
Gene Regulatory Networks and Signaling Pathways in Palatogenesis and Cleft Palate: A Comprehensive Review



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Morphologenetic and Molecular Mechanisms in Mammalian Palatogenesis
2.1. Anatomical Overview of Palatogenesis
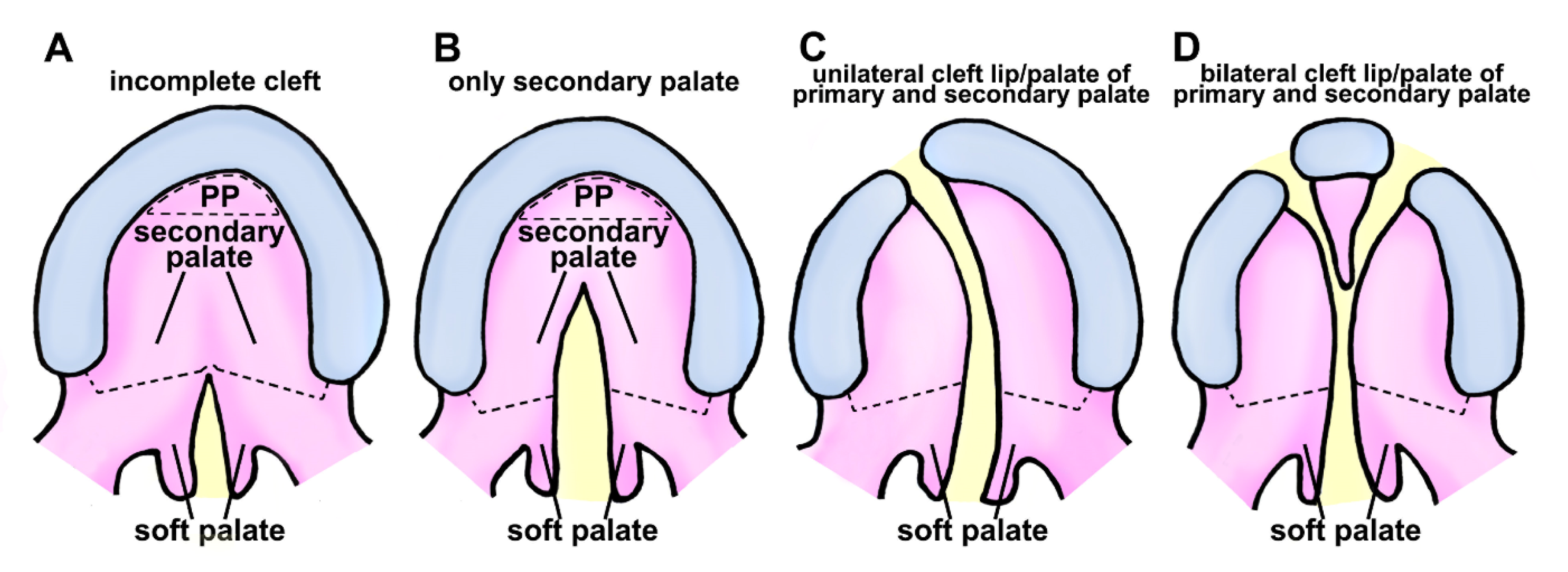
2.2. Classification of Cleft Lip and Palate in Human
2.3. Morphological and Molecular Control of Palatal Shelf Growth and Patterning
2.4. Regionalization of Anterior and Posterior Palatal Outgrowth
2.5. Patterning along the Mediolateral Axis
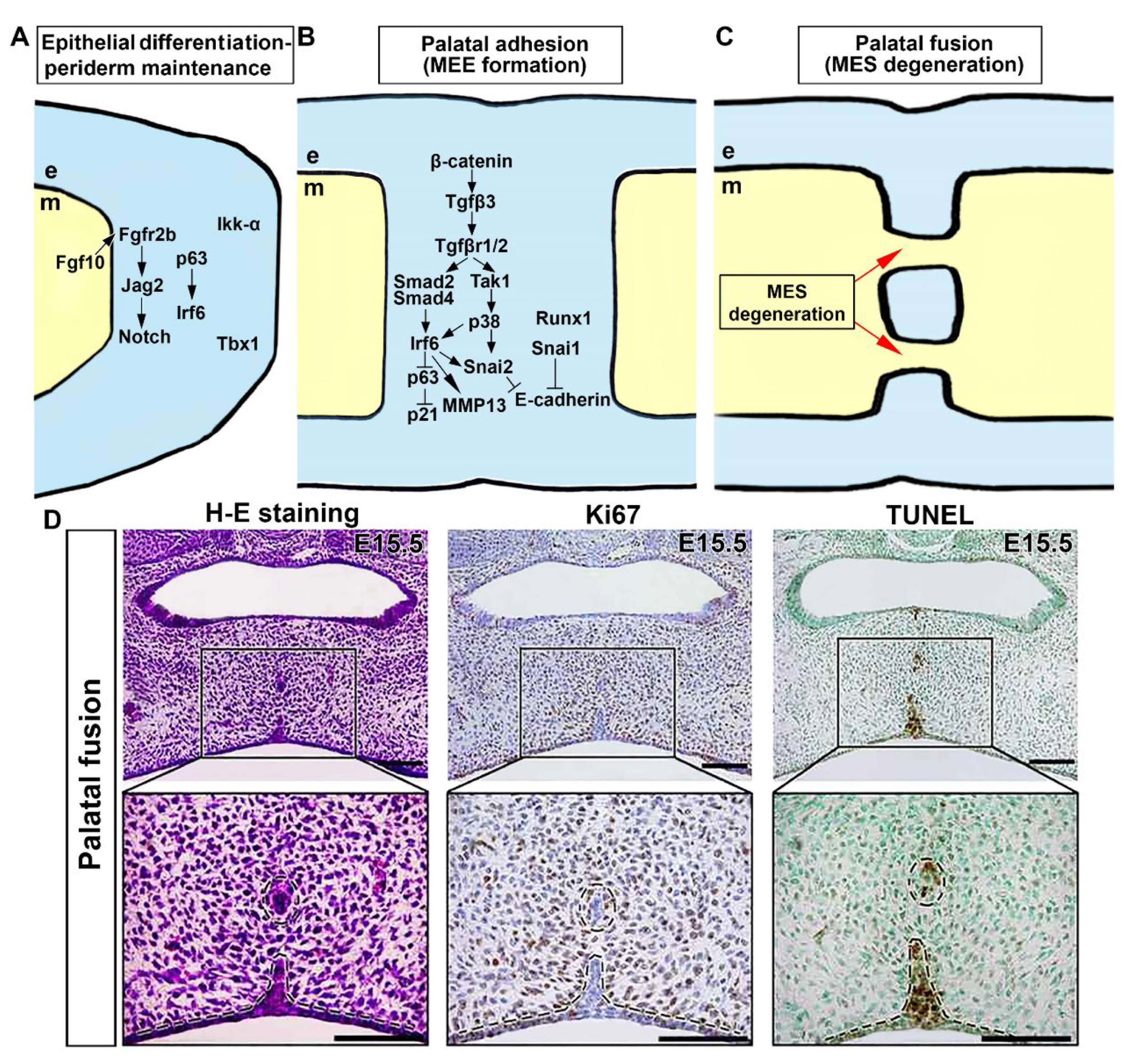
2.6. Genetic Network Controlling Palatal Shelf Adhesion and Fusion

3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parker, S.E.; Mai, C.T.; Canfield, M.A.; Rickard, R.; Wang, Y.; Meyer, R.E.; Anderson, P.; Mason, C.A.; Collins, J.S.; Kirby, R.S.; et al. Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004–2006. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Leslie, E.J.; Marazita, M.L. Genetics of Cleft Lip and Cleft Palate. Am. J. Med. Genet. C Semin. Med. Genet. 2013, 163, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Wehby, G.L.; Cassell, C.H. The impact of orofacial clefts on quality of life and healthcare use and costs. Oral Dis. 2010, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Carinci, F.; Pezzetti, F.; Scapoli, L.; Martinelli, M.; Avantaggiato, A.; Carinci, P.; Padula, E.; Baciliero, U.; Gombos, F.; Laino, G.; et al. Recent developments in orofacial cleft genetics. J. Craniofac. Surg. 2003, 14, 130–143. [Google Scholar] [CrossRef]
- Merritt, L. Part 1. Understanding the embryology and genetics of cleft lip and palate. Adv. Neonatal Care 2005, 5, 64–71. [Google Scholar] [CrossRef]
- Mossey, P.A.; Little, J.; Munger, R.G.; Dixon, M.J.; Shaw, W.C. Cleft lip and palate. Lancet 2009, 374, 1773–1785. [Google Scholar] [CrossRef]
- Lan, Y.; Jiang, R. Mouse models in palate development and orofacial cleft research: Understanding the crucial role and regulation of epithelial integrity in facial and palate morphogenesis. Curr. Top. Dev. Biol. 2022, 148, 13–50. [Google Scholar] [CrossRef]
- Hammond, N.L.; Dixon, M.J. Revisiting the embryogenesis of lip and palate development. Oral Dis. 2022, 28, 1306–1326. [Google Scholar] [CrossRef]
- Li, C.; Lan, Y.; Jiang, R. Molecular and Cellular Mechanisms of Palate Development. J. Dent. Res. 2017, 96, 1184–1191. [Google Scholar] [CrossRef]
- Patten, B.M. Patten’s Foundation of Embryology; McGraw-Hill Book Company: New York, NY, USA, 1964; pp. 423–429. [Google Scholar]
- Bush, J.O.; Jiang, R. Palatogenesis: Morphogenetic and molecular mechanisms of secondary palate development. Development 2012, 139, 231–243. [Google Scholar] [CrossRef]
- Jiang, R.; Bush, J.O.; Lidral, A.C. Development of the upper lip: Morphogenetic and molecular mechanisms. Dev. Dyn. 2006, 235, 1152–1166. [Google Scholar] [CrossRef]
- Gritli-Linde, A. Molecular control of secondary palate development. Dev. Biol. 2007, 301, 309–326. [Google Scholar] [CrossRef]
