


Understanding the Role of the Glial Scar through the Depletion of Glial Cells after Spinal Cord Injury


Abstract
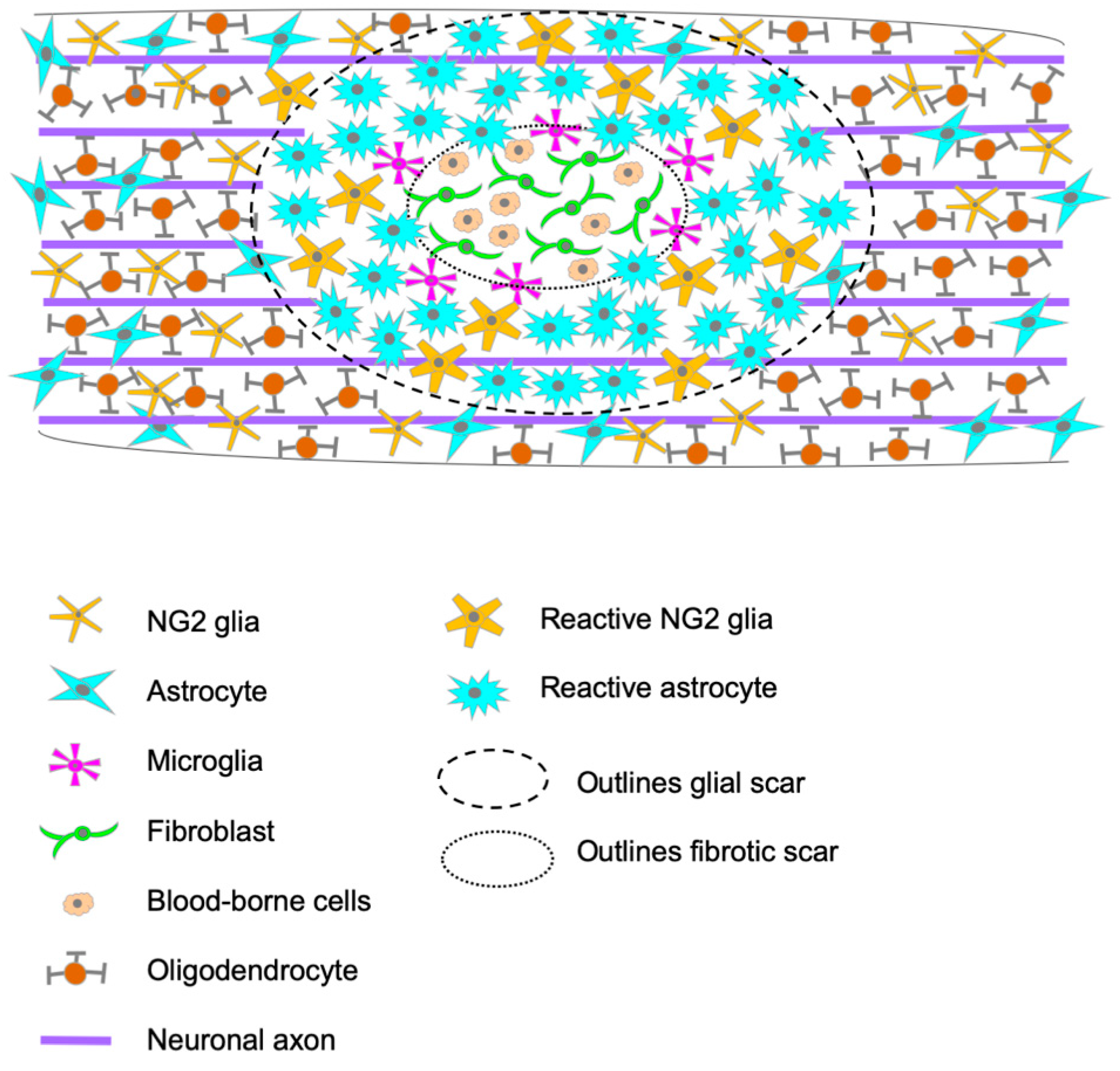
1. Glial Scar Formation and Composition
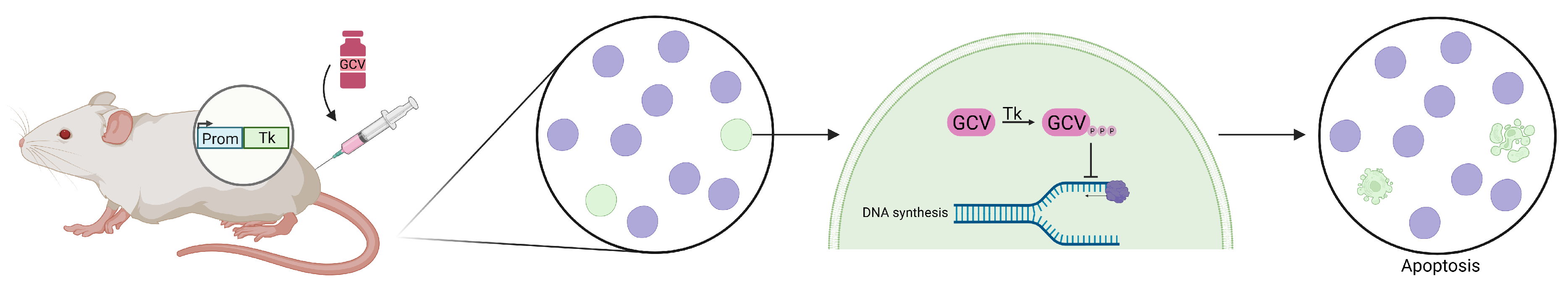
2. Conditional Cell Ablation Strategies
2.1. Genetic Approaches for Ablation of Specific Glial Cell Populations
2.2. Pharmacological Approaches for Ablation of Specific Glial Cell Populations
3. Glial Scar after SCI: What Have We Learned from Astrocytic Ablation Experiments?
4. Microglia Ablation after SCI: How Does It Affect Glial Scar Formation?
5. NG2+-Glia Ablation after SCI: What We Learnt and What Remains Unknown
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cajal, S.R. Degeneration and Regeneration of the Nervous System; Oxford University Press: London, UK, 1928. [Google Scholar]
- Reier, P.J.; Stensaas, L.J.; Guth, L. The Astrocytic Scar as an Impediment to Regeneration in the Central Nervous System. In Spinal Cord Reconstructions; Raven Press: New York, NY, USA, 1983; pp. 163–195. [Google Scholar]
- Davies, S.J.A.; Goucher, D.R.; Doller, C.; Silver, J. Robust Regeneration of Adult Sensory Axons in Degenerating White Matter of the Adult Rat Spinal Cord. J. Neurosci. 1999, 19, 5810–5822. [Google Scholar] [CrossRef]
- Asher, R.A.; Morgenstern, D.A.; Moon, L.D.; Fawcett, J.W. Chondroitin sulphate proteoglycans: Inhibitory components of the glial scar. Prog. Brain Res. 2001, 132, 611–619. [Google Scholar] [CrossRef]
- Preston, E.; Webster, J.; Small, D.; Kochanek, P.M.; Janesko, K.L.; Jenkins, L.W.; Yan, H.Q.; Kibbe, M.R.; Robichaud, P.; Wooditch, A.C.; et al. Characteristics of Sustained Blood-Brain Barrier Opening and Tissue Injury in a Model for Focal Trauma in the Rat. J. Neurotrauma 2001, 18, 83–92. [Google Scholar] [CrossRef]
- Maxwell, W.L.; Follows, R.; Ashhurst, D.E.; Berry, M. The response of the cerebral hemisphere of the rat to injury. I. The mature rat. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1990, 328, 479–500. [Google Scholar] [CrossRef]
- Kawano, H.; Kimura-Kuroda, J.; Komuta, Y.; Yoshioka, N.; Li, H.P.; Kawamura, K.; Li, Y.; Raisman, G. Role of the lesion scar in the response to damage and repair of the central nervous system. Cell Tissue Res. 2012, 349, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Emery, E.; Aldana, P.; Bunge, M.B.; Puckett, W.; Srinivasan, A.; Keane, R.W.; Bethea, J.; Levi, A.D.O. Apoptosis after traumatic human spinal cord injury. J. Neurosurg. 1998, 89, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Norenberg, M.D.; Smith, J.; Marcillo, A.; Willebrords, J.; Yanguas, S.C.; Maes, M.; Decrock, E.; Wang, N.; Leybaert, L.; Kwak, B.R.; et al. The Pathology of Human Spinal Cord Injury: Defining the Problems. J. Neurotrauma 2004, 21, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Zai, L.J.; Wrathall, J.R. Cell proliferation and replacement following contusive spinal cord injury. Glia 2005, 50, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Krishnaswamy, V.R.; Benbenishty, A.; Blinder, P.; Sagi, I. Demystifying the extracellular matrix and its proteolytic remodeling in the brain: Structural and functional insights. Cell. Mol. Life Sci. 2019, 76, 3229–3248. [Google Scholar] [CrossRef]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the brain extracellular matrix: A new target for remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Rauch, U. Brain matrix: Structure, turnover and necessity. Biochem. Soc. Trans. 2007, 35, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Haggerty, A.E.; Marlow, M.M.; Oudega, M. Extracellular matrix components as therapeutics for spinal cord injury. Neurosci. Lett. 2017, 652, 50–55. [Google Scholar] [CrossRef]
