004. 2011. Available online: https://treat-nmd.org/wp-content/uploads/2021/06/uncategorized-Wire-test.pdf (accessed on 23 February 2022).
- Bi, P.; Yue, F.; Sato, Y.; Wirbisky, S.; Liu, W.; Shan, T.; Wen, Y.; Zhou, D.; Freeman, J.; Kuang, S. Stage-specific effects of Notch activation during skeletal myogenesis. Elife 2016, 5, e17355. [Google Scholar] [CrossRef] [PubMed]
- Yue, F.; Bi, P.; Wang, C.; Li, J.; Liu, X.; Kuang, S. Conditional Loss of Pten in Myogenic Progenitors Leads to Postnatal Skeletal Muscle Hypertrophy but Age-Dependent Exhaustion of Satellite Cells. Cell Rep. 2016, 17, 2340–2353. [Google Scholar] [CrossRef] [PubMed]
- Castro, B.; Kuang, S. Evaluation of Muscle Performance in Mice by Treadmill Exhaustion Test and Whole-limb Grip Strength Assay. Bio Protoc. 2017, 7, e2237. [Google Scholar] [CrossRef]
- Grounds, M. Quantification of histopathology in Haemotoxylin and Eosin Stained Muscle Sections. In DMD_M.1.2.007. 2014. Available online: https://treat-nmd.org/wp-content/uploads/2016/08/MDX-DMD_M.1.2.007-28.pdf (accessed on 26 July 2023).
- Rodrigues, M.; Echigoya, Y.; Maruyama, R.; Lim, K.R.; Fukada, S.I.; Yokota, T. Impaired regenerative capacity and lower revertant fibre expansion in dystrophin-deficient mdx muscles on DBA/2 background. Sci. Rep. 2016, 6, 38371. [Google Scholar] [CrossRef] [PubMed]
- Mayeuf-Louchart, A.; Hardy, D.; Thorel, Q.; Roux, P.; Gueniot, L.; Briand, D.; Mazeraud, A.; Bouglé, A.; Shorte, S.L.; Staels, B.; et al. MuscleJ: A high-content analysis method to study skeletal muscle with a new Fiji tool. Skelet. Muscle 2018, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Selvais, C.M.; Davis-López de Carrizosa, M.A.; Nachit, M.; Versele, R.; Dubuisson, N.; Noel, L.; Gillard, J.; Leclercq, I.A.; Brichard, S.M.; Abou-Samra, M. AdipoRon enhances healthspan in middle-aged obese mice: Striking alleviation of myosteatosis and muscle degenerative markers. J. Cachexia Sarcopenia Muscle 2023, 14, 464–478. [Google Scholar] [CrossRef]
- Narola, J.; Pandey, S.N.; Glick, A.; Chen, Y.W. Conditional expression of TGF-β1 in skeletal muscles causes endomysial fibrosis and myofibers atrophy. PLoS ONE 2013, 8, e79356. [Google Scholar] [CrossRef]
- Morgan, J.E.; Prola, A.; Mariot, V.; Pini, V.; Meng, J.; Hourde, C.; Dumonceaux, J.; Conti, F.; Relaix, F.; Authier, F.J.; et al. Necroptosis mediates myofibre death in dystrophin-deficient mice. Nat. Commun. 2018, 9, 3655. [Google Scholar] [CrossRef]
- Grounds, M.D.; Terrill, J.R.; Al-Mshhdani, B.A.; Duong, M.N.; Radley-Crabb, H.G.; Arthur, P.G. Biomarkers for Duchenne muscular dystrophy: Myonecrosis, inflammation and oxidative stress. Dis. Model. Mech. 2020, 13, dmm043638. [Google Scholar] [CrossRef]
- Lazzarin, M.; Quintana, H.; Araújo Baptista, V.; de Oliveira, F. Lack of dystrophin influences muscle inflammation but not myogenic regulatory factors after eccentric exercise in mdx mice. Motriz. Rev. Educ. Física 2020, 26, e10200228. [Google Scholar] [CrossRef]
- Mahdy, M.A.A. Skeletal muscle fibrosis: An overview. Cell Tissue Res. 2019, 375, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Al-Rewashdy, H.; Ljubicic, V.; Lin, W.; Renaud, J.M.; Jasmin, B.J. Utrophin A is essential in mediating the functional adaptations of mdx mouse muscle following chronic AMPK activation. Hum. Mol. Genet. 2015, 24, 1243–1255. [Google Scholar] [CrossRef]
