
Targeting Prohibitins to Inhibit Melanoma Growth and Overcome Resistance to Targeted Therapies

 ,
,  , , ,
, , ,  and
and
Abstract
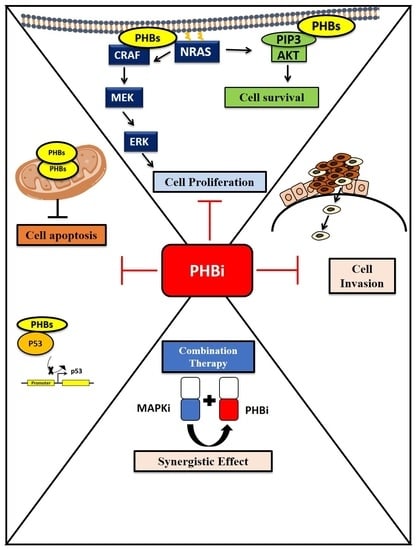
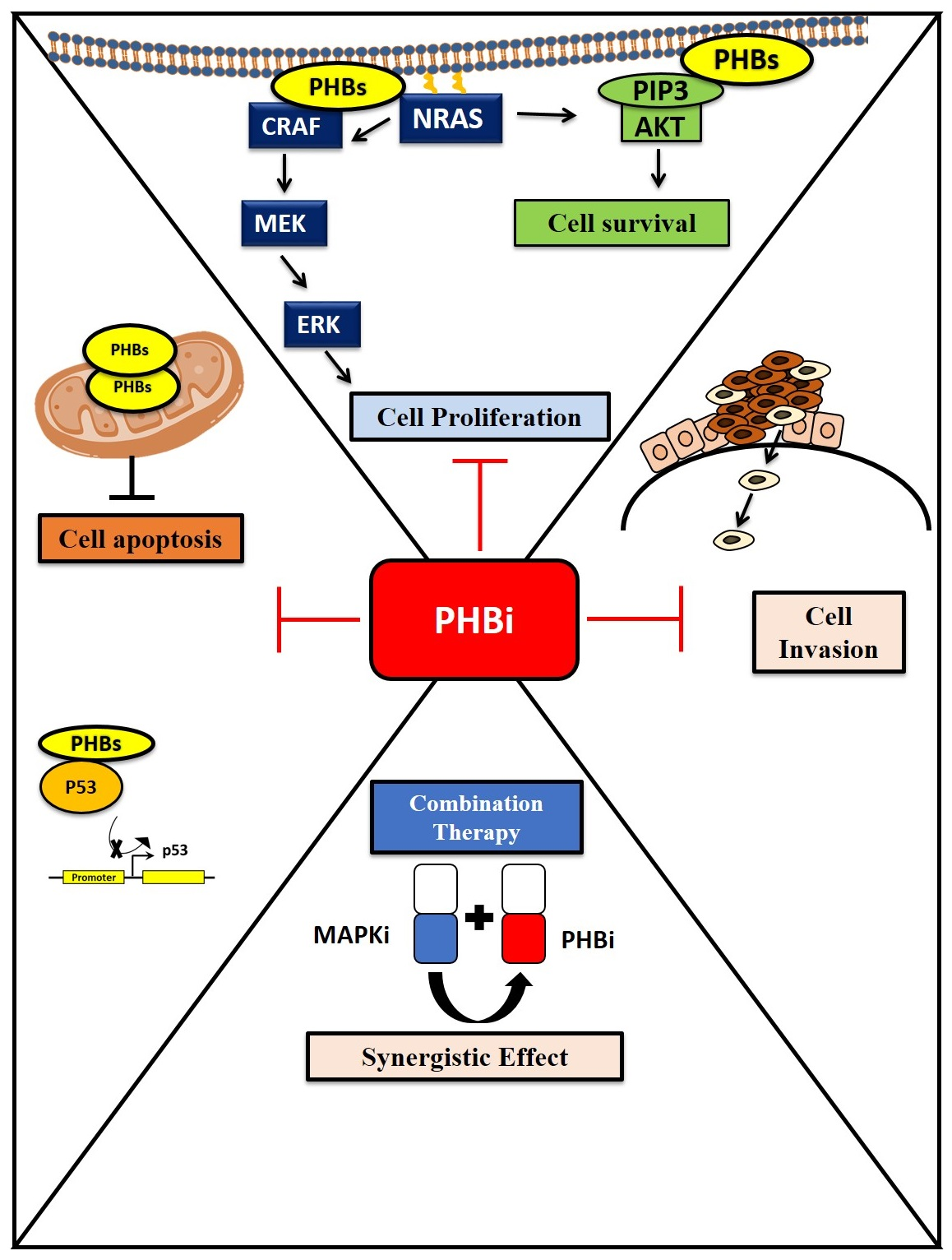
1. Introduction
2. Materials and Methods
2.1. Patients and Tissue Collection
2.2. RNA Extraction and Real-Time PCR of Patient Samples
2.3. Effectors
2.4. Melanoma Cell Culture
2.5. Proliferation Assay
2.6. Annexin V Assay/Apoptosis Determination
2.7. Measurement of Mitochondrial Membrane Potential
2.8. Caspase 3/7 Activity Assay
2.9. Evaluation of 3D Melanoma Spheroid Size and Viability
2.10. Western Blot Analysis
2.11. Autophagy Detection
2.12. Cell Migration Assay
2.13. Quantitative Real-Time PCR
2.14. Statistical Analysis
3. Results
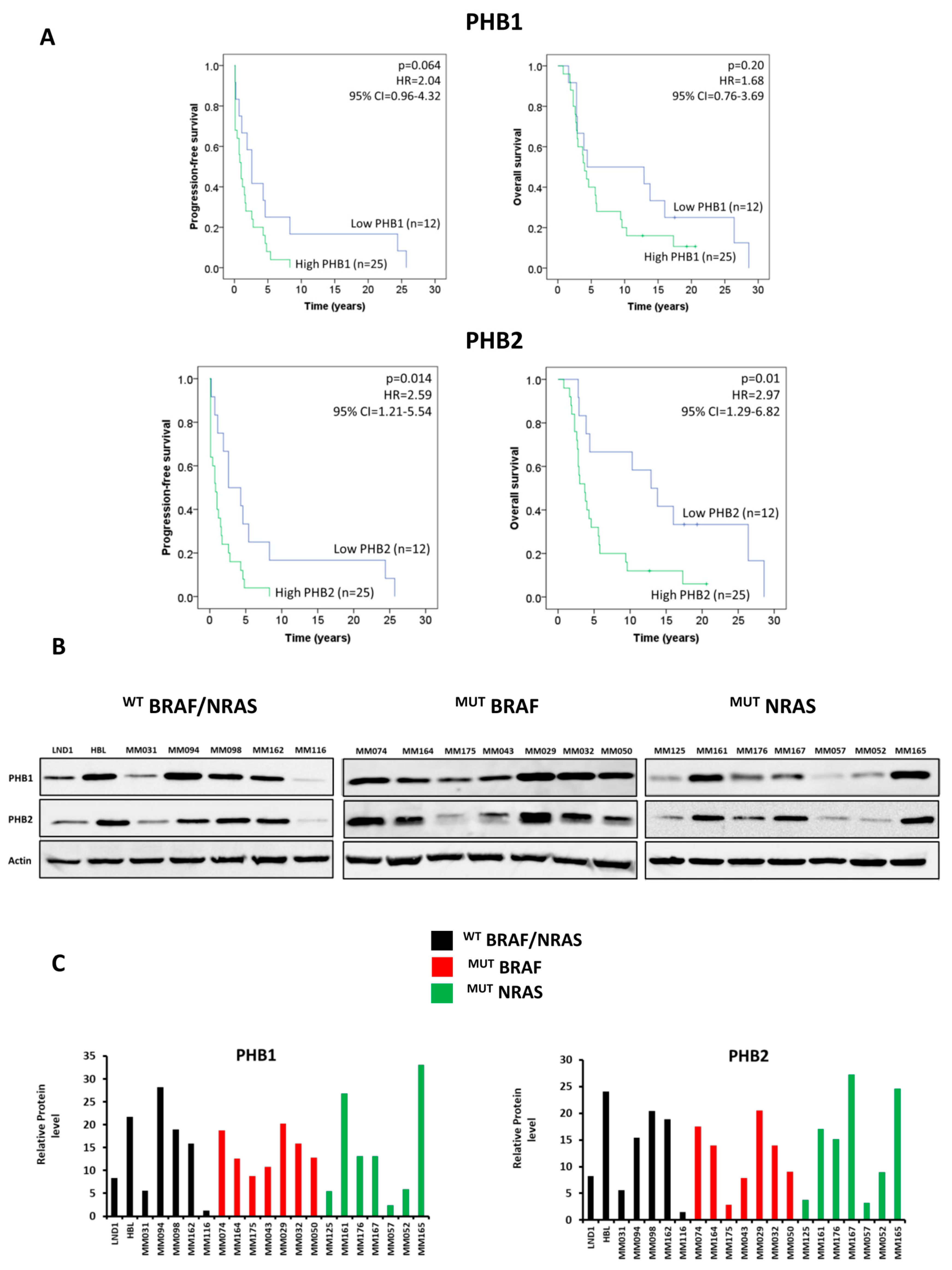
3.1. PHBs Are Associated with Short Patient Survival and Are Highly Expressed in Melanoma Lines Irrespective of BRAF/NRAS Mutational Status
3.2. PHB Ligands Inhibit Cell Proliferation in a Large Panel of Melanoma Cells
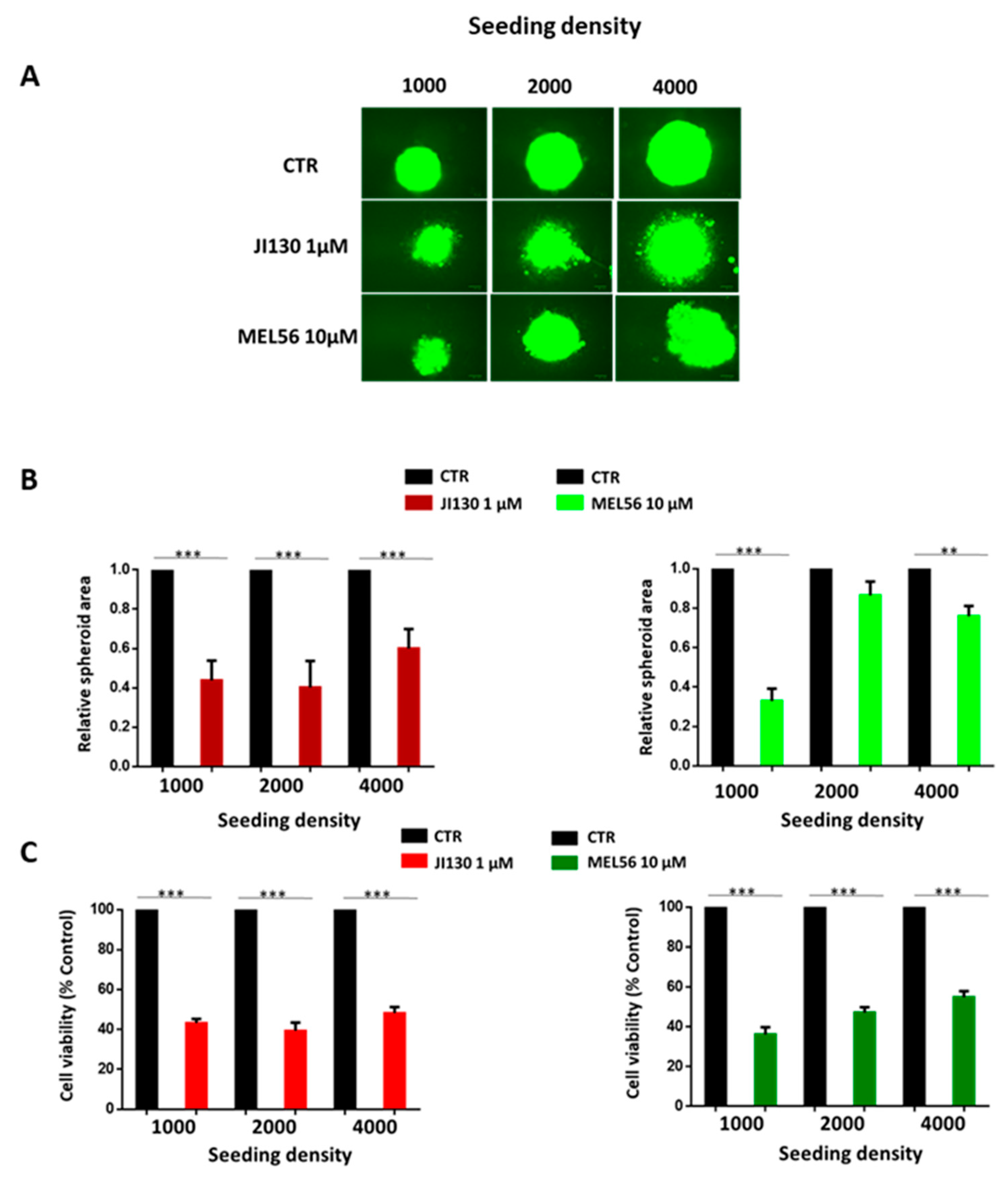
3.3. PHB Ligands Inhibit Cell Growth and Cell Viability of 3D Melanoma Spheroids
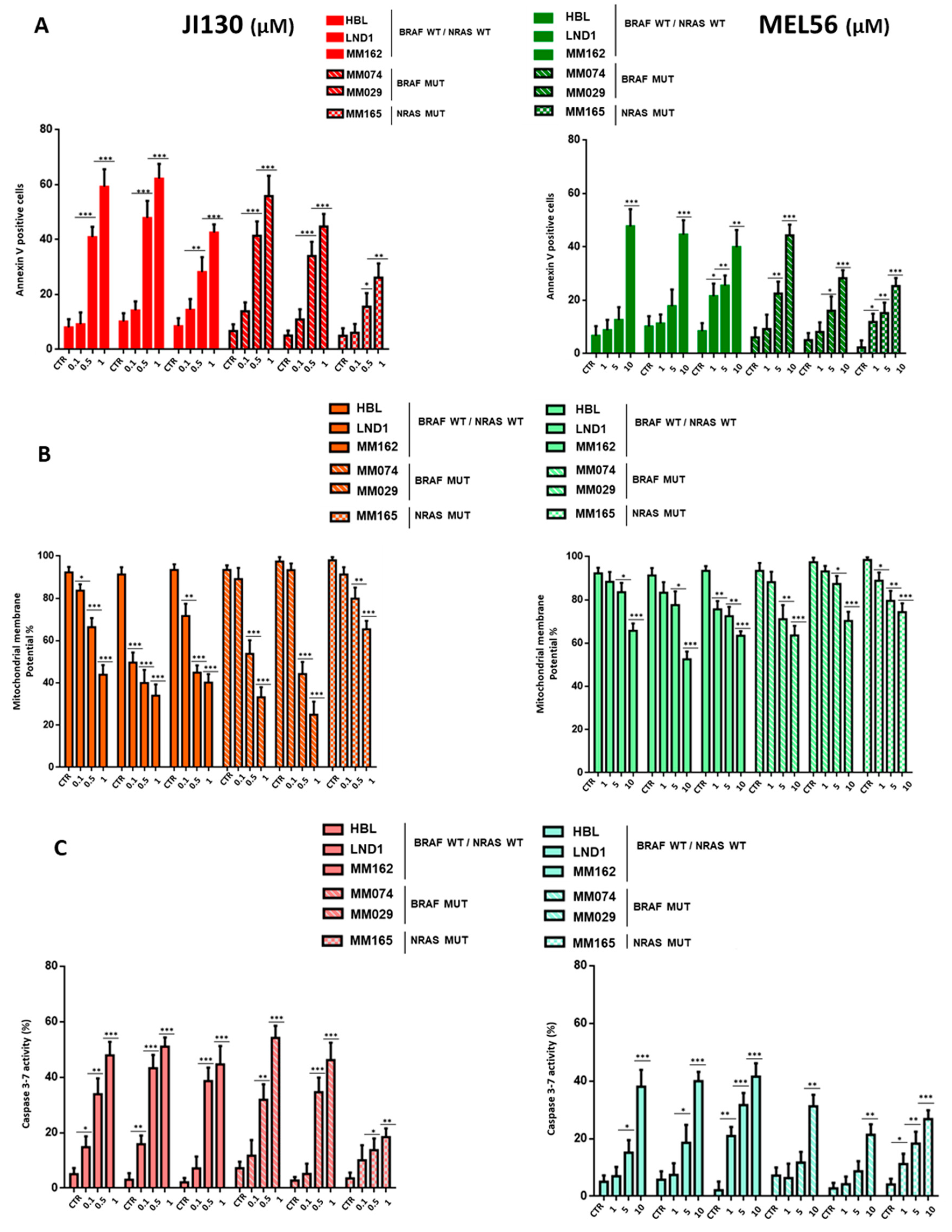
3.4. PHB Ligands Induce Cell Apoptosis via the Loss of Mitochondrial Potential (MMP) and Caspase Activation
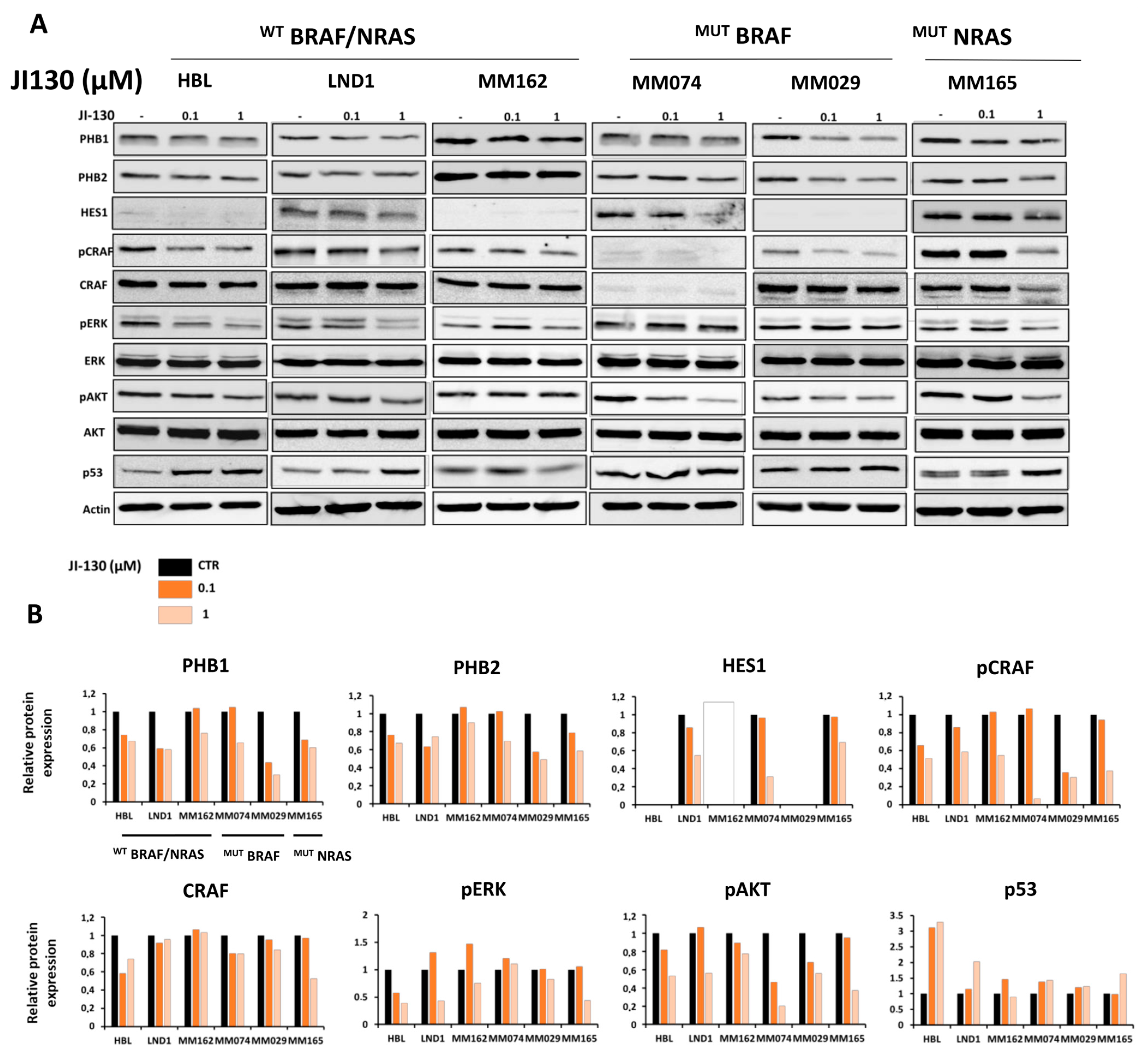
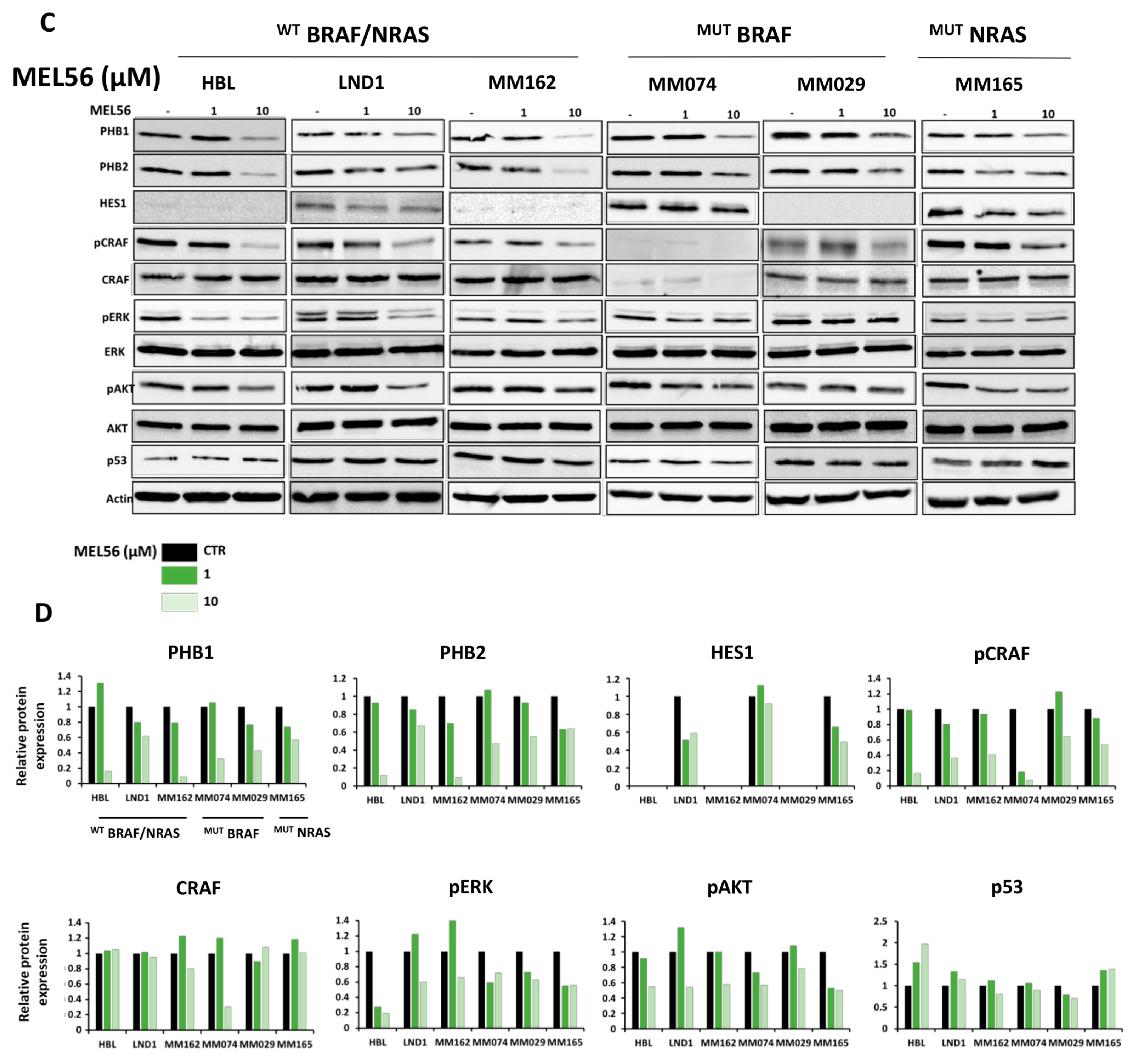
3.5. PHB Ligands Inhibit PHB Expression, the Two Main Survival Pathways MAPK and PI3K/AKT, and Promote p53 Expression in Melanoma Cells
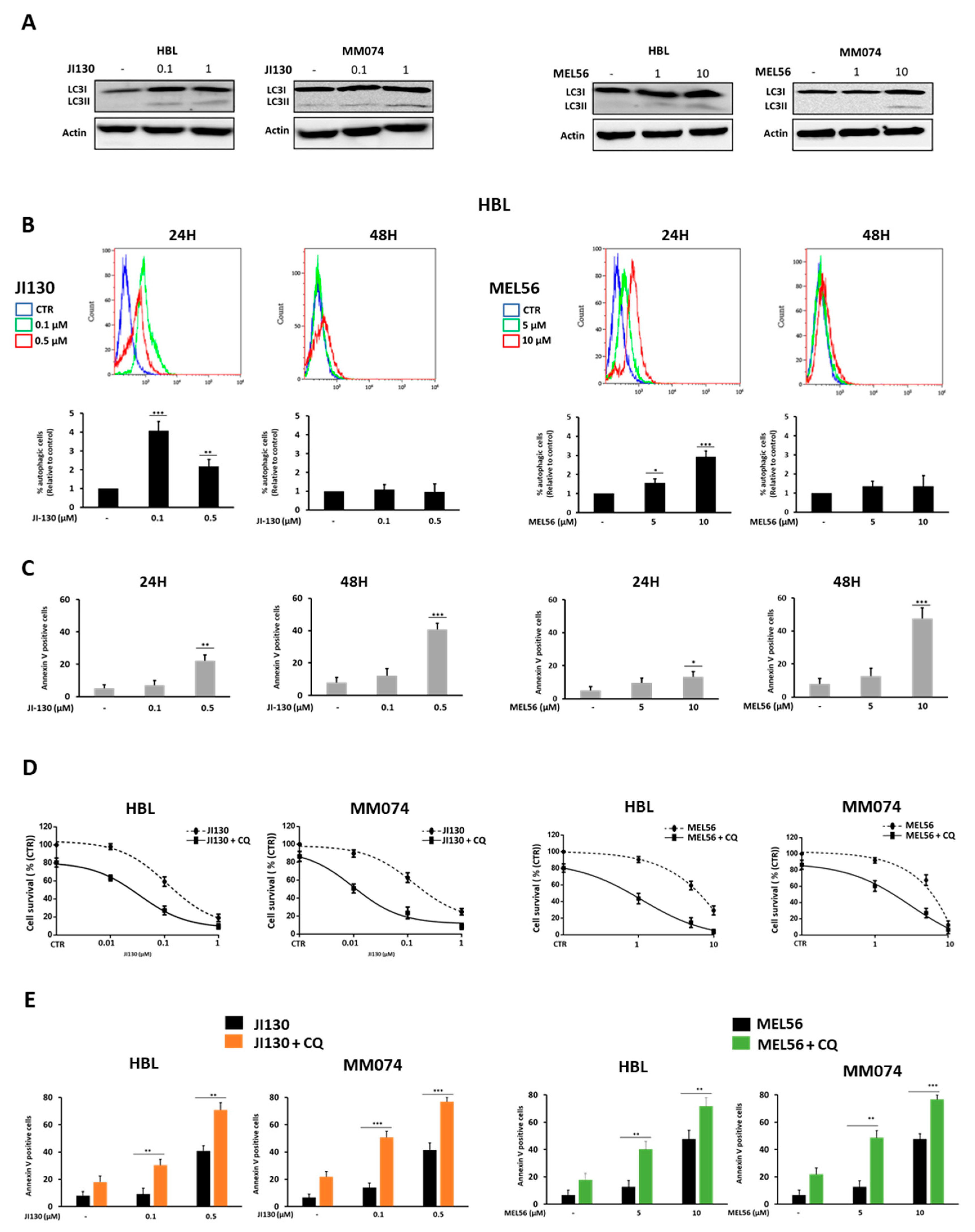
