

Botulinum Toxin Treatment of Adult Muscle Stem Cells from Children with Cerebral Palsy and hiPSC-Derived Neuromuscular Junctions

 , , , , ,
, , , , ,  , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Recruitment
2.2. Biopsy Collection
2.3. Cell Culture
2.4. Fluorescence-Activated Cell Sorting
2.5. In Vitro BoNT Treatment
2.6. Human Induced Pluripotent Stem Cell Culture
2.7. Skeletal Muscle Differentiation
2.8. Lower Motor Neuron Differentiation
2.9. Neuromuscular Junction Formation Using Microfluidic Devices
2.10. Coculture Experiments and Neuromuscular Junction Botulinum Toxin Treatment
2.11. Quantitative Real-Time PCR Analysis
2.12. Immunofluorescent Staining
2.13. Statistics
3. Results
3.1. Participants
3.2. Reduced Myotube Differentiation after In Vivo BoNT Administration
3.3. Collagen Expression Pattern of Fibroblasts after In Vitro BoNT Treatment
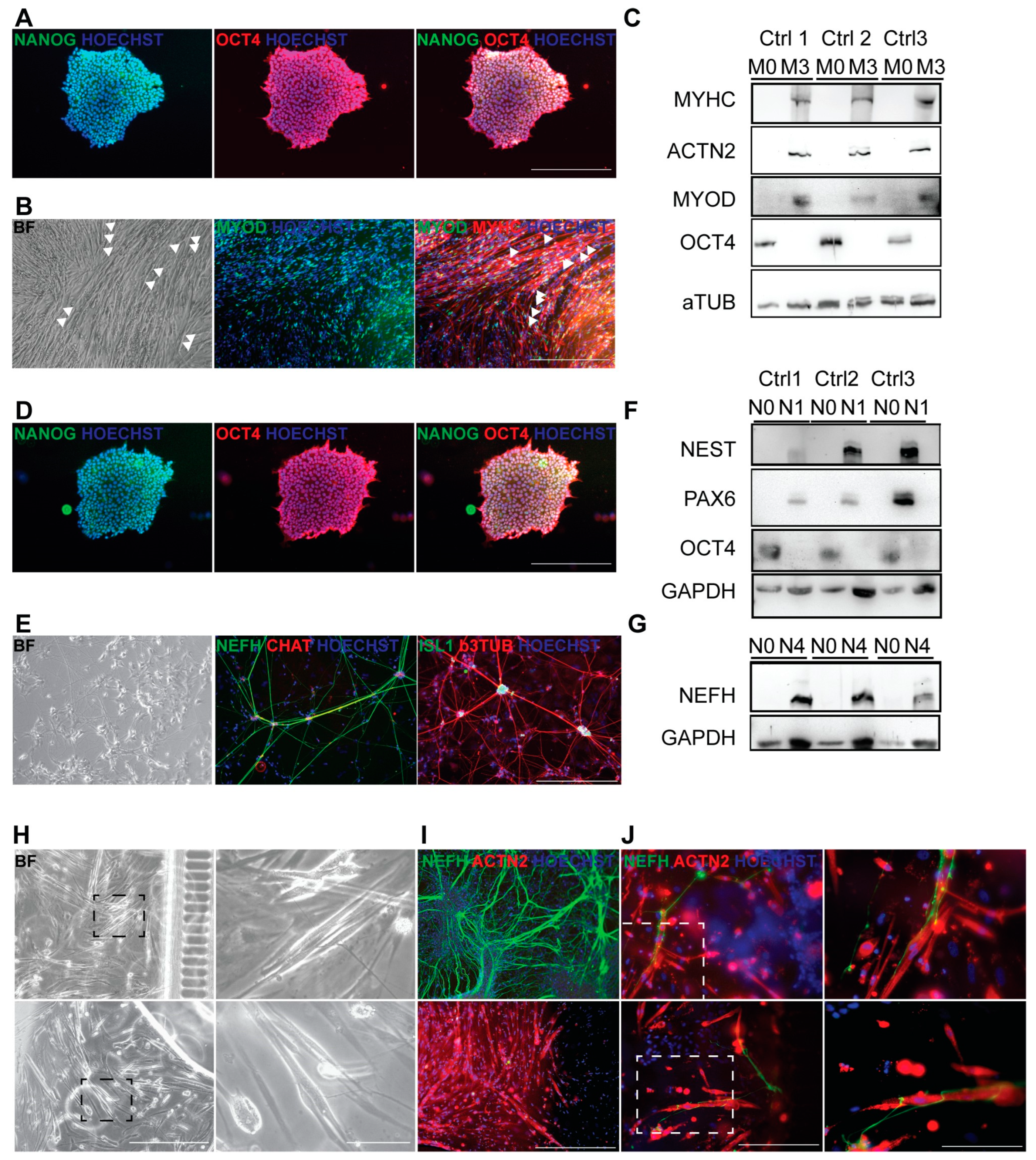
3.4. Generation of Autologous hiPSC-Based Neuromuscular Junction Model
3.5. BoNT Treatment of Neuromuscular Junctions: Morphological and Molecular Effects
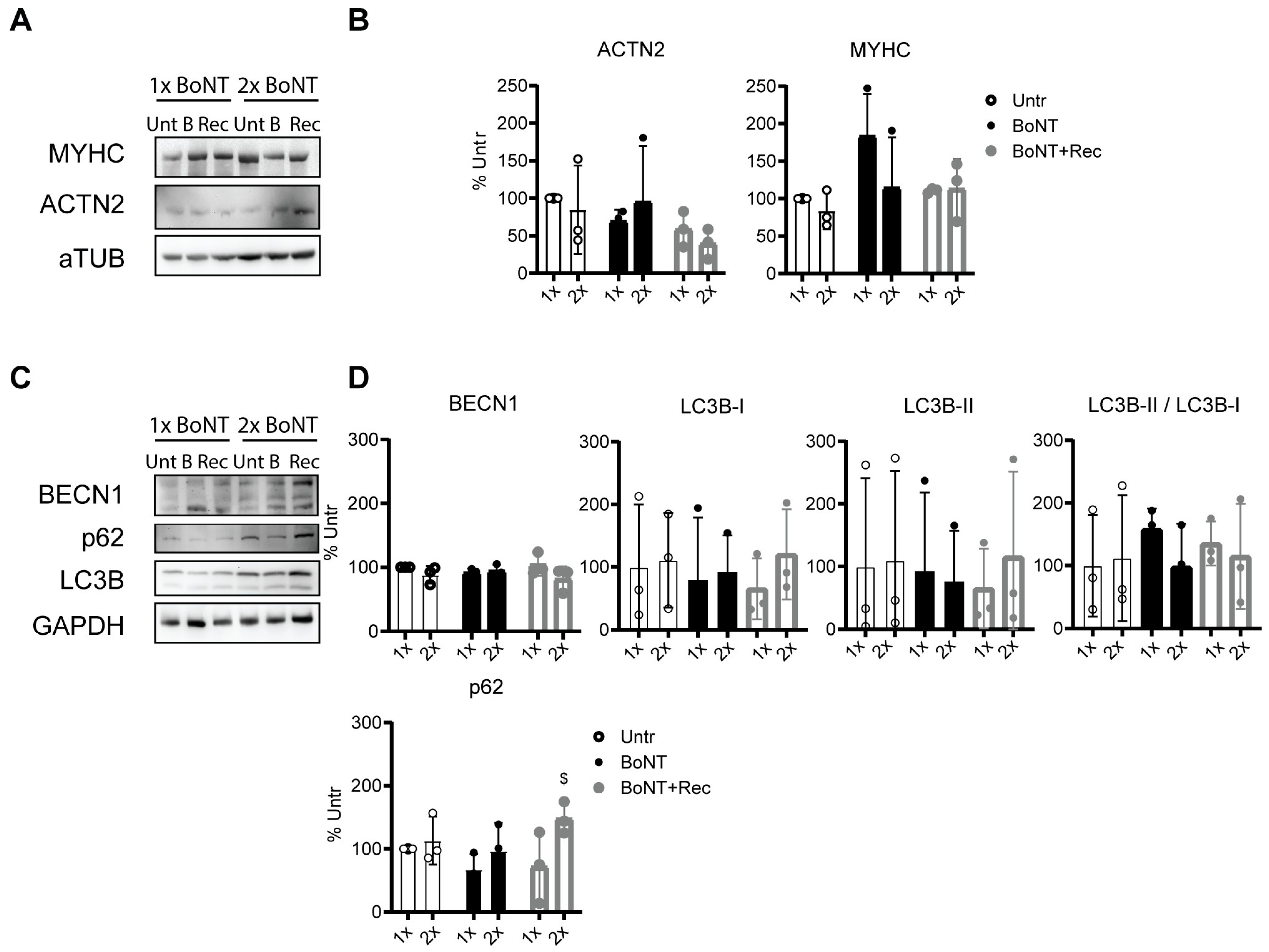
3.6. Repetitive BoNT Treatment of Neuromuscular Junctions
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Graham, H.K.; Rosenbaum, P.; Paneth, N.; Dan, B.; Lin, J.P.; Damiano, D.L.; Becher, J.G.; Gaebler-Spira, D.; Colver, A.; Reddihough, D.S.; et al. Cerebral palsy. Nat. Rev. Dis. Prim. 2016, 2, 15082. [Google Scholar] [CrossRef]
- Leonard, B.W.; Mastroeni, D.; Grover, A.; Liu, Q.; Yang, K.; Gao, M.; Wu, J.; Pootrakul, D.; van den Berge, S.A.; Hol, E.M.; et al. Subventricular zone neural progenitors from rapid brain autopsies of elderly subjects with and without neurodegenerative disease. J. Comp. Neurol. 2009, 515, 269–294. [Google Scholar] [CrossRef] [PubMed]
- Graham, H.K. Cerebral palsy prevention and cure: Vision or mirage? A personal view. J. Paediatr. Child Health 2014, 50, 89–90. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Love, S.C.; Novak, I.; Kentish, M.; Desloovere, K.; Heinen, F.; Molenaers, G.; O’Flaherty, S.; Graham, H.K.; Cerebral Palsy, I. Botulinum toxin assessment, intervention and after-care for lower limb spasticity in children with cerebral palsy: International consensus statement. Eur. J. Neurol. 2010, 17 (Suppl. 2), 9–37. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, M.; Smeulders, M.J.; Kreulen, M.; Huijing, P.A.; Jaspers, R.T. Intramuscular connective tissue differences in spastic and control muscle: A mechanical and histological study. PLoS ONE 2014, 9, e101038. [Google Scholar] [CrossRef]
- Salari, M.; Sharma, S.; Jog, M.S. Botulinum Toxin Induced Atrophy: An Uncharted Territory. Toxins 2018, 10, 313. [Google Scholar] [CrossRef]
- Barber, L.; Hastings-Ison, T.; Baker, R.; Barrett, R.; Lichtwark, G. Medial gastrocnemius muscle volume and fascicle length in children aged 2 to 5 years with cerebral palsy. Dev. Med. Child Neurol. 2011, 53, 543–548. [Google Scholar] [CrossRef]
- Malaiya, R.; McNee, A.E.; Fry, N.R.; Eve, L.C.; Gough, M.; Shortland, A.P. The morphology of the medial gastrocnemius in typically developing children and children with spastic hemiplegic cerebral palsy. J. Electromyogr. Kinesiol. 2007, 17, 657–663. [Google Scholar] [CrossRef]
- Smith, L.R.; Lee, K.S.; Ward, S.R.; Chambers, H.G.; Lieber, R.L. Hamstring contractures in children with spastic cerebral palsy result from a stiffer extracellular matrix and increased in vivo sarcomere length. J. Physiol. 2011, 589, 2625–2639. [Google Scholar] [CrossRef]