- Becker, T.; Anliker, B.; Becker, C.; Taylor, J.; Schachner, M.; Meyer, R.L.; Bartsch, U. Tenascin-R inhibits regrowth of optic fibers in vitro and persists in the optic nerve of mice after injury. Glia 2000, 29, 330–346. [Google Scholar] [CrossRef]
- Pesheva, P.; Gennarini, G.; Goridis, C.; Schachner, M. The F3/11 cell adhesion molecule mediates the repulsion of neurons by the extracellular matrix glycoprotein J1-160/180. Neuron 1993, 10, 69–82. [Google Scholar] [CrossRef]
- Lochter, A.; Schachner, M. Tenascin and extracellular matrix glycoproteins: From promotion to polarization of neurite growth in vitro. J. Neurosci. 1993, 13, 3986–4000. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Popovich, P.G. Extracellular matrix regulation of inflammation in the healthy and injured spinal cord. Exp. Neurol. 2014, 258, 24–34. [Google Scholar] [CrossRef]
- Jiang, D.; Liang, J.; Noble, P.W. Hyaluronan in Tissue Injury and Repair. Annu. Rev. Cell Dev. Biol. 2007, 23, 435–461. [Google Scholar] [CrossRef]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef]
- Asher, R.A.; Morgenstern, D.A.; Shearer, M.C.; Adcock, K.H.; Pesheva, P.; Fawcett, J.W. Versican Is Upregulated in CNS Injury and Is a Product of Oligodendrocyte Lineage Cells. J. Neurosci. 2002, 22, 2225–2236. [Google Scholar] [CrossRef]
- Jones, L.L.; Yamaguchi, Y.; Stallcup, W.B.; Tuszynski, M.H. NG2 Is a Major Chondroitin Sulfate Proteoglycan Produced after Spinal Cord Injury and Is Expressed by Macrophages and Oligodendrocyte Progenitors. J. Neurosci. 2002, 22, 2792–2803. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.L.; Margolis, R.U.; Tuszynski, M.H. The chondroitin sulfate proteoglycans neurocan, brevican, phosphacan, and versican are differentially regulated following spinal cord injury. Exp. Neurol. 2003, 182, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Buss, A.; Pech, K.; A Kakulas, B.; Martin, D.; Schoenen, J.; Noth, J.; A Brook, G. NG2 and phosphacan are present in the astroglial scar after human traumatic spinal cord injury. BMC Neurol. 2009, 9, 32. [Google Scholar] [CrossRef]
- Bradbury, E.J.; Moon, L.D.F.; Popat, R.J.; King, V.R.; Bennett, G.S.; Patel, P.N.; Fawcett, J.W.; McMahon, S.B. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 2002, 416, 636–640. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Reactive Astrocytes in Neural Repair and Protection. Neuroscientist 2005, 11, 400–407. [Google Scholar] [CrossRef]
- Bradbury, E.J.; Burnside, E.R. Moving beyond the glial scar for spinal cord repair. Nat. Commun. 2019, 10, 3879. [Google Scholar] [CrossRef]
- Faulkner, J.R.; Herrmann, J.E.; Woo, M.J.; Tansey, K.E.; Doan, N.B.; Sofroniew, M.V. Reactive Astrocytes Protect Tissue and Preserve Function after Spinal Cord Injury. J. Neurosci. 2004, 24, 2143–2155. [Google Scholar] [CrossRef]
- Gu, Y.; Cheng, X.; Huang, X.; Yuan, Y.; Qin, S.; Tan, Z.; Wang, D.; Hu, X.; He, C.; Su, Z. Conditional ablation of reactive astrocytes to dissect their roles in spinal cord injury and repair. Brain Behav. Immun. 2019, 80, 394–405. [Google Scholar] [CrossRef]
- Bellver-Landete, V.; Bretheau, F.; Mailhot, B.; Vallières, N.; Lessard, M.; Janelle, M.-E.; Vernoux, N.; Tremblay, M.; Fuehrmann, T.; Shoichet, M.S.; et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat. Commun. 2019, 10, 518. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, Z.-L.; Xie, H.; Tian, X.-B.; Xu, H.-L.; Li, W.; Yao, S. Microglial depletion impairs glial scar formation and aggravates inflammation partly by inhibiting STAT3 phosphorylation in astrocytes after spinal cord injury. Neural Regen. Res. 2023, 18, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Zhao, Y.; Hu, D.; Wang, S.; Yu, T.; Zhang, L. Depletion of microglia exacerbates injury and impairs function recovery after spinal cord injury in mice. Cell Death Dis. 2020, 11, 528. [Google Scholar] [CrossRef]