3.6. Autophagy Inhibition Enhances PHB Ligand Antitumor Efficacy in Melanoma Cells
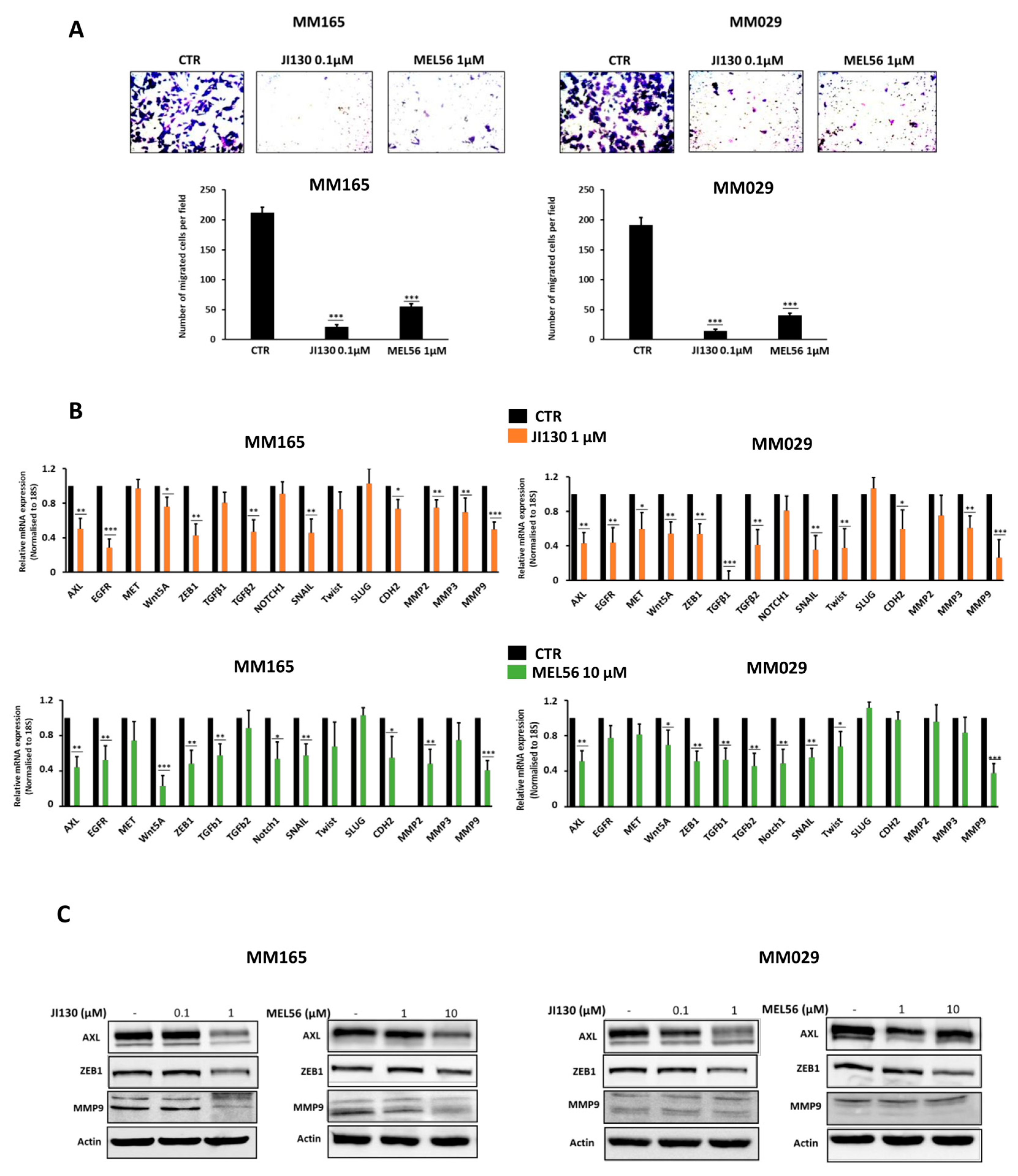
3.7. PHB Ligands Inhibit the Invasive Phenotype in Melanoma Cells
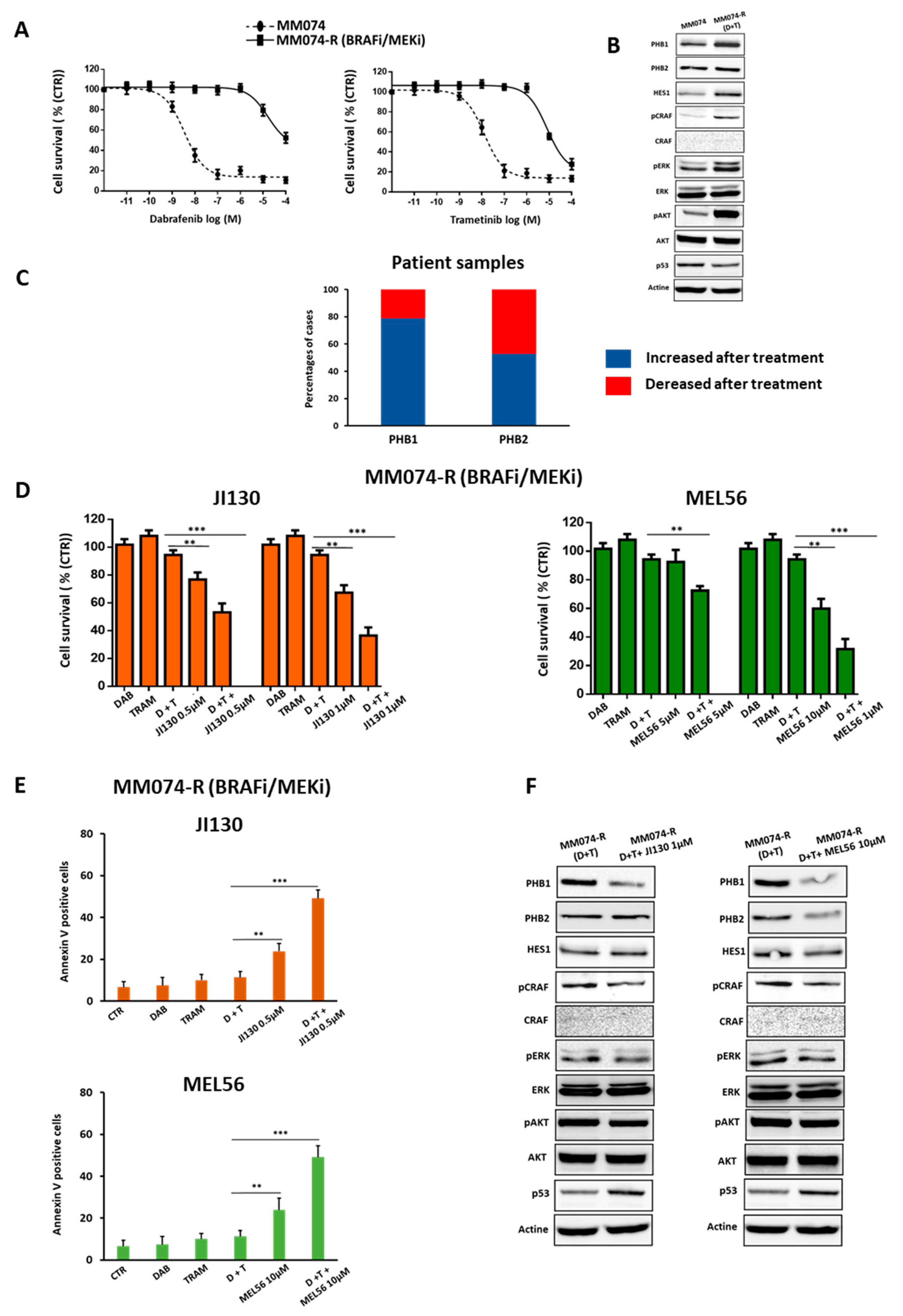
3.8. PHB Ligands Reverse the Acquired Resistance to BRAFi/MEKi Associated with an Up-Regulation of PHBs in BRAF Mutant Melanoma
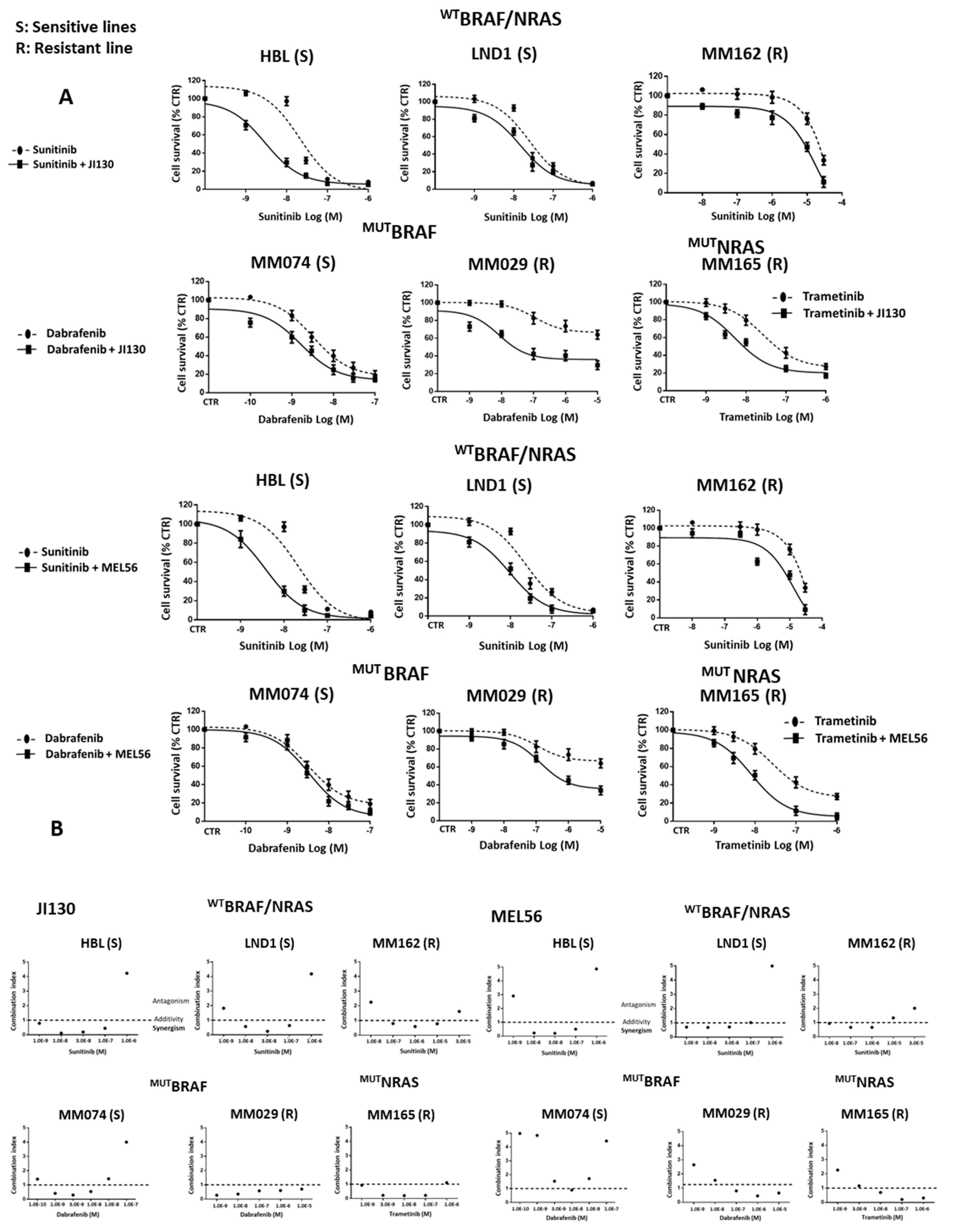
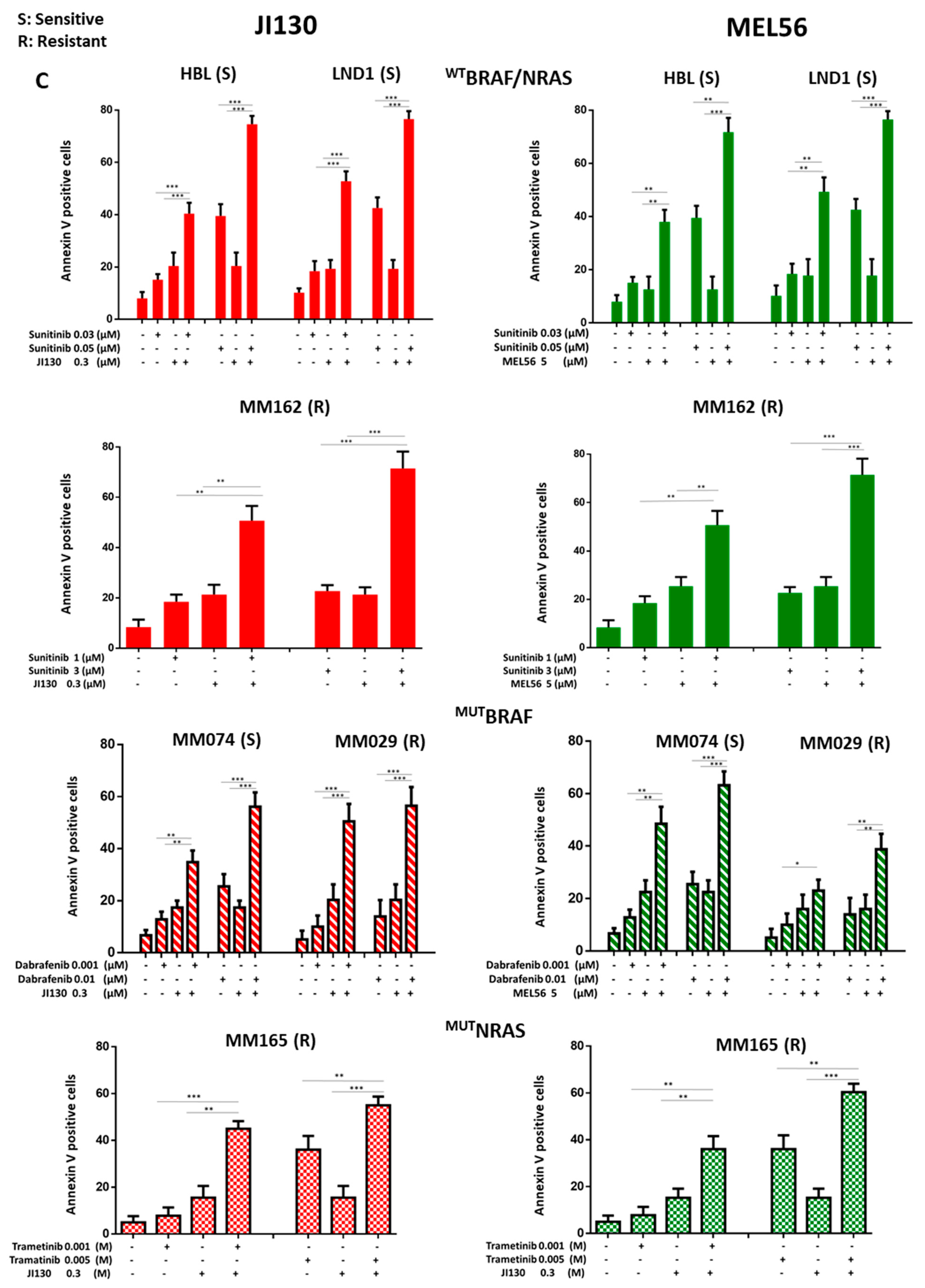
3.9. PHB Ligands Combined with MAPK Inhibitors Synergize to Inhibit Cell Proliferation and Induce Cell Apoptosis in Melanoma Cells with Different Molecular Subtypes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Najem, A.; Krayem, M.; Perdrix, A.; Kerger, J.; Awada, A.; Journe, F.; Ghanem, G. New Drug Combination Strategies in Melanoma: Current Status and Future Directions. Anticancer Res. 2017, 37, 5941–5953. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Jenkins, R.W.; Sullivan, R.J. Mechanisms of Resistance to Immune Checkpoint Blockade. Am. J. Clin. Dermatol. 2019, 20, 41–54. [Google Scholar] [CrossRef]
- Yang, J.; Li, B.; He, Q.-Y. Significance of prohibitin domain family in tumorigenesis and its implication in cancer diagnosis and treatment. Cell Death Dis. 2018, 9, 580. [Google Scholar] [CrossRef]
- Cirilo, P.D.R.; de Sousa Andrade, L.N.; Corrêa, B.R.S.; Qiao, M.; Furuya, T.K.; Chammas, R.; Penalva, L.O.F. MicroRNA-195 acts as an anti-proliferative miRNA in human melanoma cells by targeting Prohibitin 1. BMC Cancer 2017, 17, 750. [Google Scholar] [CrossRef]