- Booth, C.M.; Cortina-Borja, M.J.; Theologis, T.N. Collagen accumulation in muscles of children with cerebral palsy and correlation with severity of spasticity. Dev. Med. Child Neurol. 2001, 43, 314–320. [Google Scholar] [CrossRef]
- Stavsky, M.; Mor, O.; Mastrolia, S.A.; Greenbaum, S.; Than, N.G.; Erez, O. Cerebral Palsy-Trends in Epidemiology and Recent Development in Prenatal Mechanisms of Disease, Treatment, and Prevention. Front. Pediatr. 2017, 5, 21. [Google Scholar] [CrossRef] [PubMed]
- Montal, M. Botulinum neurotoxin: A marvel of protein design. Annu. Rev. Biochem. 2010, 79, 591–617. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, A.S.; Ertl-Wagner, B.; Britsch, S.; Schroder, J.M.; Nikolin, S.; Weis, J.; Muller-Felber, W.; Koerte, I.; Stehr, M.; Berweck, S.; et al. Muscle biopsy substantiates long-term MRI alterations one year after a single dose of botulinum toxin injected into the lateral gastrocnemius muscle of healthy volunteers. Mov. Disord. 2009, 24, 1494–1503. [Google Scholar] [CrossRef]
- Scaglione, F. Conversion Ratio between Botox(R), Dysport(R), and Xeomin(R) in Clinical Practice. Toxins 2016, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Pingel, J.; Nielsen, M.S.; Lauridsen, T.; Rix, K.; Bech, M.; Alkjaer, T.; Andersen, I.T.; Nielsen, J.B.; Feidenhansl, R. Injection of high dose botulinum-toxin A leads to impaired skeletal muscle function and damage of the fibrilar and non-fibrilar structures. Sci. Rep. 2017, 7, 14746. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, R.; Vaz, M.A.; Sawatsky, A.; Hart, D.A.; Herzog, W. A clinically relevant BTX-A injection protocol leads to persistent weakness, contractile material loss, and an altered mRNA expression phenotype in rabbit quadriceps muscles. J. Biomech. 2015, 48, 1700–1706. [Google Scholar] [CrossRef]
- Gough, M.; Fairhurst, C.; Shortland, A.P. Botulinum toxin and cerebral palsy: Time for reflection? Dev. Med. Child Neurol. 2005, 47, 709–712. [Google Scholar] [CrossRef]
- Multani, I.; Manji, J.; Hastings-Ison, T.; Khot, A.; Graham, K. Botulinum Toxin in the Management of Children with Cerebral Palsy. Pediatr. Drugs 2019, 21, 261–281. [Google Scholar] [CrossRef]
- Lippi, L.; de Sire, A.; Folli, A.; D’Abrosca, F.; Grana, E.; Baricich, A.; Carda, S.; Invernizzi, M. Multidimensional Effectiveness of Botulinum Toxin in Neuropathic Pain: A Systematic Review of Randomized Clinical Trials. Toxins 2022, 14, 308. [Google Scholar] [CrossRef]
- Nieuwenhuys, A.; Papageorgiou, E.; Pataky, T.; De Laet, T.; Molenaers, G.; Desloovere, K. Literature Review and Comparison of Two Statistical Methods to Evaluate the Effect of Botulinum Toxin Treatment on Gait in Children with Cerebral Palsy. PLoS ONE 2016, 11, e0152697. [Google Scholar] [CrossRef]
- Lee, J.; Kennedy, P.; Waugh, J.M. Experiences with CPP-Based Self Assembling Peptide Systems for Topical Delivery of Botulinum Toxin. Cell Penetrating Pept. Methods Protoc. 2015, 1324, 397–415. [Google Scholar] [CrossRef]
- Novak, I.; Graham, H.K. Botulinum toxin consensus statement—Foreword. Eur. J. Neurol. 2010, 17 (Suppl. 2), iii–iv. [Google Scholar] [CrossRef]
- Molenaers, G.; Desloovere, K.; Fabry, G.; De Cock, P. The effects of quantitative gait assessment and botulinum toxin a on musculoskeletal surgery in children with cerebral palsy. J. Bone Jt. Surg. 2006, 88, 161–170. [Google Scholar] [CrossRef]
- Peeters, N.; Papageorgiou, E.; Hanssen, B.; De Beukelaer, N.; Staut, L.; Degelaen, M.; Van den Broeck, C.; Calders, P.; Feys, H.; Van Campenhout, A.; et al. The Short-Term Impact of Botulinum Neurotoxin-A on Muscle Morphology and Gait in Children with Spastic Cerebral Palsy. Toxins 2022, 14, 676. [Google Scholar] [CrossRef] [PubMed]