- Hesp, Z.C.; Yoseph, R.Y.; Suzuki, R.; Jukkola, P.; Wilson, C.; Nishiyama, A.; McTigue, D.M. Proliferating NG2-Cell-Dependent Angiogenesis and Scar Formation Alter Axon Growth and Functional Recovery After Spinal Cord Injury in Mice. J. Neurosci. 2017, 38, 1366–1382. [Google Scholar] [CrossRef]
- Knox, R.J.; Friedlos, F.; Boland, M.P. The bioactivation of CB 1954 and its use as a prodrug in antibody-directed enzyme prodrug therapy (ADEPT). Cancer Metastasis Rev. 1993, 12, 195–212. [Google Scholar] [CrossRef]
- Clark, A.; Iwobi, M.; Cui, W.; Crompton, M.; Harold, G.; Hobbs, S.; Kamalati, T.; Knox, R.; Neil, C.; Yull, F.; et al. Selective cell ablation in transgenic mice expressing E. coli nitroreductase. Gene Ther. 1997, 4, 101–110. [Google Scholar] [CrossRef]
- Drabek, D.; Guy, J.; Craig, R.; Grosveld, F. The expression of bacterial nitroreductase in transgenic mice results in specific cell killing by the prodrug CB1954. Gene Ther. 1997, 4, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Iwawaki, T.; Taya, C.; Yonekawa, H.; Noda, M.; Inui, Y.; Mekada, E.; Kimata, Y.; Tsuru, A.; Kohno, K. Diphtheria toxin receptor–mediated conditional and targeted cell ablation in transgenic mice. Nat. Biotechnol. 2001, 19, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Han, J.; Lund, H.; Boggavarapu, N.R.; Lauschke, V.M.; Goto, S.; Cheng, H.; Wang, Y.; Tachi, A.; Xie, C.; et al. An overlooked subset of Cx3cr1 wt/wt microglia in the Cx3cr1 CreER-Eyfp/wt mouse has a repopulation advantage over Cx3cr1 CreER-Eyfp/wt microglia following microglial depletion. J. Neuroinflamm. 2022, 19, 20. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.R.P.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-Stimulating Factor 1 Receptor Signaling Is Necessary for Microglia Viability, Unmasking a Microglia Progenitor Cell in the Adult Brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef]
- Green, K.N.; Crapser, J.D.; Hohsfield, L.A. To Kill a Microglia: A Case for CSF1R Inhibitors. Trends Immunol. 2020, 41, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Benner, B.; Good, L.; Quiroga, D.; E Schultz, T.; Kassem, M.; E Carson, W.; A Cherian, M.; Sardesai, S.; Wesolowski, R. Pexidartinib, a Novel Small Molecule CSF-1R Inhibitor in Use for Tenosynovial Giant Cell Tumor: A Systematic Review of Pre-Clinical and Clinical Development. Drug Des. Dev. Ther. 2020, 14, 1693–1704. [Google Scholar] [CrossRef]
- Chang, Y.-F. Lysine Metabolism in the Rat Brain: The Pipecolic Acid-Forming Pathway. J. Neurochem. 1978, 30, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-F. Lysine metabolism in the human and the monkey: Demonstration of pipecolic acid formation in the brain and other organs. Neurochem. Res. 1982, 7, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.J.; Chang, Y.F.; Schwarcz, R.; Brookes, N. Characterization of L-α-aminoadipic acid transport in cultured rat astrocytes. Brain Res. 1996, 741, 166–173. [Google Scholar] [CrossRef]
- Khurgel, M.; Koo, A.C.; Ivy, G.O. Selective ablation of astrocytes by intracerebral injections of α- aminoadipate. Glia 1996, 16, 351–358. [Google Scholar] [CrossRef]
- Linden, J.R.; Ma, Y.; Zhao, B.; Harris, J.; Rumah, K.R.; Schaeren-Wiemers, N.; Vartanian, T. Clostridium perfringens Epsilon Toxin Causes Selective Death of Mature Oligodendrocytes and Central Nervous System Demyelination. mBio 2015, 6, e02513-14. [Google Scholar] [CrossRef]
- Zirngibl, M.; Assinck, P.; Sizov, A.; Caprariello, A.V.; Plemel, J.R. Oligodendrocyte death and myelin loss in the cuprizone model: An updated overview of the intrinsic and extrinsic causes of cuprizone demyelination. Mol. Neurodegener. 2022, 17, 34. [Google Scholar] [CrossRef]
