
Neurochemical Basis of Inter-Organ Crosstalk in Health and Obesity: Focus on the Hypothalamus and the Brainstem


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Hypothalamic and Brainstem Nuclei Involved in Appetite Control and Energy Balance
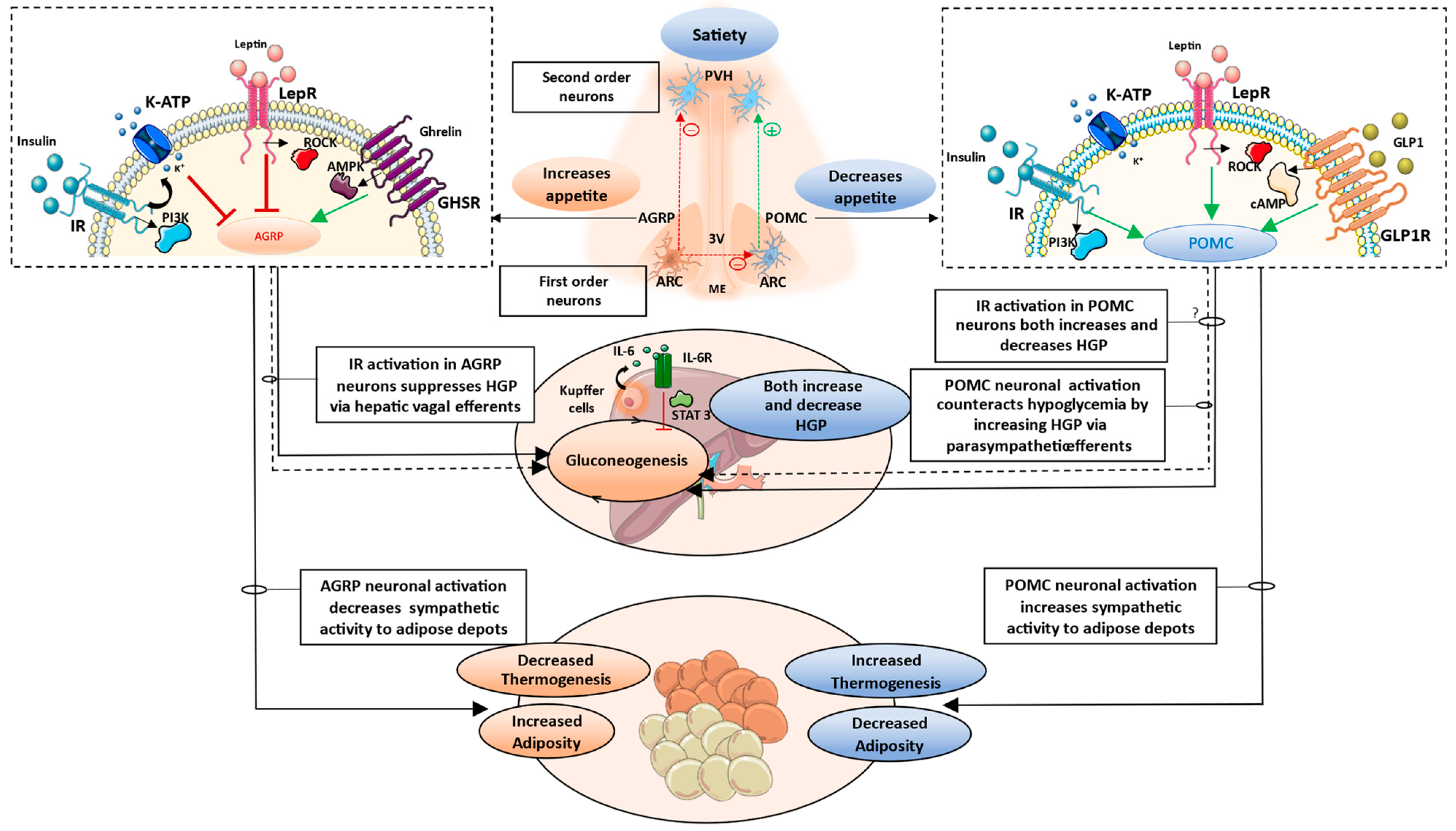
2.1. Hypothalamus
2.1.1. Arcuate Nucleus (ARC)
2.1.2. Paraventricular Nucleus (PVH)
2.1.3. Ventromedial Nucleus of the Hypothalamus (VMH)
2.1.4. Dorsomedial Hypothalamus (DMH)
2.2. Brainstem
Dorsal Vagal Complex (DVC)
3. Crosstalk between Hypothalamic and Brainstem Nuclei with Metabolic Organs to Regulate Energy and Glucose Homeostasis
3.1. Brain-Pancreas Axis and the Role of Central Insulin Signaling
3.2. Brain–Liver Axis
3.3. Brain–Adipose Tissue Axis
3.4. Gut–Brain Axis and the Role of Incretins
4. Clinical Implications
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahima, R.S.; Antwi, D.A. Brain Regulation of Appetite and Satiety. Endocrinol. Metab. Clin. N. Am. 2008, 37, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Hetherington, A.W.; Ranson, S.W. Hypothalamic Lesions and Adiposity in the Rat. Anat. Rec. 1940, 78, 149–172. [Google Scholar] [CrossRef]
- Wess, J.; Nakajima, K.; Jain, S. Novel Designer Receptors to Probe GPCR Signaling and Physiology. Trends Pharmacol. Sci. 2013, 34, 385–392. [Google Scholar] [CrossRef]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic Studies of Body Mass Index Yield New Insights for Obesity Biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef]
- Matikainen-Ankney, B.A.; Kravitz, A.V. Persistent Effects of Obesity: A Neuroplasticity Hypothesis. Ann. N. Y. Acad. Sci. 2018, 1428, 221. [Google Scholar] [CrossRef] [PubMed]
- Linehan, V.; Fang, L.Z.; Parsons, M.P.; Hirasawa, M. High-Fat Diet Induces Time-Dependent Synaptic Plasticity of the Lateral Hypothalamus. Mol. Metab. 2020, 36, 100977. [Google Scholar] [CrossRef]
- Wang, X.; Li, H. Chronic High-Fat Diet Induces Overeating and Impairs Synaptic Transmission in Feeding-Related Brain Regions. Front. Mol. Neurosci. 2022, 15, 1019446. [Google Scholar] [CrossRef]
- Parton, L.E.; Ye, C.P.; Coppari, R.; Enriori, P.J.; Choi, B.; Zhang, C.Y.; Xu, C.; Vianna, C.R.; Balthasar, N.; Lee, C.E.; et al. Glucose Sensing by POMC Neurons Regulates Glucose Homeostasis and Is Impaired in Obesity. Nature 2007, 449, 228–232. [Google Scholar] [CrossRef]
- Belgardt, B.F.; Okamura, T.; Brüning, J.C. Hormone and Glucose Signalling in POMC and AgRP Neurons. J. Physiol. 2009, 587, 5305–5314. [Google Scholar] [CrossRef]
- Roh, E.; Song, D.K.; Kim, M.S. Emerging Role of the Brain in the Homeostatic Regulation of Energy and Glucose Metabolism. Exp. Mol. Med. 2016, 48, e216. [Google Scholar] [CrossRef]