- De Beukelaer, N.; Weide, G.; Huyghe, E.; Vandekerckhove, I.; Hanssen, B.; Peeters, N.; Uytterhoeven, J.; Deschrevel, J.; Maes, K.; Corvelyn, M.; et al. Reduced Cross-Sectional Muscle Growth Six Months after Botulinum Toxin Type-A Injection in Children with Spastic Cerebral Palsy. Toxins 2022, 14, 139. [Google Scholar] [CrossRef] [PubMed]
- Schless, S.H.; Cenni, F.; Bar-On, L.; Hanssen, B.; Kalkman, B.; O’Brien, T.; Aertbelien, E.; Van Campenhout, A.; Molenaers, G.; Desloovere, K. Medial gastrocnemius volume and echo-intensity after botulinum neurotoxin A interventions in children with spastic cerebral palsy. Dev. Med. Child Neurol. 2019, 61, 783–790. [Google Scholar] [CrossRef]
- Alexander, C.; Elliott, C.; Valentine, J.; Stannage, K.; Bear, N.; Donnelly, C.J.; Shipman, P.; Reid, S. Muscle volume alterations after first botulinum neurotoxin A treatment in children with cerebral palsy: A 6-month prospective cohort study. Dev. Med. Child Neurol. 2018, 60, 1165–1171. [Google Scholar] [CrossRef]
- Park, E.S.; Sim, E.; Rha, D.W.; Jung, S. Architectural changes of the gastrocnemius muscle after botulinum toxin type A injection in children with cerebral palsy. Yonsei Med. J. 2014, 55, 1406–1412. [Google Scholar] [CrossRef]
- Kaya Keles, C.S.; Ates, F. Botulinum Toxin Intervention in Cerebral Palsy-Induced Spasticity Management: Projected and Contradictory Effects on Skeletal Muscles. Toxins 2022, 14, 772. [Google Scholar] [CrossRef]
- Kim, H.; Kolaski, K. Is botulinum toxin type A more effective and safer than other treatments for the management of lower limb spasticity in children with cerebral palsy? A Cochrane Review summary with commentary. NeuroRehabilitation 2021, 49, 161–164. [Google Scholar] [CrossRef]
- Zanotti, S.; Kapetis, D.; Gibertini, S.; Salerno, F.; Ciusani, E.; Colombo, C.; Gronchi, A.; Morandi, L.; Mantegazza, R.; Molteni, F.; et al. Botulinum toxin type A affects the transcriptome of cell cultures derived from muscle biopsies of controls and spastic patients. Toxicol. Vitr. 2018, 50, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Lamotte, J.D.; Roqueviere, S.; Gautier, H.; Raban, E.; Boure, C.; Fonfria, E.; Krupp, J.; Nicoleau, C. hiPSC-Derived Neurons Provide a Robust and Physiologically Relevant In Vitro Platform to Test Botulinum Neurotoxins. Front. Pharmacol. 2020, 11, 617867. [Google Scholar] [CrossRef] [PubMed]
- Domenighetti, A.A.; Mathewson, M.A.; Pichika, R.; Sibley, L.A.; Zhao, L.; Chambers, H.G.; Lieber, R.L. Loss of myogenic potential and fusion capacity of muscle stem cells isolated from contractured muscle in children with cerebral palsy. Am. J. Physiol. Physiol. 2018, 315, C247–C257. [Google Scholar] [CrossRef]
- Corvelyn, M.; De Beukelaer, N.; Duelen, R.; Deschrevel, J.; Van Campenhout, A.; Prinsen, S.; Gayan-Ramirez, G.; Maes, K.; Weide, G.; Desloovere, K.; et al. Muscle Microbiopsy to Delineate Stem Cell Involvement in Young Patients: A Novel Approach for Children With Cerebral Palsy. Front. Physiol. 2020, 11, 945. [Google Scholar] [CrossRef]
- Van Campenhout, A.; Molenaers, G. Localization of the motor endplate zone in human skeletal muscles of the lower limb: Anatomical guidelines for injection with botulinum toxin. Dev. Med. Child Neurol. 2011, 53, 108–119. [Google Scholar] [CrossRef]
- Boulland, P.; Favier, J.C.; Villevieille, T.; Allanic, L.; Plancade, D.; Nadaud, J.; Ruttimann, M. Premixed 50% nitrous oxide and oxygen: Theoretical recalls and practical modalities. Ann. Fr. Anesth. Rèanim. 2005, 24, 1305–1312. [Google Scholar] [CrossRef]