- Milling, M.A.; van Straelen, P. Asymmetrical cleft palate. Br. J. Plast. Surg. 1996, 49, 20–23. [Google Scholar] [CrossRef]
- Yuzuriha, S.; Oh, A.K.; Mulliken, J.B. Asymmetrical bilateral cleft lip: Complete or incomplete and contralateral lesser defect (minor-form, microform, or mini-microform). Plast. Reconstr. Surg. 2008, 122, 1494–1504. [Google Scholar] [CrossRef]
- Nasreddine, G.; El Hajj, J.; Ghassibe-Sabbagh, M. Orofacial clefts embryology, classification, epidemiology, and genetics. Mutat. Res. Rev. Mutat. Res. 2021, 787, 108373. [Google Scholar] [CrossRef]
- Dixon, M.J.; Marazita, M.L.; Beaty, T.H.; Murray, J.C. Cleft lip and palate: Understanding genetic and environmental influences. Nat. Rev. Genet. 2011, 12, 167–178. [Google Scholar] [CrossRef]
- Chai, Y.; Maxson, R.E., Jr. Recent advances in craniofacial morphogenesis. Dev. Dyn. 2006, 235, 2353–2375. [Google Scholar] [CrossRef]
- Gritli-Linde, A. The etiopathogenesis of cleft lip and cleft palate: Usefulness and caveats of mouse models. Curr. Top. Dev. Biol. 2008, 84, 37–138. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.A. Mouse resources for craniofacial research. Genesis 2011, 49, 190–199. [Google Scholar] [CrossRef]
- Ito, Y.; Yeo, J.Y.; Chytil, A.; Han, J.; Bringas, P., Jr.; Nakajima, A.; Shuler, C.F.; Moses, H.L.; Chai, Y. Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development 2003, 130, 5269–5280. [Google Scholar] [CrossRef]
- Meng, L.; Bian, Z.; Torensma, R.; Von den Hoff, J.W. Biological mechanisms in palatogenesis and cleft palate. J. Dent. Res. 2009, 88, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Zhou, J.; D’Souza, R.N. Pax9’s dual roles in modulating Wnt signaling during murine palatogenesis. Dev. Dyn. 2020, 249, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, H.; Lan, Y.; Aronow, B.J.; Kalinichenko, V.V.; Jiang, R. A Shh-Foxf-Fgf18-Shh Molecular Circuit Regulating Palate Development. PLoS Genet. 2016, 12, e1005769. [Google Scholar] [CrossRef]
- Charoenchaikorn, K.; Yokomizo, T.; Rice, D.P.; Honjo, T.; Matsuzaki, K.; Shintaku, Y.; Imai, Y.; Wakamatsu, A.; Takahashi, S.; Ito, Y.; et al. Runx1 is involved in the fusion of the primary and the secondary palatal shelves. Dev. Biol. 2009, 326, 392–402. [Google Scholar] [CrossRef]
- Kurosaka, H.; Iulianella, A.; Williams, T.; Trainor, P.A. Disrupting hedgehog and WNT signaling interactions promotes cleft lip pathogenesis. J. Clin. Investig. 2014, 124, 1660–1671. [Google Scholar] [CrossRef]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef]
- Everson, J.L.; Fink, D.M.; Yoon, J.W.; Leslie, E.J.; Kietzman, H.W.; Ansen-Wilson, L.J.; Chung, H.M.; Walterhouse, D.O.; Marazita, M.L.; Lipinski, R.J. Sonic hedgehog regulation of Foxf2 promotes cranial neural crest mesenchyme proliferation and is disrupted in cleft lip morphogenesis. Development 2017, 144, 2082–2091. [Google Scholar] [CrossRef]
- Huangfu, D.; Liu, A.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef]