- Andersson, P.-B.; Perry, V.; Gordon, S. The acute inflammatory response to lipopolysaccharide in cns parenchyma differs from that in other body tissues. Neuroscience 1992, 48, 169–186. [Google Scholar] [CrossRef]
- Smith, G.M.; Miller, R.H.; Silver, J. Changing role of forebrain astrocytes during development, regenerative failure, and induced regeneration upon transplantation. J. Comp. Neurol. 1986, 251, 23–43. [Google Scholar] [CrossRef]
- Bähr, M.; Przyrembel, C.; Bastmeyer, M. Astrocytes from adult rat optic nerves are nonpermissive for regenerating retinal ganglion cell axons. Exp. Neurol. 1995, 131, 211–220. [Google Scholar] [CrossRef]
- Barnabé-Heider, F.; Göritz, C.; Sabelström, H.; Takebayashi, H.; Pfrieger, F.W.; Meletis, K.; Frisén, J. Origin of New Glial Cells in Intact and Injured Adult Spinal Cord. Cell Stem Cell 2010, 7, 470–482. [Google Scholar] [CrossRef]
- Magnus, T.; Carmen, J.; Deleon, J.; Xue, H.; Pardo, A.C.; Lepore, A.C.; Mattson, M.P.; Rao, M.S.; Maragakis, N.J. Adult glial precursor proliferation in mutant SOD1G93A mice. Glia 2007, 56, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Wanner, I.B.; Anderson, M.A.; Song, B.; Levine, J.; Fernandez, A.; Gray-Thompson, Z.; Ao, Y.; Sofroniew, M.V. Glial Scar Borders Are Formed by Newly Proliferated, Elongated Astrocytes That Interact to Corral Inflammatory and Fibrotic Cells via STAT3-Dependent Mechanisms after Spinal Cord Injury. J. Neurosci. 2013, 33, 12870–12886. [Google Scholar] [CrossRef]
- Hara, M.; Kobayakawa, K.; Ohkawa, Y.; Kumamaru, H.; Yokota, K.; Saito, T.; Kijima, K.; Yoshizaki, S.; Harimaya, K.; Nakashima, Y.; et al. Interaction of reactive astrocytes with type I collagen induces astrocytic scar formation through the integrin–N-cadherin pathway after spinal cord injury. Nat. Med. 2017, 23, 818–828. [Google Scholar] [CrossRef]
- McKeon, R.; Schreiber, R.; Rudge, J.; Silver, J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J. Neurosci. 1991, 11, 3398–3411. [Google Scholar] [CrossRef]
- Mckeon, R.J.; Höke, A.; Silver, J. Injury-Induced Proteoglycans Inhibit the Potential for Laminin-Mediated Axon Growth on Astrocytic Scars. Exp. Neurol. 1995, 136, 32–43. [Google Scholar] [CrossRef]
- Tan, A.M.; Zhang, W.; Levine, J.M. NG2: A component of the glial scar that inhibits axon growth. J. Anat. 2005, 207, 717–725. [Google Scholar] [CrossRef]
- Uhlin-Hansen, L.; Wik, T.; Kjellen, L.; Berg, E.; Forsdahl, F.; Kolset, S.O. Proteoglycan metabolism in normal and inflammatory human macrophages. Blood 1993, 82, 2880–2889. [Google Scholar] [CrossRef]
- Bush, T.G.; Puvanachandra, N.; Horner, C.H.; Polito, A.; Ostenfeld, T.; Svendsen, C.N.; Mucke, L.; Johnson, M.H.; Sofroniew, M.V. Leukocyte Infiltration, Neuronal Degeneration, and Neurite Outgrowth after Ablation of Scar-Forming, Reactive Astrocytes in Adult Transgenic Mice. Neuron 1999, 23, 297–308. [Google Scholar] [CrossRef]
- Richardson, P.M.; Issa, V.M.K. Peripheral injury enhances central regeneration of primary sensory neurones. Nature 1984, 309, 791–793. [Google Scholar] [CrossRef]
- Neumann, S.; Woolf, C.J. Regeneration of Dorsal Column Fibers into and beyond the Lesion Site following Adult Spinal Cord Injury. Neuron 1999, 23, 83–91. [Google Scholar] [CrossRef]
- Omura, T.; Omura, K.; Tedeschi, A.; Riva, P.; Painter, M.W.; Rojas, L.; Martin, J.; Lisi, V.; Huebner, E.A.; Latremoliere, A.; et al. Robust Axonal Regeneration Occurs in the Injured CAST/Ei Mouse CNS. Neuron 2016, 90, 662. [Google Scholar] [CrossRef]