- Hennequin, M.; Collado, V.; Faulks, D.; Koscielny, S.; Onody, P.; Nicolas, E. A clinical trial of efficacy and safety of inhalation sedation with a 50% nitrous oxide/oxygen premix (Kalinox) in general practice. Clin. Oral Investig. 2012, 16, 633–642. [Google Scholar] [CrossRef]
- Birgy, C.; Trimaille, A.; Messas, N.; Ristorto, J.; Kayali, A.; Marchandot, B.; Cardi, T.; Hess, S.; Kibler, M.; Jesel, L.; et al. Impact of Opioid Analgesia and Inhalation Sedation Kalinox on Pain and Radial Artery Spasm during Transradial Coronary Angiography. J. Clin. Med. 2020, 9, 2747. [Google Scholar] [CrossRef] [PubMed]
- Illa, I.; Leon-Monzon, M.; Dalakas, M.C. Regenerating and denervated human muscle fibers and satellite cells express neural cell adhesion molecule recognized by monoclonal antibodies to natural killer cells. Ann. Neurol. 1992, 31, 46–52. [Google Scholar] [CrossRef]
- Pisani, D.F.; Dechesne, C.A.; Sacconi, S.; Delplace, S.; Belmonte, N.; Cochet, O.; Clement, N.; Wdziekonski, B.; Villageois, A.P.; Butori, C.; et al. Isolation of a highly myogenic CD34-negative subset of human skeletal muscle cells free of adipogenic potential. Stem Cells 2010, 28, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Corvelyn, M.; Meirlevede, J.; Deschrevel, J.; Huyghe, E.; De Wachter, E.; Gayan-Ramirez, G.; Sampaolesi, M.; Van Campenhout, A.; Desloovere, K.; Costamagna, D. Ex vivo adult stem cell characterization from multiple muscles in ambulatory children with cerebral palsy during early development of contractures. Differentiation 2023, 133, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Duelen, R.; Costamagna, D.; Gilbert, G.; De Waele, L.; Goemans, N.; Desloovere, K.; Verfaillie, C.M.; Sipido, K.R.; Buyse, G.M.; Sampaolesi, M. Human iPSC model reveals a central role for NOX4 and oxidative stress in Duchenne cardiomyopathy. Stem Cell Rep. 2022, 17, 352–368. [Google Scholar] [CrossRef] [PubMed]
- Costamagna, D.; Casters, V.; Beltra, M.; Sampaolesi, M.; Van Campenhout, A.; Ortibus, E.; Desloovere, K.; Duelen, R. Autologous iPSC-Derived Human Neuromuscular Junction to Model the Pathophysiology of Hereditary Spastic Paraplegia. Cells 2022, 11, 3351. [Google Scholar] [CrossRef] [PubMed]
- Coll, M.; Perea, L.; Boon, R.; Leite, S.B.; Vallverdu, J.; Mannaerts, I.; Smout, A.; El Taghdouini, A.; Blaya, D.; Rodrigo-Torres, D.; et al. Generation of Hepatic Stellate Cells from Human Pluripotent Stem Cells Enables In Vitro Modeling of Liver Fibrosis. Cell Stem Cell 2018, 23, 101–113.e7. [Google Scholar] [CrossRef]
- Rauch, C.; Feifel, E.; Kern, G.; Murphy, C.; Meier, F.; Parson, W.; Beilmann, M.; Jennings, P.; Gstraunthaler, G.; Wilmes, A. Differentiation of human iPSCs into functional podocytes. PLoS ONE 2018, 13, e0203869. [Google Scholar] [CrossRef] [PubMed]
- Paranjape, S.R.; Nagendran, T.; Poole, V.; Harris, J.; Taylor, A.M. Compartmentalization of Human Stem Cell-Derived Neurons within Pre-Assembled Plastic Microfluidic Chips. J. Vis. Exp. 2019, 147, e59250. [Google Scholar] [CrossRef]
- de Lamotte, J.D.; Polentes, J.; Roussange, F.; Lesueur, L.; Feurgard, P.; Perrier, A.; Nicoleau, C.; Martinat, C. Optogenetically controlled human functional motor endplate for testing botulinum neurotoxins. Stem Cell Res. Ther. 2021, 12, 599. [Google Scholar] [CrossRef]
- Schenke, M.; Schjeide, B.M.; Puschel, G.P.; Seeger, B. Analysis of Motor Neurons Differentiated from Human Induced Pluripotent Stem Cells for the Use in Cell-Based Botulinum Neurotoxin Activity Assays. Toxins 2020, 12, 276. [Google Scholar] [CrossRef]