- Shin, J.O.; Song, J.; Choi, H.S.; Lee, J.; Lee, K.; Ko, H.W.; Bok, J. Activation of sonic hedgehog signaling by a Smoothened agonist restores congenital defects in mouse models of endocrine-cerebro-osteodysplasia syndrome. eBioMedicine 2019, 49, 305–317. [Google Scholar] [CrossRef]
- Rice, R.; Spencer-Dene, B.; Connor, E.C.; Gritli-Linde, A.; McMahon, A.P.; Dickson, C.; Thesleff, I.; Rice, D.P. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J. Clin. Investig. 2004, 113, 1692–1700. [Google Scholar] [CrossRef]
- Hosokawa, R.; Deng, X.; Takamori, K.; Xu, X.; Urata, M.; Bringas, P., Jr.; Chai, Y. Epithelial-specific requirement of FGFR2 signaling during tooth and palate development. J. Exp. Zool. B Mol. Dev. Evol. 2009, 312, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Jiang, R. Sonic hedgehog signaling regulates reciprocal epithelial-mesenchymal interactions controlling palatal outgrowth. Development 2009, 136, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Ovitt, C.E.; Cho, E.S.; Maltby, K.M.; Wang, Q.; Jiang, R. Odd-skipped related 2 (Osr2) encodes a key intrinsic regulator of secondary palate growth and morphogenesis. Development 2004, 131, 3207–3216. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Gao, Y.; Lan, Y.; Jia, S.; Jiang, R. Pax9 regulates a molecular network involving Bmp4, Fgf10, Shh signaling and the Osr2 transcription factor to control palate morphogenesis. Development 2013, 140, 4709–4718. [Google Scholar] [CrossRef]
- Han, J.; Mayo, J.; Xu, X.; Li, J.; Bringas, P., Jr.; Maas, R.L.; Rubenstein, J.L.; Chai, Y. Indirect modulation of Shh signaling by Dlx5 affects the oral-nasal patterning of palate and rescues cleft palate in Msx1-null mice. Development 2009, 136, 4225–4233. [Google Scholar] [CrossRef]
- Cesario, J.M.; Landin Malt, A.; Deacon, L.J.; Sandberg, M.; Vogt, D.; Tang, Z.; Zhao, Y.; Brown, S.; Rubenstein, J.L.; Jeong, J. Lhx6 and Lhx8 promote palate development through negative regulation of a cell cycle inhibitor gene, p57Kip2. Hum. Mol. Genet. 2015, 24, 5024–5039. [Google Scholar] [CrossRef]
- Iwata, J.; Suzuki, A.; Yokota, T.; Ho, T.V.; Pelikan, R.; Urata, M.; Sanchez-Lara, P.A.; Chai, Y. TGFbeta regulates epithelial-mesenchymal interactions through WNT signaling activity to control muscle development in the soft palate. Development 2014, 141, 909–917. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, Y.; Zhao, X.; Zhang, X.; Fermin, C.; Chen, Y. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development 2002, 129, 4135–4146. [Google Scholar] [CrossRef]
- Liu, W.; Sun, X.; Braut, A.; Mishina, Y.; Behringer, R.R.; Mina, M.; Martin, J.F. Distinct functions for Bmp signaling in lip and palate fusion in mice. Development 2005, 132, 1453–1461. [Google Scholar] [CrossRef]
- Xiong, W.; He, F.; Morikawa, Y.; Yu, X.; Zhang, Z.; Lan, Y.; Jiang, R.; Cserjesi, P.; Chen, Y. Hand2 is required in the epithelium for palatogenesis in mice. Dev. Biol. 2009, 330, 131–141. [Google Scholar] [CrossRef]
- Andl, T.; Ahn, K.; Kairo, A.; Chu, E.Y.; Wine-Lee, L.; Reddy, S.T.; Croft, N.J.; Cebra-Thomas, J.A.; Metzger, D.; Chambon, P.; et al. Epithelial Bmpr1a regulates differentiation and proliferation in postnatal hair follicles and is essential for tooth development. Development 2004, 131, 2257–2268. [Google Scholar] [CrossRef]