- Yoon, N.A.; Diano, S. Hypothalamic Glucose-Sensing Mechanisms. Diabetologia 2021, 64, 985–993. [Google Scholar] [CrossRef]
- Yang, Y.; Atasoy, D.; Su, H.H.; Sternson, S.M. Hunger States Switch a Flip-Flop Memory Circuit via a Synaptic AMPK-Dependent Positive Feedback Loop. Cell 2011, 146, 992–1003. [Google Scholar] [CrossRef]
- Millington, G.W.M. The Role of Proopiomelanocortin (POMC) Neurones in Feeding Behaviour. Nutr. Metab. 2007, 4, 1–16. [Google Scholar] [CrossRef]
- Rau, A.R.; Hentges, S.T. GABAergic Inputs to POMC Neurons Originating from the Dorsomedial Hypothalamus Are Regulated by Energy State. J. Neurosci. 2019, 39, 6449–6459. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, D.; Morgan, D.G.A.; Meeran, K.; Edwards, C.M.B.; Turton, M.D.; Choi, S.J.; Heath, M.M.; Gunn, I.; Taylor, G.M.; Howard, J.K.; et al. Neuropeptide Y Induced Feeding in the Rat Is Mediated by a Novel Receptor. Endocrinology 1997, 138, 196–202. [Google Scholar] [CrossRef]
- Nijenhuis, W.A.J.; Oosterom, J.; Adan, R.A.H. AgRP(83-132) Acts as an Inverse Agonist on the Human-Melanocortin-4 Receptor. Mol. Endocrinol. 2001, 15, 164–171. [Google Scholar] [CrossRef]
- Luquet, S.; Perez, F.A.; Hnasko, T.S.; Palmiter, R.D. NPY/AgRP Neurons Are Essential for Feeding in Adult Mice but Can Be Ablated in Neonates. Science 2005, 310, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Krashes, M.J.; Koda, S.; Ye, C.P.; Rogan, S.C.; Adams, A.C.; Cusher, D.S.; Maratos-Flier, E.; Roth, B.L.; Lowell, B.B. Rapid, Reversible Activation of AgRP Neurons Drives Feeding Behavior in Mice. J. Clin. Investig. 2011, 121, 1424–1428. [Google Scholar] [CrossRef]
- Joly-Amado, A.; Denis, R.G.P.; Castel, J.; Lacombe, A.; Cansell, C.; Rouch, C.; Kassis, N.; Dairou, J.; Cani, P.D.; Ventura-Clapier, R.; et al. Hypothalamic AgRP-Neurons Control Peripheral Substrate Utilization and Nutrient Partitioning. EMBO J. 2012, 31, 4276–4288. [Google Scholar] [CrossRef]
- Nakajima, K.I.; Cui, Z.; Li, C.; Meister, J.; Cui, Y.; Fu, O.; Smith, A.S.; Jain, S.; Lowell, B.B.; Krashes, M.J.; et al. Gs-Coupled GPCR Signalling in AgRP Neurons Triggers Sustained Increase in Food Intake. Nat. Commun. 2016, 7, 10268. [Google Scholar] [CrossRef] [PubMed]
- Krashes, M.J.; Shah, B.P.; Koda, S.; Lowell, B.B. Rapid versus Delayed Stimulation of Feeding by the Endogenously Released AgRP Neuron Mediators GABA, NPY, and AgRP. Cell Metab. 2013, 18, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Lee, N.J.; Ip, C.K.; Enriquez, R.; Tasan, R.; Zhang, L.; Herzog, H. NPY Derived from AGRP Neurons Controls Feeding via Y1 and Energy Expenditure and Food Foraging Behaviour via Y2 Signalling. Mol. Metab. 2022, 59, 101455. [Google Scholar] [CrossRef] [PubMed]