- Houston, F.E.; Hain, B.A.; Dodd, S.L. Inhibition of the proteasome partially attenuates atrophy in botulinum neurotoxin treated skeletal muscle. Toxicon 2018, 144, 48–54. [Google Scholar] [CrossRef]
- Shi, Z.; Lv, J.; Xiaoyu, L.; Zheng, L.W.; Yang, X.W. Condylar Degradation from Decreased Occlusal Loading following Masticatory Muscle Atrophy. BioMed Res. Int. 2018, 2018, 6947612. [Google Scholar] [CrossRef]
- Balanta-Melo, J.; Toro-Ibacache, V.; Torres-Quintana, M.A.; Kupczik, K.; Vega, C.; Morales, C.; Hernandez-Moya, N.; Arias-Calderon, M.; Beato, C.; Buvinic, S. Early molecular response and microanatomical changes in the masseter muscle and mandibular head after botulinum toxin intervention in adult mice. Ann. Anat. 2018, 216, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Stoklund Dittlau, K.; Krasnow, E.N.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Kerstens, A.; Giacomazzi, G.; Pavie, B.; Rossaert, E.; Beckers, J.; et al. Human motor units in microfluidic devices are impaired by FUS mutations and improved by HDAC6 inhibition. Stem Cell Rep. 2021, 16, 2213–2227. [Google Scholar] [CrossRef] [PubMed]
- Havlicek, S.; Kohl, Z.; Mishra, H.K.; Prots, I.; Eberhardt, E.; Denguir, N.; Wend, H.; Plotz, S.; Boyer, L.; Marchetto, M.C.; et al. Gene dosage-dependent rescue of HSP neurite defects in SPG4 patients’ neurons. Hum. Mol. Genet. 2014, 23, 2527–2541. [Google Scholar] [CrossRef] [PubMed]
- Denton, K.R.; Lei, L.; Grenier, J.; Rodionov, V.; Blackstone, C.; Li, X.J. Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells 2014, 32, 414–423. [Google Scholar] [CrossRef] [PubMed]
- De Pace, R.; Skirzewski, M.; Damme, M.; Mattera, R.; Mercurio, J.; Foster, A.M.; Cuitino, L.; Jarnik, M.; Hoffmann, V.; Morris, H.D.; et al. Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLoS Genet. 2018, 14, e1007363. [Google Scholar] [CrossRef]
- Becker, K.A.; Uerschels, A.K.; Goins, L.; Doolen, S.; McQuerry, K.J.; Bielawski, J.; Sure, U.; Bieberich, E.; Taylor, B.K.; Gulbins, E.; et al. Role of 1-Deoxysphingolipids in docetaxel neurotoxicity. J. Neurochem. 2020, 154, 662–672. [Google Scholar] [CrossRef]
- Luningschror, P.; Werner, G.; Stroobants, S.; Kakuta, S.; Dombert, B.; Sinske, D.; Wanner, R.; Lullmann-Rauch, R.; Wefers, B.; Wurst, W.; et al. The FTLD Risk Factor TMEM106B Regulates the Transport of Lysosomes at the Axon Initial Segment of Motoneurons. Cell Rep. 2020, 30, 3506–3519.e6. [Google Scholar] [CrossRef]
- Marchi, P.M.; Marrone, L.; Brasseur, L.; Coens, A.; Webster, C.P.; Bousset, L.; Destro, M.; Smith, E.F.; Walther, C.G.; Alfred, V.; et al. C9ORF72-derived poly-GA DPRs undergo endocytic uptake in iAstrocytes and spread to motor neurons. Life Sci. Alliance 2022, 5, e202101276. [Google Scholar] [CrossRef]
- Suzuki, N.; Akiyama, T.; Warita, H.; Aoki, M. Omics Approach to Axonal Dysfunction of Motor Neurons in Amyotrophic Lateral Sclerosis (ALS). Front. Neurosci. 2020, 14, 194. [Google Scholar] [CrossRef]
- Wali, G.; Liyanage, E.; Blair, N.F.; Sutharsan, R.; Park, J.S.; Mackay-Sim, A.; Sue, C.M. Oxidative Stress-Induced Axon Fragmentation Is a Consequence of Reduced Axonal Transport in Hereditary Spastic Paraplegia SPAST Patient Neurons. Front. Neurosci. 2020, 14, 401. [Google Scholar] [CrossRef]
- Sasaki, Y.; Vohra, B.P.; Baloh, R.H.; Milbrandt, J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J. Neurosci. 2009, 29, 6526–6534. [Google Scholar] [CrossRef]
- Beirowski, B.; Morreale, G.; Conforti, L.; Mazzola, F.; Di Stefano, M.; Wilbrey, A.; Babetto, E.; Janeckova, L.; Magni, G.; Coleman, M.P. WldS can delay Wallerian degeneration in mice when interaction with valosin-containing protein is weakened. Neuroscience 2010, 166, 201–211. [Google Scholar] [CrossRef]