- Webster, L.R.; Provan, P.J.; Graham, D.J.; Byth, K.; Walker, R.L.; Davis, S.; Salisbury, E.L.; Morey, A.L.; Ward, R.L.; Hawkins, N.J.; et al. Prohibitin expression is associated with high grade breast cancer but is not a driver of amplification at 17q21.33. Pathology 2013, 45, 629–636. [Google Scholar] [CrossRef]
- Yurugi, H.; Marini, F.; Weber, C.; David, K.; Zhao, Q.; Binder, H.; Désaubry, L.; Rajalingam, K. Targeting prohibitins with chemical ligands inhibits KRAS-mediated lung tumours. Oncogene 2017, 36, 4778–4789. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, I.; Bei, Y.; Garcia, H.D.; Ortiz, M.V.; Toedling, J.; Klironomos, F.; Rolff, J.; Eggert, A.; Schulte, J.H.; Kentsis, A.; et al. Prohibitin promotes dedifferentiation and is a potential therapeutic target in neuroblastoma. J. Clin. Investig. 2019, 4, e127130. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, S.; Li, Y.; Liu, Z.; Mi, L.; Cai, Y.; Wang, X.; Chen, L.; Ran, H.; Xiao, D.; et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nat. Commun. 2021, 12, 3720. [Google Scholar] [CrossRef]
- Wu, B.; Chang, N.; Xi, H.; Xiong, J.; Zhou, Y.; Wu, Y.; Wu, S.; Wang, N.; Yi, H.; Song, Y.; et al. PHB2 promotes tumorigenesis via RACK1 in non-small cell lung cancer. Theranostics 2021, 11, 3150–3166. [Google Scholar] [CrossRef]
- Cao, Y.; Liang, H.; Zhang, F.; Luan, Z.; Zhao, S.; Wang, X.-A.; Liu, S.; Bao, R.; Shu, Y.; Ma, Q.; et al. Prohibitin overexpression predicts poor prognosis and promotes cell proliferation and invasion through ERK pathway activation in gallbladder cancer. J. Exp. Clin. Cancer Res. 2016, 35, 68. [Google Scholar] [CrossRef]