- Gordon, B.S.; Delgado Diaz, D.C.; Kostek, M.C. Resveratrol decreases inflammation and increases utrophin gene expression in the mdx mouse model of Duchenne muscular dystrophy. Clin. Nutr. 2013, 32, 104–111. [Google Scholar] [CrossRef] [PubMed]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. Dis. Model. Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Hathout, Y.; Marathi, R.L.; Rayavarapu, S.; Zhang, A.; Brown, K.J.; Seol, H.; Gordish-Dressman, H.; Cirak, S.; Bello, L.; Nagaraju, K.; et al. Discovery of serum protein biomarkers in the mdx mouse model and cross-species comparison to Duchenne muscular dystrophy patients. Hum. Mol. Genet. 2014, 23, 6458–6469. [Google Scholar] [CrossRef]
- Hathout, Y.; Liang, C.; Ogundele, M.; Xu, G.; Tawalbeh, S.M.; Dang, U.J.; Hoffman, E.P.; Gordish-Dressman, H.; Conklin, L.S.; van den Anker, J.N.; et al. Disease-specific and glucocorticoid-responsive serum biomarkers for Duchenne Muscular Dystrophy. Sci. Rep. 2019, 9, 12167. [Google Scholar] [CrossRef]
- Okada-Iwabu, M.; Yamauchi, T.; Iwabu, M.; Honma, T.; Hamagami, K.; Matsuda, K.; Yamaguchi, M.; Tanabe, H.; Kimura-Someya, T.; Shirouzu, M.; et al. A small-molecule AdipoR agonist for type 2 diabetes and short life in obesity. Nature 2013, 503, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Morris, G.E.; Wilton, S.D.; Ly, T.; Artem’yeva, O.V.; Strong, P.; Partridge, T.A. Massive idiosyncratic exon skipping corrects the nonsense mutation in dystrophic mouse muscle and produces functional revertant fibers by clonal expansion. J. Cell Biol. 2000, 148, 985–996. [Google Scholar] [CrossRef]
- Yokota, T.; Lu, Q.L.; Morgan, J.E.; Davies, K.E.; Fisher, R.; Takeda, S.; Partridge, T.A. Expansion of revertant fibers in dystrophic mdx muscles reflects activity of muscle precursor cells and serves as an index of muscle regeneration. J. Cell Sci. 2006, 119, 2679–2687. [Google Scholar] [CrossRef]
- Biferali, B.; Proietti, D.; Mozzetta, C.; Madaro, L. Fibro-Adipogenic Progenitors Cross-Talk in Skeletal Muscle: The Social Network. Front. Physiol. 2019, 10, 1074. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Wehling-Henricks, M. Shifts in macrophage cytokine production drive muscle fibrosis. Nat. Med. 2015, 21, 665–666. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Chen, Z.; Yu, Y.; Zhang, N.; Jiang, H.; Zhang, G.; Zhang, Z.; Zhang, B. Current Pharmacological Strategies for Duchenne Muscular Dystrophy. Front. Cell Dev. Biol. 2021, 9, 689533. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Hyttinen, J.M.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.R.; Damsker, J.M.; Yu, Q.; Dillingham, B.C.; Huynh, T.; Van der Meulen, J.H.; Sali, A.; Miller, B.K.; Phadke, A.; Scheffer, L.; et al. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol. Med. 2013, 5, 1569–1585. [Google Scholar] [CrossRef]
- Siegel, A.L.; Bledsoe, C.; Lavin, J.; Gatti, F.; Berge, J.; Millman, G.; Turin, E.; Winders, W.T.; Rutter, J.; Palmeiri, B.; et al. Treatment with inhibitors of the NF-kappaB pathway improves whole body tension development in the mdx mouse. Neuromuscul. Disord. 2009, 19, 131–139. [Google Scholar] [CrossRef]