- Li, L.; Lin, M.; Wang, Y.; Cserjesi, P.; Chen, Z.; Chen, Y. BmprIa is required in mesenchymal tissue and has limited redundant function with BmprIb in tooth and palate development. Dev. Biol. 2011, 349, 451–461. [Google Scholar] [CrossRef]
- Baek, J.A.; Lan, Y.; Liu, H.; Maltby, K.M.; Mishina, Y.; Jiang, R. Bmpr1a signaling plays critical roles in palatal shelf growth and palatal bone formation. Dev. Biol. 2011, 350, 520–531. [Google Scholar] [CrossRef]
- He, F.; Xiong, W.; Wang, Y.; Matsui, M.; Yu, X.; Chai, Y.; Klingensmith, J.; Chen, Y. Modulation of BMP signaling by Noggin is required for the maintenance of palatal epithelial integrity during palatogenesis. Dev. Biol. 2010, 347, 109–121. [Google Scholar] [CrossRef][Green Version]
- Li, C.; Lan, Y.; Krumlauf, R.; Jiang, R. Modulating Wnt Signaling Rescues Palate Morphogenesis in Pax9 Mutant Mice. J. Dent. Res. 2017, 96, 1273–1281. [Google Scholar] [CrossRef]
- Jia, S.; Zhou, J.; Wee, Y.; Mikkola, M.L.; Schneider, P.; D’Souza, R.N. Anti-EDAR Agonist Antibody Therapy Resolves Palate Defects in Pax9−/− Mice. J. Dent. Res. 2017, 96, 1282–1289. [Google Scholar] [CrossRef]
- Jia, S.; Zhou, J.; Fanelli, C.; Wee, Y.; Bonds, J.; Schneider, P.; Mues, G.; D’Souza, R.N. Small-molecule Wnt agonists correct cleft palates in Pax9 mutant mice in utero. Development 2017, 144, 3819–3828. [Google Scholar] [CrossRef]
- Headon, D.J.; Overbeek, P.A. Involvement of a novel Tnf receptor homologue in hair follicle induction. Nat. Genet. 1999, 22, 370–374. [Google Scholar] [CrossRef]
- Hilliard, S.A.; Yu, L.; Gu, S.; Zhang, Z.; Chen, Y.P. Regional regulation of palatal growth and patterning along the anterior-posterior axis in mice. J. Anat. 2005, 207, 655–667. [Google Scholar] [CrossRef]
- Li, Q.; Ding, J. Gene expression analysis reveals that formation of the mouse anterior secondary palate involves recruitment of cells from the posterior side. Int. J. Dev. Biol. 2007, 51, 167–172. [Google Scholar] [CrossRef]
- Welsh, I.C.; O’Brien, T.P. Signaling integration in the rugae growth zone directs sequential SHH signaling center formation during the rostral outgrowth of the palate. Dev. Biol. 2009, 336, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Pantalacci, S.; Prochazka, J.; Martin, A.; Rothova, M.; Lambert, A.; Bernard, L.; Charles, C.; Viriot, L.; Peterkova, R.; Laudet, V. Patterning of palatal rugae through sequential addition reveals an anterior/posterior boundary in palatal development. BMC Dev. Biol. 2008, 8, 116. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Gu, S.; Alappat, S.; Song, Y.; Yan, M.; Zhang, X.; Zhang, G.; Jiang, Y.; Zhang, Z.; Zhang, Y.; et al. Shox2-deficient mice exhibit a rare type of incomplete clefting of the secondary palate. Development 2005, 132, 4397–4406. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lan, Y.; Pauws, E.; Meester-Smoor, M.A.; Stanier, P.; Zwarthoff, E.C.; Jiang, R. The Mn1 transcription factor acts upstream of Tbx22 and preferentially regulates posterior palate growth in mice. Development 2008, 135, 3959–3968. [Google Scholar] [CrossRef]