- Luan, Z.; He, Y.; Alattar, M.; Chen, Z.; He, F. Targeting the prohibitin scaffold-CRAF kinase interaction in RAS-ERK-driven pancreatic ductal adenocarcinoma. Mol. Cancer 2014, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-F.; Ho, M.-Y.; Peng, J.-M.; Hung, S.-W.; Lee, W.-H.; Liang, C.-M.; Liang, S.-M. Raf activation by Ras and promotion of cellular metastasis require phosphorylation of prohibitin in the raft domain of the plasma membrane. Oncogene 2013, 32, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Krayem, M.; Journe, F.; Wiedig, M.; Morandini, R.; Najem, A.; Salès, F.; van Kempen, L.C.; Sibille, C.; Awada, A.; Marine, J.-C.; et al. p53 Reactivation by PRIMA-1Met (APR-246) sensitises V600E/KBRAF melanoma to vemurafenib. Eur. J. Cancer 2016, 55, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Tortelli, T.C.; de Godoy, L.M.F.; de Souza, G.A.; Bonatto, D.; Otake, A.H.; de Freitas Saito, R.; Rosa, J.C.; Greene, L.J.; Chammas, R. Accumulation of prohibitin is a common cellular response to different stressing stimuli and protects melanoma cells from ER stress and chemotherapy-induced cell death. Oncotarget 2017, 8, 43114–43129. [Google Scholar] [CrossRef]
- Ho, M.-Y.; Liang, C.-M.; Liang, S.-M. MIG-7 and phosphorylated prohibitin coordinately regulate lung cancer invasion/metastasis. Oncotarget 2014, 6, 381–393. [Google Scholar] [CrossRef]
- Ma, L.-L.; Shen, L.; Tong, G.-H.; Tang, N.; Luo, Y.; Guo, L.-L.; Hu, C.-T.; Huang, Y.-X.; Huang, G.; Jing, F.-Y.; et al. Prohibitin, relocated to the front ends, can control the migration directionality of colorectal cancer cells. Oncotarget 2017, 8, 76340–76356. [Google Scholar] [CrossRef]