- Tremblay, M. The role of microglia at synapses in the healthy CNS: Novel insights from recent imaging studies. Neuron Glia Biol. 2011, 7, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Neuroscience: Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Hanisch, U.-K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Tremblay, M.-È.; Stevens, B.; Sierra, A.; Wake, H.; Bessis, A.; Nimmerjahn, A. The Role of Microglia in the Healthy Brain. J. Neurosci. 2011, 31, 16064–16069. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Schwab, J.M.; Frei, E.; Klusman, I.; Schnell, L.; E Schwab, M.; Schluesener, H.J. AIF-1 expression defines a proliferating and alert microglial/macrophage phenotype following spinal cord injury in rats. J. Neuroimmunol. 2001, 119, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Pineau, I.; Lacroix, S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: Multiphasic expression pattern and identification of the cell types involved. J. Comp. Neurol. 2006, 500, 267–285. [Google Scholar] [CrossRef]
- Xu, J.; Kim, G.-M.; Chen, S.; Yan, P.; Ahmed, S.H.; Ku, G.; Beckman, J.S.; Xu, X.M.; Hsu, C.Y. iNOS and Nitrotyrosine Expression After Spinal Cord Injury. J. Neurotrauma 2001, 18, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed]
- Jordão, M.J.; Sankowski, R.; Brendecke, S.M.; Sagar; Locatelli, G.; Tai, Y.H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; et al. Neuroimmunology: Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019, 363, eaat7554. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; Van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Hakim, R.; Zachariadis, V.; Sankavaram, S.R.; Han, J.; Harris, R.A.; Brundin, L.; Enge, M.; Svensson, M. Spinal Cord Injury Induces Permanent Reprogramming of Microglia into a Disease-Associated State Which Contributes to Functional Recovery. J. Neurosci. 2021, 41, 8441–8459. [Google Scholar] [CrossRef]
- Yamamoto, S.; Nakajima, K.; Kohsaka, S. Macrophage-colony stimulating factor as an inducer of microglial proliferation in axotomized rat facial nucleus. J. Neurochem. 2010, 115, 1057–1067. [Google Scholar] [CrossRef]
- Smith, A.M.; Gibbons, H.M.; Oldfield, R.L.; Bergin, P.M.; Mee, E.W.; A Curtis, M.; Faull, R.L.M.; Dragunow, M. M-CSF increases proliferation and phagocytosis while modulating receptor and transcription factor expression in adult human microglia. J. Neuroinflamm. 2013, 10, 859. [Google Scholar] [CrossRef]
- Han, J.; Fan, Y.; Zhou, K.; Blomgren, K.; Harris, R.A. Uncovering sex differences of rodent microglia. J. Neuroinflamm. 2021, 18, 74. [Google Scholar] [CrossRef]
- Dawson, M.R.; Polito, A.; Levine, J.M.; Reynolds, R. NG2-expressing glial progenitor cells: An abundant and widespread population of cycling cells in the adult rat CNS. Mol. Cell. Neurosci. 2003, 24, 476–488. [Google Scholar] [CrossRef]
- Hughes, E.G.; Orthmann-Murphy, J.L.; Langseth, A.J.; Bergles, D.E. Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat. Neurosci. 2018, 21, 696–706. [Google Scholar] [CrossRef] [PubMed]
- A Hill, R.; Patel, K.D.; Goncalves, C.M.; Grutzendler, J.; Nishiyama, A. Modulation of oligodendrocyte generation during a critical temporal window after NG2 cell division. Nat. Neurosci. 2014, 17, 1518–1527. [Google Scholar] [CrossRef]
- Hughes, E.G.; Kang, S.H.; Fukaya, M.; E Bergles, D. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat. Neurosci. 2013, 16, 668–676. [Google Scholar] [CrossRef]
- Bergles, D.E.; Roberts, J.D.B.; Somogyi, P.; Jahr, C.E. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 2000, 405, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Kula, B.; Chen, T.J.; Kukley, M. Glutamatergic signaling between neurons and oligodendrocyte lineage cells: Is it synaptic or non-synaptic? Glia 2019, 67, 2071–2091. [Google Scholar] [CrossRef]
- Butt, A.M.; Kiff, J.; Hubbard, P.; Berry, M. Synantocytes: New functions for novel NG2 expressing glia. J. Neurocytol. 2002, 31, 551–565. [Google Scholar] [CrossRef]