- Pauws, E.; Moore, G.E.; Stanier, P. A functional haplotype variant in the TBX22 promoter is associated with cleft palate and ankyloglossia. J. Med. Genet. 2009, 46, 555–561. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, M.; Zhao, W.; Yuan, X.; Yang, X.; Li, Y.; Qiu, M.; Zhu, X.J.; Zhang, Z. Gpr177-mediated Wnt Signaling Is Required for Secondary Palate Development. J. Dent. Res. 2015, 94, 961–967. [Google Scholar] [CrossRef]
- He, F.; Xiong, W.; Yu, X.; Espinoza-Lewis, R.; Liu, C.; Gu, S.; Nishita, M.; Suzuki, K.; Yamada, G.; Minami, Y.; et al. Wnt5a regulates directional cell migration and cell proliferation via Ror2-mediated noncanonical pathway in mammalian palate development. Development 2008, 135, 3871–3879. [Google Scholar] [CrossRef]
- Nishihara, H.; Kobayashi, N.; Kimura-Yoshida, C.; Yan, K.; Bormuth, O.; Ding, Q.; Nakanishi, A.; Sasaki, T.; Hirakawa, M.; Sumiyama, K.; et al. Coordinately Co-opted Multiple Transposable Elements Constitute an Enhancer for wnt5a Expression in the Mammalian Secondary Palate. PLoS Genet. 2016, 12, e1006380. [Google Scholar] [CrossRef]
- Almaidhan, A.; Cesario, J.; Landin Malt, A.; Zhao, Y.; Sharma, N.; Choi, V.; Jeong, J. Neural crest-specific deletion of Ldb1 leads to cleft secondary palate with impaired palatal shelf elevation. BMC Dev. Biol. 2014, 14, 3. [Google Scholar] [CrossRef]
- Gao, Y.; Lan, Y.; Ovitt, C.E.; Jiang, R. Functional equivalence of the zinc finger transcription factors Osr1 and Osr2 in mouse development. Dev. Biol. 2009, 328, 200–209. [Google Scholar] [CrossRef]
- Fu, X.; Xu, J.; Chaturvedi, P.; Liu, H.; Jiang, R.; Lan, Y. Identification of Osr2 Transcriptional Target Genes in Palate Development. J. Dent. Res. 2017, 96, 1451–1458. [Google Scholar] [CrossRef]
- Guo, L.; Degenstein, L.; Fuchs, E. Keratinocyte growth factor is required for hair development but not for wound healing. Genes Dev. 1996, 10, 165–175. [Google Scholar] [CrossRef]
- Ferguson, M.W. Palate development. Development 1988, 103, 41–60. [Google Scholar] [CrossRef]
- Alappat, S.R.; Zhang, Z.; Suzuki, K.; Zhang, X.; Liu, H.; Jiang, R.; Yamada, G.; Chen, Y. The cellular and molecular etiology of the cleft secondary palate in Fgf10 mutant mice. Dev. Biol. 2005, 277, 102–113. [Google Scholar] [CrossRef]
- Casey, L.M.; Lan, Y.; Cho, E.S.; Maltby, K.M.; Gridley, T.; Jiang, R. Jag2-Notch1 signaling regulates oral epithelial differentiation and palate development. Dev. Dyn. 2006, 235, 1830–1844. [Google Scholar] [CrossRef]
- Ingraham, C.R.; Kinoshita, A.; Kondo, S.; Yang, B.; Sajan, S.; Trout, K.J.; Malik, M.I.; Dunnwald, M.; Goudy, S.L.; Lovett, M.; et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat. Genet. 2006, 38, 1335–1340. [Google Scholar] [CrossRef]
- Peyrard-Janvid, M.; Leslie, E.J.; Kousa, Y.A.; Smith, T.L.; Dunnwald, M.; Magnusson, M.; Lentz, B.A.; Unneberg, P.; Fransson, I.; Koillinen, H.K.; et al. Dominant mutations in GRHL3 cause Van der Woude Syndrome and disrupt oral periderm development. Am. J. Hum. Genet. 2014, 94, 23–32. [Google Scholar] [CrossRef]