- Fang, C.-H.; Lin, Y.-T.; Liang, C.-M.; Liang, S.-M. A novel c-Kit/phospho-prohibitin axis enhances ovarian cancer stemness and chemoresistance via Notch3—PBX1 and β-catenin—ABCG2 signaling. J. Biomed. Sci. 2020, 27, 42. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-H.; Dai, X.-M.; Du, B. Hes1: A key role in stemness, metastasis and multidrug resistance. Cancer Biol. Ther. 2015, 16, 353–359. [Google Scholar] [CrossRef]
- Wang, S.-C.; Lin, X.-L.; Wang, H.-Y.; Qin, Y.-J.; Chen, L.; Li, J.; Jia, J.-S.; Shen, H.-F.; Yang, S.; Xie, R.-Y.; et al. Hes1 triggers epithelial-mesenchymal transition (EMT)-like cellular marker alterations and promotes invasion and metastasis of nasopharyngeal carcinoma by activating the PTEN/AKT pathway. Oncotarget 2015, 6, 36713–36730. [Google Scholar] [CrossRef]
- Wang, D.; Tabti, R.; Elderwish, S.; Abou-Hamdan, H.; Djehal, A.; Yu, P.; Yurugi, H.; Rajalingam, K.; Nebigil, C.G.; Désaubry, L. Prohibitin ligands: A growing armamentarium to tackle cancers, osteoporosis, inflammatory, cardiac and neurological diseases. Cell. Mol. Life Sci. 2020, 77, 3525–3546. [Google Scholar] [CrossRef]
- Basmadjian, C.; Thuaud, F.; Ribeiro, N.; Désaubry, L. Flavaglines: Potent anticancer drugs that target prohibitins and the helicase eIF4A. Futur. Med. Chem. 2013, 5, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, R.; Yildirim, O.; Gasser, A.; Basmadjian, C.; Zhao, Q.; Wilmet, J.