- Giovarelli, M.; Zecchini, S.; Catarinella, G.; Moscheni, C.; Sartori, P.; Barbieri, C.; Roux-Biejat, P.; Napoli, A.; Vantaggiato, C.; Cervia, D.; et al. Givinostat as metabolic enhancer reverting mitochondrial biogenesis deficit in Duchenne Muscular Dystrophy. Pharmacol. Res. 2021, 170, 105751. [Google Scholar] [CrossRef]
- Selsby, J.T.; Morine, K.J.; Pendrak, K.; Barton, E.R.; Sweeney, H.L. Rescue of dystrophic skeletal muscle by PGC-1α involves a fast to slow fiber type shift in the mdx mouse. PLoS ONE 2012, 7, e30063. [Google Scholar] [CrossRef] [PubMed]
- Squire, S.; Raymackers, J.M.; Vandebrouck, C.; Potter, A.; Tinsley, J.; Fisher, R.; Gillis, J.M.; Davies, K.E. Prevention of pathology in mdx mice by expression of utrophin: Analysis using an inducible transgenic expression system. Hum. Mol. Genet. 2002, 11, 3333–3344. [Google Scholar] [CrossRef]
- Marangoni, R.G.; Masui, Y.; Fang, F.; Korman, B.; Lord, G.; Lee, J.; Lakota, K.; Wei, J.; Scherer, P.E.; Otvos, L.; et al. Adiponectin is an endogenous anti-fibrotic mediator and therapeutic target. Sci. Rep. 2017, 7, 4397. [Google Scholar] [CrossRef] [PubMed]
- Darmawan, C.C.; Montenegro, S.E.; Jo, G.; Kusumaningrum, N.; Lee, S.H.; Chung, J.H.; Mun, J.H. Adiponectin-Based Peptide (ADP355) Inhibits Transforming Growth Factor-β1-Induced Fibrosis in Keloids. Int. J. Mol. Sci. 2020, 21, 2833. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, L.; Gao, Y.; Chen, Y.; Tian, Q.; Son, J.S.; Chae, S.A.; de Avila, J.M.; Zhu, M.J.; Du, M. AMP-activated protein kinase inhibition in fibro-adipogenic progenitors impairs muscle regeneration and increases fibrosis. J. Cachexia Sarcopenia Muscle 2023, 14, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Juban, G.; Saclier, M.; Yacoub-Youssef, H.; Kernou, A.; Arnold, L.; Boisson, C.; Ben Larbi, S.; Magnan, M.; Cuvellier, S.; Théret, M.; et al. AMPK Activation Regulates LTBP4-Dependent TGF-β1 Secretion by Pro-inflammatory Macrophages and Controls Fibrosis in Duchenne Muscular Dystrophy. Cell Rep. 2018, 25, 2163–2176.e6. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Dong, L.Q. Adiponectin signaling and function in insulin target tissues. J. Mol. Cell Biol. 2016, 8, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Gamberi, T.; Magherini, F.; Modesti, A.; Fiaschi, T. Adiponectin Signaling Pathways in Liver Diseases. Biomedicines 2018, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M.; et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 2010, 464, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, H.; Zhang, R.; Xiang, Y.; Lu, J.; Xia, B.; Peng, L.; Wu, J. AdipoRon exerts opposing effects on insulin sensitivity via fibroblast growth factor 21-mediated time-dependent mechanisms. J. Biol. Chem. 2022, 298, 101641. [Google Scholar] [CrossRef]
- Rodrigues, M.; Echigoya, Y.; Fukada, S.I.; Yokota, T. Current Translational Research and Murine Models For Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2016, 3, 29–48. [Google Scholar] [CrossRef]