- Gensert, J.M.; E Goldman, J. Endogenous Progenitors Remyelinate Demyelinated Axons in the Adult CNS. Neuron 1997, 19, 197–203. [Google Scholar] [CrossRef]
- Zawadzka, M.; Rivers, L.E.; Fancy, S.P.; Zhao, C.; Tripathi, R.; Jamen, F.; Young, K.; Goncharevich, A.; Pohl, H.; Rizzi, M.; et al. CNS-Resident Glial Progenitor/Stem Cells Produce Schwann Cells as well as Oligodendrocytes during Repair of CNS Demyelination. Cell Stem Cell 2010, 6, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Hackett, A.R.; Lee, D.-H.; Dawood, A.; Rodriguez, M.; Funk, L.; Tsoulfas, P.; Lee, J.K. STAT3 and SOCS3 regulate NG2 cell proliferation and differentiation after contusive spinal cord injury. Neurobiol. Dis. 2016, 89, 10–22. [Google Scholar] [CrossRef]
- Seo, J.H.; Miyamoto, N.; Hayakawa, K.; Pham, L.D.D.; Maki, T.; Ayata, C.; Kim, K.W.; Lo, E.H.; Arai, K. OPCs induce early blood-brain barrier opening. J. Clin. Investig. 2013, 123, 782–786. [Google Scholar]
- McTigue, D.M.; Wei, P.; Stokes, B.T. Proliferation of NG2-Positive Cells and Altered Oligodendrocyte Numbers in the Contused Rat Spinal Cord. J. Neurosci. 2001, 21, 3392–3400. [Google Scholar] [CrossRef]
- Tripathi, R.; McTigue, D.M. Prominent oligodendrocyte genesis along the border of spinal contusion lesions. Glia 2007, 55, 698–711. [Google Scholar] [CrossRef]
- Hesp, Z.C.; Goldstein, E.A.; Miranda, C.J.; Kaspar, B.K.; McTigue, D.M. Chronic Oligodendrogenesis and Remyelination after Spinal Cord Injury in Mice and Rats. J. Neurosci. 2015, 35, 1274–1290. [Google Scholar] [CrossRef] [PubMed]
- Hackett, A.R.; Lee, J.K. Understanding the NG2 Glial Scar after Spinal Cord Injury. Front. Neurol. 2016, 7, 199. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cheng, X.; He, Q.; Zheng, Y.; Kim, D.H.; Whittemore, S.R.; Cao, Q.L. Astrocytes from the Contused Spinal Cord Inhibit Oligodendrocyte Differentiation of Adult Oligodendrocyte Precursor Cells by Increasing the Expression of Bone Morphogenetic Proteins. J. Neurosci. 2011, 31, 6053–6058. [Google Scholar] [CrossRef]
- Levine, J. The reactions and role of NG2 glia in spinal cord injury. Brain Res. 2016, 1638, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Sellers, D.L.; Maris, D.O.; Horner, P.J. Postinjury Niches Induce Temporal Shifts in Progenitor Fates to Direct Lesion Repair after Spinal Cord Injury. J. Neurosci. 2009, 29, 6722–6733. [Google Scholar] [CrossRef]
- Hackett, A.R.; Yahn, S.L.; Lyapichev, K.; Dajnoki, A.; Lee, D.-H.; Rodriguez, M.; Cammer, N.; Pak, J.; Mehta, S.T.; Bodamer, O.; et al. Injury type-dependent differentiation of NG2 glia into heterogeneous astrocytes. Exp. Neurol. 2018, 308, 72–79. [Google Scholar] [CrossRef]
- Assinck, P.; Duncan, G.J.; Plemel, J.R.; Lee, M.; Stratton, J.A.; Manesh, S.B.; Liu, J.; Ramer, L.M.; Kang, S.H.; Bergles, D.E.; et al. Myelinogenic Plasticity of Oligodendrocyte Precursor Cells following Spinal Cord Contusion Injury. J. Neurosci. 2017, 37, 8635–8654. [Google Scholar] [CrossRef]
- Crawford, A.H.; Tripathi, R.B.; Foerster, S.; McKenzie, I.; Kougioumtzidou, E.; Grist, M.; Richardson, W.D.; Franklin, R.J. Pre-Existing Mature Oligodendrocytes Do Not Contribute to Remyelination following Toxin-Induced Spinal Cord Demyelination. Am. J. Pathol. 2016, 186, 511–516. [Google Scholar] [CrossRef]
- Keirstead, H.S.; Levine, J.M.; Blakemore, W.F. Response of the oligodendrocyte progenitor cell population (Defined by NG2 labelling) to demyelination of the adult spinal cord. Glia 1998, 22, 161–170. [Google Scholar] [CrossRef]