- Richardson, R.J.; Dixon, J.; Malhotra, S.; Hardman, M.J.; Knowles, L.; Boot-Handford, R.P.; Shore, P.; Whitmarsh, A.; Dixon, M.J. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat. Genet. 2006, 38, 1329–1334. [Google Scholar] [CrossRef]
- Jiang, R.; Lan, Y.; Chapman, H.D.; Shawber, C.; Norton, C.R.; Serreze, D.V.; Weinmaster, G.; Gridley, T. Defects in limb, craniofacial, and thymic development in Jagged2 mutant mice. Genes Dev. 1998, 12, 1046–1057. [Google Scholar] [CrossRef]
- Fitchett, J.E.; Hay, E.D. Medial edge epithelium transforms to mesenchyme after embryonic palatal shelves fuse. Dev. Biol. 1989, 131, 455–474. [Google Scholar] [CrossRef]
- Richardson, R.J.; Dixon, J.; Jiang, R.; Dixon, M.J. Integration of IRF6 and Jagged2 signalling is essential for controlling palatal adhesion and fusion competence. Hum. Mol. Genet. 2009, 18, 2632–2642. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.A.; Zheng, B.; Wang, X.J.; Vogel, H.; Roop, D.R.; Bradley, A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Thomason, H.A.; Zhou, H.; Kouwenhoven, E.N.; Dotto, G.P.; Restivo, G.; Nguyen, B.C.; Little, H.; Dixon, M.J.; van Bokhoven, H.; Dixon, J. Cooperation between the transcription factors p63 and IRF6 is essential to prevent cleft palate in mice. J. Clin. Investig. 2010, 120, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Candi, E.; Rufini, A.; Terrinoni, A.; Giamboi-Miraglia, A.; Lena, A.M.; Mantovani, R.; Knight, R.; Melino, G. DeltaNp63 regulates thymic development through enhanced expression of FgfR2 and Jag2. Proc. Natl. Acad. Sci. USA 2007, 104, 11999–12004. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Ishida, S.; Morimoto, I.; Yamashita, T.; Kojima, T.; Kihara, C.; Tanaka, T.; Imai, K.; Nakamura, Y.; Tokino, T. The p53 family member genes are involved in the Notch signal pathway. J. Biol. Chem. 2002, 277, 719–724. [Google Scholar] [CrossRef]
- Richardson, R.J.; Hammond, N.L.; Coulombe, P.A.; Saloranta, C.; Nousiainen, H.O.; Salonen, R.; Berry, A.; Hanley, N.; Headon, D.; Karikoski, R.; et al. Periderm prevents pathological epithelial adhesions during embryogenesis. J. Clin. Investig. 2014, 124, 3891–3900. [Google Scholar] [CrossRef]
- Funato, N.; Nakamura, M.; Richardson, J.A.; Srivastava, D.; Yanagisawa, H. Tbx1 regulates oral epithelial adhesion and palatal development. Hum. Mol. Genet. 2012, 21, 2524–2537. [Google Scholar] [CrossRef]
- Vaziri Sani, F.; Hallberg, K.; Harfe, B.D.; McMahon, A.P.; Linde, A.; Gritli-Linde, A. Fate-mapping of the epithelial seam during palatal fusion rules out epithelial-mesenchymal transformation. Dev. Biol. 2005, 285, 490–495. [Google Scholar] [CrossRef]
- Xu, X.; Han, J.; Ito, Y.; Bringas, P., Jr.; Urata, M.M.; Chai, Y. Cell autonomous requirement for Tgfbr2 in the disappearance of medial edge epithelium during palatal fusion. Dev. Biol. 2006, 297, 238–248. [Google Scholar] [CrossRef]