- Luo, N.; Marcelin, G.; Liu, S.M.; Schwartz, G.; Chua, S. Neuropeptide Y and Agouti-Related Peptide Mediate Complementary Functions of Hyperphagia and Reduced Energy Expenditure in Leptin Receptor Deficiency. Endocrinology 2011, 152, 883–889. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tong, Q.; Ye, C.P.; Jones, J.E.; Elmquist, J.K.; Lowell, B.B. Synaptic Release of GABA by AgRP Neurons Is Required for Normal Regulation of Energy Balance. Nat. Neurosci. 2008, 11, 998–1000. [Google Scholar] [CrossRef]
- Deem, J.D.; Faber, C.L.; Morton, G.J. AgRP Neurons: Regulators of Feeding, Energy Expenditure, and Behavior. FEBS J 2022, 289, 2362–2381. [Google Scholar] [CrossRef]
- D’agostino, G.; Diano, S. Alpha-Melanocyte Stimulating Hormone: Production and Degradation. J. Mol. Med. 2010, 88, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Greenman, Y.; Kuperman, Y.; Drori, Y.; Asa, S.L.; Navon, I.; Forkosh, O.; Gil, S.; Stern, N.; Chen, A. Postnatal Ablation of POMC Neurons Induces an Obese Phenotype Characterized by Decreased Food Intake and Enhanced Anxiety-like Behavior. Mol. Endocrinol. 2013, 27, 1091–1102. [Google Scholar] [CrossRef]
- Zhan, C.; Zhou, J.; Feng, Q.; Zhang, J.-e.; Lin, S.; Bao, J.; Wu, P.; Luo, M. Acute and Long-Term Suppression of Feeding Behavior by POMC Neurons in the Brainstem and Hypothalamus, Respectively. J. Neurosci. 2013, 33, 3624–3632. [Google Scholar] [CrossRef]
- Toda, C.; Santoro, A.; Kim, J.D.; Diano, S. POMC Neurons: From Birth to Death. Annu. Rev. Physiol. 2017, 79, 209–236. [Google Scholar] [CrossRef]
- Quarta, C.; Claret, M.; Zeltser, L.M.; Williams, K.W.; Yeo, G.S.H.; Tschöp, M.H.; Diano, S.; Brüning, J.C.; Cota, D. POMC Neuronal Heterogeneity in Energy Balance and beyond: An Integrated View. Nat. Metab. 2021, 3, 299–308. [Google Scholar] [CrossRef]
- Saucisse, N.; Mazier, W.; Simon, V.; Binder, E.; Catania, C.; Bellocchio, L.; Romanov, R.A.; Léon, S.; Matias, I.; Zizzari, P.; et al. Functional Heterogeneity of POMC Neurons Relies on MTORC1 Signaling. Cell Rep. 2021, 37, 109800. [Google Scholar] [CrossRef]
- Wu, Q.; Howell, M.P.; Cowley, M.A.; Palmiter, R.D. Starvation after AgRP Neuron Ablation Is Independent of Melanocortin Signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 2687–2692. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.R.; Chen, H.; Zhou, J.J.; Pradhan, G.; Sun, Y.; Pan, H.L.; Li, D.P. Ghrelin Receptors Mediate Ghrelin-Induced Excitation of Agouti-Related Protein/Neuropeptide Y but Not pro-Opiomelanocortin Neurons. J. Neurochem. 2017, 142, 512–520. [Google Scholar] [CrossRef]