- Cao, Q.; Xu, X.-M.; DeVries, W.H.; Enzmann, G.U.; Ping, P.; Tsoulfas, P.; Wood, P.M.; Bunge, M.B.; Whittemore, S.R. Functional Recovery in Traumatic Spinal Cord Injury after Transplantation of Multineurotrophin-Expressing Glial-Restricted Precursor Cells. J. Neurosci. 2005, 25, 6947–6957. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, A.; Tsuji, O.; Shibata, S.; Nori, S.; Takano, M.; Kobayashi, Y.; Takahashi, Y.; Fujiyoshi, K.; Hara, C.M.; Miyawaki, A.; et al. Significance of Remyelination by Neural Stem/Progenitor Cells Transplanted into the Injured Spinal Cord. Stem Cells 2011, 29, 1983–1994. [Google Scholar] [CrossRef]
- Hawryluk, G.W.J.; Spano, S.; Chew, D.; Wang, S.; Erwin, M.; Chamankhah, M.; Forgione, N.; Fehlings, M.G. An Examination of the Mechanisms by which Neural Precursors Augment Recovery following Spinal Cord Injury: A Key Role for Remyelination. Cell Transplant. 2014, 23, 365–380. [Google Scholar] [CrossRef]
- Plemel, J.R.; Keough, M.B.; Duncan, G.J.; Sparling, J.S.; Yong, V.W.; Stys, P.K.; Tetzlaff, W. Remyelination after spinal cord injury: Is it a target for repair? Prog. Neurobiol. 2014, 117, 54–72. [Google Scholar] [CrossRef]
- Duncan, G.J.; Manesh, S.B.; Hilton, B.J.; Assinck, P.; Plemel, J.R.; Tetzlaff, W. The fate and function of oligodendrocyte progenitor cells after traumatic spinal cord injury. Glia 2019, 68, 227–245. [Google Scholar] [CrossRef]
- Duncan, G.J.; Manesh, S.B.; Hilton, B.J.; Assinck, P.; Liu, J.; Moulson, A.; Plemel, J.R.; Tetzlaff, W. Locomotor recovery following contusive spinal cord injury does not require oligodendrocyte remyelination. Nat. Commun. 2018, 9, 3066. [Google Scholar] [CrossRef] [PubMed]
- Dou, C.; Levine, J. Inhibition of neurite growth by the NG2 chondroitin sulfate proteoglycan. J. Neurosci. 1994, 14, 7616–7628. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.M.; Colletti, M.; Rorai, A.T.; Skene, J.H.P.; Levine, J.M. Antibodies against the NG2 Proteoglycan Promote the Regeneration of Sensory Axons within the Dorsal Columns of the Spinal Cord. J. Neurosci. 2006, 26, 4729–4739. [Google Scholar] [CrossRef]
- Petrosyan, H.A.; Hunanyan, A.S.; Alessi, V.; Schnell, L.; Levine, J.; Arvanian, V.L. Neutralization of Inhibitory Molecule NG2 Improves Synaptic Transmission, Retrograde Transport, and Locomotor Function after Spinal Cord Injury in Adult Rats. J. Neurosci. 2013, 33, 4032–4043. [Google Scholar] [CrossRef]
- McTigue, D.M.; Tripathi, R.; Wei, P. NG2 Colocalizes with Axons and Is Expressed by a Mixed Cell Population in Spinal Cord Lesions. J. Neuropathol. Exp. Neurol. 2006, 65, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, A.; Skuba, A.; Himes, B.T.; Bhagat, S.L.; Hyun, J.K.; Tessler, A.; Bishop, D.; Son, Y.-J. In Vivo Imaging of Dorsal Root Regeneration: Rapid Immobilization and Presynaptic Differentiation at the CNS/PNS Border. J. Neurosci. 2011, 31, 4569–4582. [Google Scholar] [CrossRef] [PubMed]
- Filous, A.R.; Tran, A.; Howell, C.J.; Busch, S.A.; Evans, T.A.; Stallcup, W.B.; Kang, S.H.; Bergles, D.E.; Lee, S.-I.; Levine, J.M.; et al. Entrapment via Synaptic-Like Connections between NG2 Proteoglycan+ Cells and Dystrophic Axons in the Lesion Plays a Role in Regeneration Failure after Spinal Cord Injury. J. Neurosci. 2014, 34, 16369–16384. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.J. Synapsing with NG2 cells (polydendrocytes), unappreciated barrier to axon regeneration? Neural Regen. Res. 2015, 10, 346–348. [Google Scholar] [CrossRef] [PubMed]
- Hirrlinger, J.; Scheller, A.; Hirrlinger, P.G.; Kellert, B.; Tang, W.; Wehr, M.C.; Goebbels, S.; Reichenbach, A.; Sprengel, R.; Rossner, M.J.; et al. Split-Cre Complementation Indicates Coincident Activity of Different Genes In Vivo. PLoS ONE 2009, 4, e4286. [Google Scholar] [CrossRef]
- Göritz, C.; Dias, D.O.; Tomilin, N.; Barbacid, M.; Shupliakov, O.; Frisén, J. A Pericyte Origin of Spinal Cord Scar Tissue. Science 2011, 333, 238–242. [Google Scholar] [CrossRef]