- Liu, X.; Wu, H.; Byrne, M.; Krane, S.; Jaenisch, R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 1852–1856. [Google Scholar] [CrossRef]
- Tracy, L.E.; Minasian, R.A.; Caterson, E.J. Extracellular Matrix and Dermal Fibroblast Function in the Healing Wound. Adv. Wound Care 2016, 5, 119–136. [Google Scholar] [CrossRef] [PubMed]
- Chapman, M.A.; Meza, R.; Lieber, R.L. Skeletal muscle fibroblasts in health and disease. Differentiation 2016, 92, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Csapo, R.; Gumpenberger, M.; Wessner, B. Skeletal Muscle Extracellular Matrix—What Do We Know About Its Composition, Regulation, and Physiological Roles? A Narrative Review. Front. Physiol. 2020, 11, 253. [Google Scholar] [CrossRef]
- Kovanen, V. Intramuscular extracellular matrix: Complex environment of muscle cells. Exerc. Sport Sci. Rev. 2002, 30, 20–25. [Google Scholar] [CrossRef] [PubMed]
- McKee, T.J.; Perlman, G.; Morris, M.; Komarova, S.V. Extracellular matrix composition of connective tissues: A systematic review and meta-analysis. Sci. Rep. 2019, 9, 10542. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; von Maltzahn, J.; Soleimani, V.D.; Yin, H.; Rudnicki, M.A. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell 2013, 12, 75–87. [Google Scholar] [CrossRef]
- Lieber, R.L.; Domenighetti, A.A. Commentary: Muscle Microbiopsy to Delineate Stem Cell Involvement in Young Patients: A Novel Approach for Children with Cerebral Palsy. Front. Physiol. 2021, 12, 642366. [Google Scholar] [CrossRef]
- Franceschini, M.; Iocco, M.; Molteni, F.; Santamato, A.; Smania, N.; Italian Spasticity Study, G. Management of stroke patients submitted to botulinum toxin type A therapy: A Delphi survey of an Italian expert panel of specialist injectors. Eur. J. Phys. Rehabil. Med. 2014, 50, 525–533. [Google Scholar] [PubMed]
- Baricich, A.; Carda, S.; Bertoni, M.; Maderna, L.; Cisari, C. A single-blinded, randomized pilot study of botulinum toxin type A combined with non-pharmacological treatment for spastic foot. J. Rehabil. Med. 2008, 40, 870–872. [Google Scholar] [CrossRef] [PubMed]
- Baghdadi, M.B.; Firmino, J.; Soni, K.; Evano, B.; Di Girolamo, D.; Mourikis, P.; Castel, D.; Tajbakhsh, S. Notch-Induced miR-708 Antagonizes Satellite Cell Migration and Maintains Quiescence. Cell Stem Cell 2018, 23, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Lieber, R.L.; Friden, J. Muscle contracture and passive mechanics in cerebral palsy. J. Appl. Physiol. 2019, 126, 1492–1501. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.R.; Chambers, H.G.; Subramaniam, S.; Lieber, R.L. Transcriptional abnormalities of hamstring muscle contractures in children with cerebral palsy. PLoS ONE 2012, 7, e40686. [Google Scholar] [CrossRef]
- Smith, L.R.; Pichika, R.; Meza, R.C.; Gillies, A.R.; Baliki, M.N.; Chambers, H.G.; Lieber, R.L. Contribution of extracellular matrix components to the stiffness of skeletal muscle contractures in patients with cerebral palsy. Connect. Tissue Res. 2021, 62, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular pathways of motor neuron injury in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Soares, P.; Silva, C.; Chavarria, D.; Silva, F.S.G.; Oliveira, P.J.; Borges, F. Drug discovery and amyotrophic lateral sclerosis: Emerging challenges and therapeutic opportunities. Ageing Res. Rev. 2023, 83, 101790. [Google Scholar] [CrossRef]