-P.; Désaubry, L.; Nebigil, C.G. FL3, a Synthetic Flavagline and Ligand of Prohibitins, Protects Cardiomyocytes via STAT3 from Doxorubicin Toxicity. PLoS ONE 2015, 10, e0141826. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Moyama, C.; Taniguchi, K.; Ando, K.; Matsuda, R.; Ando, S.; Ii, H.; Kageyama, S.; Kawauchi, A.; Chouha, N.; et al. Fluorizoline Blocks the Interaction between Prohibitin-2 and γ-Glutamylcyclotransferase and Induces P21Waf1/Cip1 Expression in MCF7 Breast Cancer Cells. Mol. Pharmacol. 2022, 101, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Perron, A.; Nishikawa, Y.; Iwata, J.; Shimojo, H.; Takaya, J.; Kobayashi, K.; Imayoshi, I.; Mbenza, N.M.; Takenoya, M.; Kageyama, R.; et al. Small-molecule screening yields a compound that inhibits the cancer-associated transcription factor Hes1 via the PHB2 chaperone. J. Biol. Chem. 2018, 293, 8285–8294. [Google Scholar] [CrossRef] [PubMed]
- Djehal, A.; Krayem, M.; Najem, A.; Hammoud, H.; Cresteil, T.; Nebigil, C.G.; Wang, D.; Yu, P.; Bentouhami, E.; Ghanem, G.E.; et al. Targeting prohibitin with small molecules to promote melanogenesis and apoptosis in melanoma cells. Eur. J. Med. Chem. 2018, 155, 880–888. [Google Scholar] [CrossRef]
- Najem, A.; Wouters, J.; Krayem, M.; Rambow, F.; Sabbah, M.; Sales, F.; Awada, A.; Aerts, S.; Journe, F.; Marine, J.-C.; et al. Tyrosine-Dependent Phenotype Switching Occurs Early in Many Primary Melanoma Cultures Limiting Their Translational Value. Front. Oncol. 2021, 11, 780654. [Google Scholar] [CrossRef]