- Dias, D.O.; Kim, H.; Holl, D.; Solnestam, B.W.; Lundeberg, J.; Carlén, M.; Göritz, C.; Frisén, J. Reducing Pericyte-Derived Scarring Promotes Recovery after Spinal Cord Injury. Cell 2018, 173, 153–165.e22. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Ablated Cell Type | Cell Ablation Technique | Species/ Strain | Animal Age | Time-Point of Cell Ablation | Time-Point of Tissue Collection | Consequences of Cell Ablation | Reference |
|---|---|---|---|---|---|---|---|
| Astrocytes | GFAP-Tk mice. Tk-GCV system | Transgenic mice on C57BL/6J background | Not specified | from 0 to 7 dpi | 14 dpi | Tissue degeneration. Increased infiltration of pro-inflammatory cells. Failed repair of the BBB. Reduced oligodendrocyte and neuronal survival. Increased demyelination. Impaired locomotor recovery. | [29] |
| Astrocytes | GFAP-Tk mice. Tk-GCV system | Transgenic mice on C57BL/6J background (females) | 8–16 weeks | from 0 to 7 dpi | 14–56 dpi | Lesion expansion. Unchanged axonal regrowth and extended axonal dieback. Maintained CSPGs production. Ablation of astrocytes eliminates the beneficial effect of conditioning lesions and neurotrophic factors on axonal regrowth. | [21] |
| loxP-DTR mice. DTA-DTR system | loxP-DTR mice on C57BL/6 background (females) | 8–16 weeks | from 35 to 45 dpi | 70 dpi | Tissue degeneration and lesion expansion. Axon regrowth was not enhanced. | ||
| Astrocytes | lentivirus-mediated Tk/GCV system | C57BL/6 mice (females) | 8–12 weeks | from 1 to 8 dpi | 14, 28, and 42 dpi | Enhanced infiltration of proinflammatory cells and tissue damage. Augmented neuronal death. Impaired locomotor recovery | [30] |
| Microglia | PLX5622 Feeding | C57BL/6N mice | 8–10 weeks | from 21 d before injury to 35 dpi; from 0 to 35 dpi; from 21 d before injury to 0 dpi | 1, 4, 7, 14 and 35 dpi | Disorganization of the glial scar. Enhanced infiltration of immune cells. Reduced oligodendrocyte and neuronal survival. Impaired locomotor recovery | [31] |
| Microglia | PLX3397 Intragastric | C57BL/6 mice (females) | 8–10 weeks | 14 days pre-injury until 7 or 14 dpi | 7 and 14 dpi | Disorganization of the glial scar. Reduced astrocytic proliferation. Increased proinflammatory factors. Reduced neuronal survival. Impaired locomotor recovery. | [32] |
| Microglia | PLX3397 Feeding | C57BL/6 mice (females) | 6 weeks | from 7 days before injury to 28 dpi | 7, 14, 28, 30, and 56 dpi | Disorganization of the glial scar. Acute expansion of the lesion. Increased axon dieback. Impaired locomotor recovery | [33] |
| NG2 cells | NG2-Tk mice. Tk-GCV system | Transgenic mice on C57BL/6J background | 12 weeks | from 0 dpi to 7 or 14 dpi | 7, 14, and 21 dpi | Edema and swelling. Reduced intra-lesion laminin. Disorganization of astrocytes and the glial scar. Increased macrophage infiltration. Enhanced axon regrowth. Impaired locomotor recovery. | [34] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez-Gianmarco, L.; Kukley, M. Understanding the Role of the Glial Scar through the Depletion of Glial Cells after Spinal Cord Injury. Cells 2023, 12, 1842. https://doi.org/10.3390/cells12141842
Perez-Gianmarco L, Kukley M. Understanding the Role of the Glial Scar through the Depletion of Glial Cells after Spinal Cord Injury. Cells. 2023; 12(14):1842. https://doi.org/10.3390/cells12141842
Chicago/Turabian StylePerez-Gianmarco, Lucila, and Maria Kukley. 2023. "Understanding the Role of the Glial Scar through the Depletion of Glial Cells after Spinal Cord Injury" Cells 12, no. 14: 1842. https://doi.org/10.3390/cells12141842
APA StylePerez-Gianmarco, L., & Kukley, M. (2023). Understanding the Role of the Glial Scar through the Depletion of Glial Cells after Spinal Cord Injury. Cells, 12(14), 1842. https://doi.org/10.3390/cells12141842