- Rau, A.R.; Hentges, S.T. The Relevance of AgRP Neuron-Derived GABA Inputs to POMC Neurons Differs for Spontaneous and Evoked Release. J. Neurosci. 2017, 37, 7362–7372. [Google Scholar] [CrossRef]
- Dutia, R.; Meece, K.; Dighe, S.; Kim, A.J.; Wardlaw, S.L. β-Endorphin Antagonizes the Effects of α-MSH on Food Intake and Body Weight. Endocrinology 2012, 153, 4246–4255. [Google Scholar] [CrossRef]
- Kelly, M.J.; Loose, M.D.; Ronnekleiv, O.K. Opioids Hyperpolarize Beta-Endorphin Neurons via Mu-Receptor Activation of a Potassium Conductance. Neuroendocrinology 1990, 52, 268–275. [Google Scholar] [CrossRef]
- Koch, M.; Varela, L.; Kim, J.G.; Kim, J.D.; Hernández-Nuño, F.; Simonds, S.E.; Castorena, C.M.; Vianna, C.R.; Elmquist, J.K.; Morozov, Y.M.; et al. Hypothalamic POMC Neurons Promote Cannabinoid-Induced Feeding. Nature 2015, 519, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Tolentino, L.; Iqbal, A.; Rahman, S.; Lutfy, K. The Role of Beta-Endorphin in Food Deprivation-Mediated Increases in Food Intake and Binge-Eating. Brain Sciences 2023, 13, 212. [Google Scholar] [CrossRef]
- Panigrahi, S.K.; Meece, K.; Wardlaw, S.L. Effects of Naltrexone on Energy Balance and Hypothalamic Melanocortin Peptides in Male Mice Fed a High-Fat Diet. J. Endocr. Soc. 2019, 3, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.J.; Panigrahi, S.K.; Meece, K.; Atalayer, D.; Smiley, R.; Wardlaw, S.L. Effects of Opioid Antagonism on Cerebrospinal Fluid Melanocortin Peptides and Cortisol Levels in Humans. J. Endocr. Soc. 2017, 1, 1235–1246. [Google Scholar] [CrossRef]
- Elias, C.F.; Kelly, J.F.; Lee, C.E.; Ahima, R.S.; Drucker, D.J.; Saper, C.B.; Elmquist, J.K. Chemical Characterization of Leptin-Activated Neurons in the Rat Brain. J. Comp. Neurol. 2000, 423, 261–281. [Google Scholar] [CrossRef]
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdán, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin Activates Anorexigenic POMC Neurons through a Neural Network in the Arcuate Nucleus. Nature 2001, 411, 480–484. [Google Scholar] [CrossRef]
- Korner, J.; Savontaus, E.; Chua, S.C.; Leibel, R.L.; Wardlaw, S.L. Leptin Regulation of Agrp and Npy MRNA in the Rat Hypothalamus. J. Neuroendocrinol. 2001, 13, 959–966. [Google Scholar] [CrossRef]
- Takahashi, K.A.; Cone, R.D. Fasting Induces a Large, Leptin-Dependent Increase in the Intrinsic Action Potential Frequency of Orexigenic Arcuate Nucleus Neuropeptide Y/Agouti-Related Protein Neurons. Endocrinology 2005, 146, 1043–1047. [Google Scholar] [CrossRef]