- Najem, A.; Krayem, M.; Salès, F.; Hussein, N.; Badran, B.; Robert, C.; Awada, A.; Journe, F.; Ghanem, G.E. P53 and MITF/Bcl-2 identified as key pathways in the acquired resistance of NRAS-mutant melanoma to MEK inhibition. Eur. J. Cancer 2017, 83, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, K.; Wang, H.; Dai, Y. Inhibition of Autophagy by Chloroquine Enhances the Antitumor Efficacy of Sorafenib in Glioblastoma. Cell. Mol. Neurobiol. 2016, 36, 1197–1208. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Peng, Y.-T.; Chen, P.; Ouyang, R.-Y.; Song, L. Multifaceted role of prohibitin in cell survival and apoptosis. Apoptosis 2015, 20, 1135–1149. [Google Scholar] [CrossRef]
- Belser, M.; Walker, D.W. Role of Prohibitins in Aging and Therapeutic Potential Against Age-Related Diseases. Front. Genet. 2021, 12, 714228. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef]
- Wirawan, E.; Vanden Berghe, T.; Lippens, S.; Agostinis, P.; Vandenabeele, P. Autophagy: For better or for worse. Cell Res. 2012, 22, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, Z.; Diao, X.; Huang, S. Induction of autophagy-dependent apoptosis by the survivin suppressant YM155 in prostate cancer cells. Cancer Lett. 2011, 302, 29–36. [Google Scholar] [CrossRef]
- He, L.; Han, J.; Li, B.; Huang, L.; Ma, K.; Chen, Q.; Liu, X.; Bao, L.; Liu, H. Identification of a new cyathane diterpene that induces mitochondrial and autophagy-dependent apoptosis and shows a potent in vivo anti-colorectal cancer activity. Eur. J. Med. Chem. 2016, 111, 183–192. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | Resistance to MAPKi | IC50 (µM) JI130 | IC50 (µM) MEL56 | |