- Haberecht-Muller, S.; Kruger, E.; Fielitz, J. Out of Control: The Role of the Ubiquitin Proteasome System in Skeletal Muscle during Inflammation. Biomolecules 2021, 11, 1327. [Google Scholar] [CrossRef]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef]
- Multani, I.; Manji, J.; Tang, M.J.; Herzog, W.; Howard, J.J.; Graham, H.K. Sarcopenia, Cerebral Palsy, and Botulinum Toxin Type A. JBJS Rev. 2019, 7, e4. [Google Scholar] [CrossRef] [PubMed]
- Tintignac, L.A.; Lagirand, J.; Batonnet, S.; Sirri, V.; Leibovitch, M.P.; Leibovitch, S.A. Degradation of MyoD mediated by the SCF (MAFbx) ubiquitin ligase. J. Biol. Chem. 2005, 280, 2847–2856. [Google Scholar] [CrossRef] [PubMed]
- Lagirand-Cantaloube, J.; Offner, N.; Csibi, A.; Leibovitch, M.P.; Batonnet-Pichon, S.; Tintignac, L.A.; Segura, C.T.; Leibovitch, S.A. The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J. 2008, 27, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Lokireddy, S.; Wijesoma, I.W.; Teng, S.; Bonala, S.; Gluckman, P.D.; McFarlane, C.; Sharma, M.; Kambadur, R. The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metab. 2012, 16, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Toldi, J.; Escobar, J.; Brown, A. Cerebral Palsy: Sport and Exercise Considerations. Curr. Sports Med. Rep. 2021, 20, 19–25. [Google Scholar] [CrossRef]
- Verschuren, O.; Hulst, R.Y.; Voorman, J.; Pillen, S.; Luitwieler, N.; Dudink, J.; Gorter, J.W. 24-hour activity for children with cerebral palsy: A clinical practice guide. Dev. Med. Child Neurol. 2021, 63, 54–59. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Antibody Name (#Catalog Number) | Provider | FACS | IF | WB |
|---|---|---|---|---|---|
| ACTN2 | Anti-SARCOMERIC ALPHA ACTININ (EA-53) (Mouse monoclonal) (#ab9465) | Abcam, Cambridge, UK | 1:200 | 1:500 | |
| ALKALINE PHOSPATASE-PE | Human/Mouse/Rat Alkaline Phosphatase PE MAb (Cl B4-78), Mouse IgG1 (FAB1448P-100) | R&D systems, Minneapolis, MN, USA | 1.2 µL/106 cells | ||
| ATROGIN1 | Atrogin-1 (Rabbit polyclonal) (AP 2041) | ECM Bioscences, Aurora, CO, USA | 1:200 | ||
| BECLIN-1 | Anti-Beclin1-BH3 Domain (Rabbit polyclonal) (SAB1306537) | Merck, Darmstadt, Germany | 1:1000 | ||
| CD56-APC | APC anti-human CD56 (NCAM) Mouse IgG1 (clone MEM-188) (#304610) | BioLegend, San Diego, CA, USA | 0.1 µL/106 cells | ||
| COL1A1 | COL1A1 (E8F4L) XP (Rabbit monoclonal Ab) (#72026) | Cell Signaling, Danvers, MA, USA | 1:1000 | ||
| COL3A1 | COL3A1 (E8D7R) XP (Rabbit monoclonal Ab) (#63034) | Cell Signaling, Danvers, MA, USA | 1:500 | ||
| COL5A1 | COL5A1 (E6U9W) (Rabbit monoclonal Ab) (#86903) | Cell Signaling, Danvers, MA, USA | 1:500 | ||
| FN1 | Fibronectin1 (E5H6X) (Rabbit monoclonal Ab) (#26836) | Cell Signaling, Danvers, MA, USA | 1:500 | ||
| GAPDH | Anti-GAPDH (Rabbit polyclonal) (G9545) | Merck, Darmstadt, Germany | 1:1000 | ||
| ISL1 | Recombinant Anti-ISLET 1 (EP4182)—Neural Stem Cell Marker (Rabbit monoclonal) (#ab109517) | Abcam, Cambridge, UK | 1:500 | 1:60 | |
| LC3B | Anti-LC3B (Rabbit polyclonal) (L7543) | Merck, Darmstadt, Germany | 1:1000 | ||
| MF20 | MYHC1 (MF20) (Mouse monoclonal) | Develop Studies Hybridoma Bank (DSHB), IOWA, USA | 1:300 | 1:3 | |
| MYOD | MYOD1 (D8G3) XP Rabbit mAb (Rabbit monoclonal) (#13812) | Cell Signaling, Danvers, MA, USA | 1:300 | 1:1000 | |
| NANOG | NANOG (Rabbit polyclonal) (#PA1-097) | Thermo Fisher Scientific, Waltham, MA, USA | 1:200 | ||
| NEFH | Anti-NEUROFILAMENT HEAVY POLYPEPTIDE antibody (Rabbit polyclonal) (#ab8135) | Abcam, Cambridge, UK | 1:1000 | 1:100 | |
| NEST | Purified anti-NESTIN (10C2) (Mouse monoclonal) (#656801) | BioLegend, San Diego, CA, USA | 1:250 | ||
| PAX6 | PAX6 (D3A9V) XP Rabbit mAb (Rabbit monoclonal) (#60433) | Cell Signaling, Danvers, MA, USA | 1:150 | ||