- Baver, S.B.; Hope, K.; Guyot, S.; Bjørbaek, C.; Kaczorowski, C.; O’Connell, K.M.S. Leptin Modulates the Intrinsic Excitability of AgRP/NPY Neurons in the Arcuate Nucleus of the Hypothalamus. J. Neurosci. 2014, 34, 5486–5496. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Kong, D.; Byun, K.H.; Ye, C.; Koda, S.; Lee, D.H.; Oh, B.C.; Lee, S.W.; Lee, B.; Zabolotny, J.M.; et al. Rho-Kinase Regulates Energy Balance by Targeting Hypothalamic Leptin Receptor Signaling. Nat. Neurosci. 2012, 15, 1391–1398. [Google Scholar] [CrossRef]
- Huang, H.; Lee, S.H.; Ye, C.; Lima, I.S.; Oh, B.C.; Lowell, B.B.; Zabolotny, J.M.; Kim, Y.B. ROCK1 in AgRP Neurons Regulates Energy Expenditure and Locomotor Activity in Male Mice. Endocrinology 2013, 154, 3660–3670. [Google Scholar] [CrossRef] [PubMed]
- Koch, L.; Wunderlich, F.T.; Seibler, J.; Könner, A.C.; Hampel, B.; Irlenbusch, S.; Brabant, G.; Kahn, C.R.; Schwenk, F.; Brüning, J.C. Central Insulin Action Regulates Peripheral Glucose and Fat Metabolism in Mice. J. Clin. Investig. 2008, 118, 2132–2147. [Google Scholar] [CrossRef]
- Dodd, G.T.; Kim, S.J.; Méquinion, M.; Xirouchaki, C.E.; Brüning, J.C.; Andrews, Z.B.; Tiganis, T. Insulin Signaling in AgRP Neurons Regulates Meal Size to Limit Glucose Excursions and Insulin Resistance. Sci. Adv. 2021, 7, eabf4100. [Google Scholar] [CrossRef] [PubMed]
- Balthasar, N.; Dalgaard, L.T.; Lee, C.E.; Yu, J.; Funahashi, H.; Williams, T.; Ferreira, M.; Tang, V.; McGovern, R.A.; Kenny, C.D.; et al. Divergence of Melanocortin Pathways in the Control of Food Intake and Energy Expenditure. Cell 2005, 123, 493–505. [Google Scholar] [CrossRef]
- Könner, A.C.; Janoschek, R.; Plum, L.; Jordan, S.D.; Rother, E.; Ma, X.; Xu, C.; Enriori, P.; Hampel, B.; Barsh, G.S.; et al. Insulin Action in AgRP-Expressing Neurons Is Required for Suppression of Hepatic Glucose Production. Cell Metab. 2007, 5, 438–449. [Google Scholar] [CrossRef]
- Andrews, Z.B.; Liu, Z.W.; Walllingford, N.; Erion, D.M.; Borok, E.; Friedman, J.M.; Tschöp, M.H.; Shanabrough, M.; Cline, G.; Shulman, G.I.; et al. UCP2 Mediates Ghrelin’s Action on NPY/AgRP Neurons by Lowering Free Radicals. Nature 2008, 454, 846–851. [Google Scholar] [CrossRef]
- Guan, X.; Shi, X.; Li, X.; Chang, B.; Wang, Y.; Li, D.; Chan, L. GLP-2 Receptor in POMC Neurons Suppresses Feeding Behavior and Gastric Motility. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E853. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Alhadeff, A.L.; Betley, J.N. Nutritive, Post-Ingestive Signals Are the Primary Regulators of AgRP Neuron Activity. Cell Rep. 2017, 21, 2724–2736. [Google Scholar] [CrossRef] [PubMed]