|---|---|---|---|---|
| Wild-type BRAF/NRAS cell lines | HBL | sensitive | 0.10 | 6.1 |
| LND1 | sensitive | 0.08 | 4.7 | |
| MM162 | NA | 0.16 | 8.4 | |
| BRAF mutant cell lines | MM074 | sensitive | 0.09 | 5.0 |
| MM164 | intermediate | 0.11 | 9.3 | |
| MM029 | intrinsic | 0.17 | 12.6 | |
| MM032 | intrinsic | 0.20 | 10.2 | |
| NRAS mutant cell lines | MM161 | sensitive | 0.25 | 15.5 |
| MM165 | Low sensitivity | 0.18 | 13.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najem, A.; Krayem, M.; Sabbah, S.; Pesetti, M.; Journe, F.; Awada, A.; Désaubry, L.; Ghanem, G.E. Targeting Prohibitins to Inhibit Melanoma Growth and Overcome Resistance to Targeted Therapies. Cells 2023, 12, 1855. https://doi.org/10.3390/cells12141855
Najem A, Krayem M, Sabbah S, Pesetti M, Journe F, Awada A, Désaubry L, Ghanem GE. Targeting Prohibitins to Inhibit Melanoma Growth and Overcome Resistance to Targeted Therapies. Cells. 2023; 12(14):1855. https://doi.org/10.3390/cells12141855
Chicago/Turabian StyleNajem, Ahmad, Mohammad Krayem, Serena Sabbah, Matilde Pesetti, Fabrice Journe, Ahmad Awada, Laurent Désaubry, and Ghanem E. Ghanem. 2023. "Targeting Prohibitins to Inhibit Melanoma Growth and Overcome Resistance to Targeted Therapies" Cells 12, no. 14: 1855. https://doi.org/10.3390/cells12141855
APA StyleNajem, A., Krayem, M., Sabbah, S., Pesetti, M., Journe, F., Awada, A., Désaubry, L., & Ghanem, G. E. (2023). Targeting Prohibitins to Inhibit Melanoma Growth and Overcome Resistance to Targeted Therapies. Cells, 12(14), 1855. https://doi.org/10.3390/cells12141855