- Chamberlain, J.S.; Metzger, J.; Reyes, M.; Townsend, D.; Faulkner, J.A. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 2007, 21, 2195–2204. [Google Scholar] [CrossRef]
- Lecompte, S.; Abou-Samra, M.; Boursereau, R.; Noel, L.; Brichard, S.M. Skeletal muscle secretome in Duchenne muscular dystrophy: A pivotal anti-inflammatory role of adiponectin. Cell. Mol. Life Sci. 2017, 74, 2487–2501. [Google Scholar] [CrossRef]
- Yamashita, T.; Lakota, K.; Taniguchi, T.; Yoshizaki, A.; Sato, S.; Hong, W.; Zhou, X.; Sodin-Semrl, S.; Fang, F.; Asano, Y.; et al. An orally-active adiponectin receptor agonist mitigates cutaneous fibrosis, inflammation and microvascular pathology in a murine model of systemic sclerosis. Sci. Rep. 2018, 8, 11843. [Google Scholar] [CrossRef] [PubMed]
- Sha, M.; Gao, Y.; Deng, C.; Wan, Y.; Zhuang, Y.; Hu, X.; Wang, Y. Therapeutic effects of AdipoRon on liver inflammation and fibrosis induced by CCl(4) in mice. Int. Immunopharmacol. 2020, 79, 106157. [Google Scholar] [CrossRef]
- Li, Y.; Song, B.; Ruan, C.; Xue, W.; Zhao, J. AdipoRon Attenuates Hypertension-Induced Epithelial-Mesenchymal Transition and Renal Fibrosis via Promoting Epithelial Autophagy. J. Cardiovasc. Transl. Res. 2021, 14, 538–545. [Google Scholar] [CrossRef]
- Jones, D. Duchenne muscular dystrophy awaits gene therapy. Nat. Biotechnol. 2019, 37, 335–337. [Google Scholar] [CrossRef]
- Elangkovan, N.; Dickson, G. Gene Therapy for Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2021, 8, S303–S316. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2016, CD003725. [Google Scholar] [CrossRef] [PubMed]
- Da Eira, D.; Jani, S.; Sung, H.; Sweeney, G.; Ceddia, R.B. Effects of the adiponectin mimetic compound ALY688 on glucose and fat metabolism in visceral and subcutaneous rat adipocytes. Adipocyte 2020, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubuisson, N.; Versele, R.; Davis-López de Carrizosa, M.A.; Selvais, C.M.; Noel, L.; Planchon, C.; Van den Bergh, P.Y.K.; Brichard, S.M.; Abou-Samra, M. The Adiponectin Receptor Agonist, ALY688: A Promising Therapeutic for Fibrosis in the Dystrophic Muscle. Cells 2023, 12, 2101. https://doi.org/10.3390/cells12162101
Dubuisson N, Versele R, Davis-López de Carrizosa MA, Selvais CM, Noel L, Planchon C, Van den Bergh PYK, Brichard SM, Abou-Samra M. The Adiponectin Receptor Agonist, ALY688: A Promising Therapeutic for Fibrosis in the Dystrophic Muscle. Cells. 2023; 12(16):2101. https://doi.org/10.3390/cells12162101
Chicago/Turabian StyleDubuisson, Nicolas, Romain Versele, Maria A. Davis-López de Carrizosa, Camille M. Selvais, Laurence Noel, Chloé Planchon, Peter Y. K. Van den Bergh, Sonia M. Brichard, and Michel Abou-Samra. 2023. "The Adiponectin Receptor Agonist, ALY688: A Promising Therapeutic for Fibrosis in the Dystrophic Muscle" Cells 12, no. 16: 2101. https://doi.org/10.3390/cells12162101
APA StyleDubuisson, N., Versele, R., Davis-López de Carrizosa, M. A., Selvais, C. M., Noel, L., Planchon, C., Van den Bergh, P. Y. K., Brichard, S. M., & Abou-Samra, M. (2023). The Adiponectin Receptor Agonist, ALY688: A Promising Therapeutic for Fibrosis in the Dystrophic Muscle. Cells, 12(16), 2101. https://doi.org/10.3390/cells12162101