- Lutz, T.A.; Coester, B.; Whiting, L.; Dunn-Meynell, A.A.; Boyle, C.N.; Bouret, S.G.; Levin, B.E.; Le Foll, C. Amylin Selectively Signals onto POMC Neurons in the Arcuate Nucleus of the Hypothalamus. Diabetes 2018, 67, 805–817. [Google Scholar] [CrossRef]
- Gouveia, A.; de Oliveira Beleza, R.; Steculorum, S.M. AgRP Neuronal Activity across Feeding-Related Behaviours. Eur. J. Neurosci. 2021, 54, 7458–7475. [Google Scholar] [CrossRef] [PubMed]
- Üner, A.G.; Keçik, O.; Quaresma, P.G.F.; De Araujo, T.M.; Lee, H.; Li, W.; Kim, H.J.; Chung, M.; Bjørbæk, C.; Kim, Y.B. Role of POMC and AgRP Neuronal Activities on Glycaemia in Mice. Sci. Rep. 2019, 9, 13068. [Google Scholar] [CrossRef] [PubMed]
- Iwen, K.A.; Oelkrug, R.; Brabant, G. Effects of Thyroid Hormones on Thermogenesis and Energy Partitioning. J. Mol. Endocrinol. 2018, 60, R157–R170. [Google Scholar] [CrossRef]
- Fekete, C.; Kelly, J.; Mihály, E.; Sarkar, S.; Rand, W.M.; Légrádi, G.; Emerson, C.H.; Lechan, R.M. Neuropeptide Y Has a Central Inhibitory Action on the Hypothalamic-Pituitary-Thyroid Axis. Endocrinology 2001, 142, 2606–2613. [Google Scholar] [CrossRef]
- Kim, M.S.; Small, C.J.; Stanley, S.A.; Morgan, D.G.A.; Seal, L.J.; Kong, W.M.; Edwards, C.M.B.; Abusnana, S.; Sunter, D.; Ghatei, M.A.; et al. The Central Melanocortin System Affects the Hypothalamo-Pituitary Thyroid Axis and May Mediate the Effect of Leptin. J. Clin. Investig. 2000, 105, 1005–1011. [Google Scholar] [CrossRef]
- Fekete, C.; Sarkar, S.; Rand, W.M.; Harney, J.W.; Emerson, C.H.; Bianco, A.C.; Lechan, R.M. Agouti-Related Protein (AGRP) Has a Central Inhibitory Action on the Hypothalamic-Pituitary-Thyroid (HPT) Axis; Comparisons between the Effect of AGRP and Neuropeptide Y on Energy Homeostasis and the HPT Axis. Endocrinology 2002, 143, 3846–3853. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martin, N.M.; Smith, K.L.; Bloom, S.R.; Small, C.J. Interactions between the Melanocortin System and the Hypothalamo–Pituitary–Thyroid Axis. Peptides 2006, 27, 333–339. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, C.; He, Y.; Xu, P.; Yang, Y.; Saito, K.; Xia, Y.; Yan, X.; Hinton, A.; Yan, C.; Ding, H.; et al. TAp63 Contributes to Sexual Dimorphism in POMC Neuron Functions and Energy Homeostasis. Nat. Commun. 2018, 9, 1544. [Google Scholar] [CrossRef]
- Xu, Y.; Nedungadi, T.P.; Zhu, L.; Sobhani, N.; Irani, B.G.; Davis, K.E.; Zhang, X.; Zou, F.; Gent, L.M.; Hahner, L.D.; et al. Distinct Hypothalamic Neurons Mediate Estrogenic Effects on Energy Homeostasis and Reproduction. Cell Metab. 2011, 14, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; van der Klaauw, A.A.; Zhu, L.; Cacciottolo, T.M.; He, Y.; Stadler, L.K.J.; Wang, C.; Xu, P.; Saito, K.; Hinton, A.; et al. Steroid Receptor Coactivator-1 Modulates the Function of Pomc Neurons and Energy Homeostasis. Nat. Commun. 2019, 10, 1718. [Google Scholar] [CrossRef]