- Jin, J.Z.; Ding, J. Analysis of cell migration, transdifferentiation and apoptosis during mouse secondary palate fusion. Development 2006, 133, 3341–3347. [Google Scholar] [CrossRef]
- Cecconi, F.; Alvarez-Bolado, G.; Meyer, B.I.; Roth, K.A.; Gruss, P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 1998, 94, 727–737. [Google Scholar] [CrossRef]
- Cuervo, R.; Covarrubias, L. Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Development 2004, 131, 15–24. [Google Scholar] [CrossRef]
- Martinez-Alvarez, C.; Blanco, M.J.; Perez, R.; Rabadan, M.A.; Aparicio, M.; Resel, E.; Martinez, T.; Nieto, M.A. Snail family members and cell survival in physiological and pathological cleft palates. Dev. Biol. 2004, 265, 207–218. [Google Scholar] [CrossRef]
- Jin, J.Z.; Ding, J. Analysis of Meox-2 mutant mice reveals a novel postfusion-based cleft palate. Dev. Dyn. 2006, 235, 539–546. [Google Scholar] [CrossRef]
- Proetzel, G.; Pawlowski, S.A.; Wiles, M.V.; Yin, M.; Boivin, G.P.; Howles, P.N.; Ding, J.; Ferguson, M.W.; Doetschman, T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 1995, 11, 409–414. [Google Scholar] [CrossRef]
- Kaartinen, V.; Voncken, J.W.; Shuler, C.; Warburton, D.; Bu, D.; Heisterkamp, N.; Groffen, J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 1995, 11, 415–421. [Google Scholar] [CrossRef]
- Kaartinen, V.; Cui, X.M.; Heisterkamp, N.; Groffen, J.; Shuler, C.F. Transforming growth factor-beta3 regulates transdifferentiation of medial edge epithelium during palatal fusion and associated degradation of the basement membrane. Dev. Dyn. 1997, 209, 255–260. [Google Scholar] [CrossRef]
- Cui, X.M.; Shiomi, N.; Chen, J.; Saito, T.; Yamamoto, T.; Ito, Y.; Bringas, P.; Chai, Y.; Shuler, C.F. Overexpression of Smad2 in Tgf-beta3-null mutant mice rescues cleft palate. Dev. Biol. 2005, 278, 193–202. [Google Scholar] [CrossRef]
- Shiomi, N.; Cui, X.M.; Yamamoto, T.; Saito, T.; Shuler, C.F. Inhibition of SMAD2 expression prevents murine palatal fusion. Dev. Dyn. 2006, 235, 1785–1793. [Google Scholar] [CrossRef]
- Xu, X.; Han, J.; Ito, Y.; Bringas, P., Jr.; Deng, C.; Chai, Y. Ectodermal Smad4 and p38 MAPK are functionally redundant in mediating TGF-beta/BMP signaling during tooth and palate development. Dev. Cell 2008, 15, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Iwata, J.; Suzuki, A.; Pelikan, R.C.; Ho, T.V.; Sanchez-Lara, P.A.; Urata, M.; Dixon, M.J.; Chai, Y. Smad4-Irf6 genetic interaction and TGFbeta-mediated IRF6 signaling cascade are crucial for palatal fusion in mice. Development 2013, 140, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.O.; Lee, J.M.; Bok, J.; Jung, H.S. Inhibition of the Zeb family prevents murine palatogenesis through regulation of apoptosis and the cell cycle. Biochem. Biophys. Res. Commun. 2018, 506, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.Z.; Warner, D.R.; Lu, Q.; Pisano, M.M.; Greene, R.M.; Ding, J. Deciphering TGF-beta3 function in medial edge epithelium specification and fusion during mouse secondary palate development. Dev. Dyn. 2014, 243, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Xiong, W.; Wang, Y.; Li, L.; Liu, C.; Yamagami, T.; Taketo, M.M.; Zhou, C.; Chen, Y. Epithelial Wnt/beta-catenin signaling regulates palatal shelf fusion through regulation of Tgfbeta3 expression. Dev. Biol. 2011, 350, 511–519. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Murray, S.A.; Oram, K.F.; Gridley, T. Multiple functions of Snail family genes during palate development in mice. Development 2007, 134, 1789–1797. [Google Scholar] [CrossRef]