| PDGFRa-APC | APC anti-human CD140a (PDGFRα) Mouse IgG1 (clone 16A1) (#323512) | BioLegend, San Diego, CA, USA | 0.1 µL/106 cells | ||
| p62 | Anti-p62 (Mouse monoclonal) | BD Bioscences, NJ, USA | 1:9000 | ||
| SNAP25 | Anti-SNAP-25 (Rabbit polyclonal) (S9684) | Merck, Darmstadt, Germany | 1:200 | ||
| TUBA4A | Anti-a-TUBULIN (B-5-1-2) (Mouse monoclonal) (#T6074) | Merck, Darmstadt, Germany | 1:1000 | ||
| TUBB3 | Anti-beta III TUBULIN (2G10)—Neuronal Marker (Mouse monoclonal) (#ab78078) | Abcam, Cambridge, UK | 1:500 | 1:300 |
| Gene | Primer Direction | Primer Sequence (5′ > 3′) |
|---|---|---|
| COL1A1 | Forward | CCTGGATGCCATCAAAGTCT |
| Reverse | TCTTGTCCTTGGGGTTCTTG | |
| COL3A1 | Forward | AAGAATTTGGTGTGGACGTTG |
| Reverse | TTTTGTCGGTCACTTGCACT | |
| COL4A1 | Forward | CCAGGATTTCAAGGTCCAAA |
| Reverse | CTC CCCTTTGATGATGTCGT | |
| COL5A1 | Forward | CCTGACCCTGGACAGTGAAG |
| Reverse | GGCTCCTTCCCTCTGTTCTC | |
| COL5A2 | Forward | TCAAAAGAAGCCTCCCAGAA |
| Reverse | TCTAAGTCATTTGCCCCTTTG | |
| COL8A1 | Forward | ACCACCCCAGGGAGAGTATC |
| Reverse | AATGCAGGCATCTCATAGGC | |
| COL11A1 | Forward | GCATTTTGATGCTTTATTCAAGG |
| Reverse | CACACATTTCCCTGTCCAAA | |
| ACTB | Forward | GGACCTGACTGACTACCTCAT |
| Reverse | CGTAGCACAGCTTCTCCTTAAT |
| TD (n = 7) | CP t0 (n = 10) GMFCS I n = 4 GMFCS II n = 4 GMFCS III n = 2 | CP t1 (n = 6) GMFCS I n = 3 GMFCS II n = 2 GMFCS III n = 1 | CP t2 (n = 9) GMFCS I n = 3 GMFCS II n = 4 GMFCS III n = 2 | ||
|---|---|---|---|---|---|
| Age of first biopsy (year) | Mean (SD) | 6.19 (1.92) | 4.11 (1.66) | 5.2 (1.7) | 6.1 (2.0) |
| Range | 4–7 | 2–7 | 3.1–7.5 | 3.7–8.9 | |
| Gender (m-f) | n | 5–2 | 5–5 | ||
| Body mass (kg) | Mean (SD) | 22.18 (7.06) | 15.60 (6.52) | 17.4 (2.8) | 19.5 (4.6) |
| Height (cm) | Mean (SD) | 118.36 (14.70) | 100.15 (14.62) | 104.5 (10.4) | 113.6 (15.5) |
| Involvement (unilat-bilat.) | n | - | 5–5 | ||
| Physiotherapy (min/week) | Mean (SD) | - | 139.7 (60.1) | 123 (60.4) | 158.6 (56.7) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costamagna, D.; Bastianini, V.; Corvelyn, M.; Duelen, R.; Deschrevel, J.; De Beukelaer, N.; De Houwer, H.; Sampaolesi, M.; Gayan-Ramirez, G.; Campenhout, A.V.; et al. Botulinum Toxin Treatment of Adult Muscle Stem Cells from Children with Cerebral Palsy and hiPSC-Derived Neuromuscular Junctions. Cells 2023, 12, 2072. https://doi.org/10.3390/cells12162072
Costamagna D, Bastianini V, Corvelyn M, Duelen R, Deschrevel J, De Beukelaer N, De Houwer H, Sampaolesi M, Gayan-Ramirez G, Campenhout AV, et al. Botulinum Toxin Treatment of Adult Muscle Stem Cells from Children with Cerebral Palsy and hiPSC-Derived Neuromuscular Junctions. Cells. 2023; 12(16):2072. https://doi.org/10.3390/cells12162072
Chicago/Turabian StyleCostamagna, Domiziana, Valeria Bastianini, Marlies Corvelyn, Robin Duelen, Jorieke Deschrevel, Nathalie De Beukelaer, Hannah De Houwer, Maurilio Sampaolesi, Ghislaine Gayan-Ramirez, Anja Van Campenhout, and et al. 2023. "Botulinum Toxin Treatment of Adult Muscle Stem Cells from Children with Cerebral Palsy and hiPSC-Derived Neuromuscular Junctions" Cells 12, no. 16: 2072. https://doi.org/10.3390/cells12162072
APA StyleCostamagna, D., Bastianini, V., Corvelyn, M., Duelen, R., Deschrevel, J., De Beukelaer, N., De Houwer, H., Sampaolesi, M., Gayan-Ramirez, G., Campenhout, A. V., & Desloovere, K. (2023). Botulinum Toxin Treatment of Adult Muscle Stem Cells from Children with Cerebral Palsy and hiPSC-Derived Neuromuscular Junctions. Cells, 12(16), 2072. https://doi.org/10.3390/cells12162072