- Ke, C.Y.; Xiao, W.L.; Chen, C.M.; Lo, L.J.; Wong, F.H. IRF6 is the mediator of TGFbeta3 during regulation of the epithelial mesenchymal transition and palatal fusion. Sci. Rep. 2015, 5, 12791. [Google Scholar] [CrossRef]
- Serrano, M.J.; Liu, J.; Svoboda, K.K.; Nawshad, A.; Benson, M.D. Ephrin reverse signaling mediates palatal fusion and epithelial-to-mesenchymal transition independently of Tgfss3. J. Cell. Physiol. 2015, 230, 2961–2972. [Google Scholar] [CrossRef]
- Mima, J.; Koshino, A.; Oka, K.; Uchida, H.; Hieda, Y.; Nohara, K.; Kogo, M.; Chai, Y.; Sakai, T. Regulation of the epithelial adhesion molecule CEACAM1 is important for palate formation. PLoS ONE 2013, 8, e61653. [Google Scholar] [CrossRef]
- Shin, J.O.; Nakagawa, E.; Kim, E.J.; Cho, K.W.; Lee, J.M.; Cho, S.W.; Jung, H.S. miR-200b regulates cell migration via Zeb family during mouse palate development. Histochem. Cell Biol. 2012, 137, 459–470. [Google Scholar] [CrossRef]
- Shin, J.O.; Lee, J.M.; Cho, K.W.; Kwak, S.; Kwon, H.J.; Lee, M.J.; Cho, S.W.; Kim, K.S.; Jung, H.S. MiR-200b is involved in Tgf-beta signaling to regulate mammalian palate development. Histochem. Cell Biol. 2012, 137, 67–78. [Google Scholar] [CrossRef]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef]
- Li, J.; Rodriguez, G.; Han, X.; Janeckova, E.; Kahng, S.; Song, B.; Chai, Y. Regulatory Mechanisms of Soft Palate Development and Malformations. J. Dent. Res. 2019, 98, 959–967. [Google Scholar] [CrossRef]
- Li, H.; Jones, K.L.; Hooper, J.E.; Williams, T. The molecular anatomy of mammalian upper lip and primary palate fusion at single cell resolution. Development 2019, 146, dev174888. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Won, H.-J.; Kim, J.-W.; Won, H.-S.; Shin, J.-O. Gene Regulatory Networks and Signaling Pathways in Palatogenesis and Cleft Palate: A Comprehensive Review. Cells 2023, 12, 1954. https://doi.org/10.3390/cells12151954
Won H-J, Kim J-W, Won H-S, Shin J-O. Gene Regulatory Networks and Signaling Pathways in Palatogenesis and Cleft Palate: A Comprehensive Review. Cells. 2023; 12(15):1954. https://doi.org/10.3390/cells12151954
Chicago/Turabian StyleWon, Hyung-Jin, Jin-Woo Kim, Hyung-Sun Won, and Jeong-Oh Shin. 2023. "Gene Regulatory Networks and Signaling Pathways in Palatogenesis and Cleft Palate: A Comprehensive Review" Cells 12, no. 15: 1954. https://doi.org/10.3390/cells12151954
APA StyleWon, H.-J., Kim, J.-W., Won, H.-S., & Shin, J.-O. (2023). Gene Regulatory Networks and Signaling Pathways in Palatogenesis and Cleft Palate: A Comprehensive Review. Cells, 12(15), 1954. https://doi.org/10.3390/cells12151954