- Xu, A.W.; Ste-Marie, L.; Kaelin, C.B.; Barsh, G.S. Inactivation of Signal Transducer and Activator of Transcription 3 in Proopiomelanocortin (Pomc) Neurons Causes Decreased Pomc Expression, Mild Obesity, and Defects in Compensatory Refeeding. Endocrinology 2007, 148, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bean, J.C.; Liu, H.; He, Y.; Yang, Y.; Cai, X.; Yu, K.; Pei, Z.; Liu, H.; Tu, L.; et al. SK3 in POMC Neurons Plays a Sexually Dimorphic Role in Energy and Glucose Homeostasis. Cell Biosci. 2022, 12, 170. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; He, Y.; Bean, J.C.; Yang, Y.; Liu, H.; Yu, M.; Yu, K.; Hyseni, I.; Cai, X.; Liu, H.; et al. Gabra5 Plays a Sexually Dimorphic Role in POMC Neuron Activity and Glucose Balance. Front. Endocrinol. 2022, 13, 889122. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, S.; Du, D.; Li, Q.; Xie, K.; Chen, L.; Feng, X.; Wu, X.; Sun, Z.; Zhou, J.; et al. TLR4 in POMC Neurons Regulates Thermogenesis in a Sex-Dependent Manner. J. Lipid Res. 2023, 64, 100368. [Google Scholar] [CrossRef]
- Nuzzaci, D.; Laderrière, A.; Lemoine, A.; Nédélec, E.; Pénicaud, L.; Rigault, C.; Benani, A. Plasticity of the Melanocortin System: Determinants and Possible Consequences on Food Intake. Front. Endocrinol. 2015, 6, 143. [Google Scholar] [CrossRef]
- Benani, A.; Hryhorczuk, C.; Gouazé, A.; Fioramonti, X.; Brenachot, X.; Guissard, C.; Krezymon, A.; Duparc, T.; Colom, A.; Nédélec, E.; et al. Food Intake Adaptation to Dietary Fat Involves PSA-Dependent Rewiring of the Arcuate Melanocortin System in Mice. J. Neurosci. 2012, 32, 11970–11979. [Google Scholar] [CrossRef]
- Liu, T.; Kong, D.; Shah, B.P.; Ye, C.; Koda, S.; Saunders, A.; Ding, J.B.; Yang, Z.; Sabatini, B.L.; Lowell, B.B. Fasting Activation of AgRP Neurons Requires NMDA Receptors and Involves Spinogenesis and Increased Excitatory Tone. Neuron 2012, 73, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Nuzzaci, D.; Cansell, C.; Liénard, F.; Nédélec, E.; Ben Fradj, S.; Castel, J.; Foppen, E.; Denis, R.; Grouselle, D.; Laderrière, A.; et al. Postprandial Hyperglycemia Stimulates Neuroglial Plasticity in Hypothalamic POMC Neurons after a Balanced Meal. Cell Rep. 2020, 30, 3067–3078.e5. [Google Scholar] [CrossRef] [PubMed]
- McNay, D.E.G.; Briançon, N.; Kokoeva, M.V.; Maratos-Flier, E.; Flier, J.S. Remodeling of the Arcuate Nucleus Energy-Balance Circuit Is Inhibited in Obese Mice. J. Clin. Investig. 2012, 122, 142. [Google Scholar] [CrossRef]
- Horvath, T.L.; Sarman, B.; García-Cáceres, C.; Enriori, P.J.; Sotonyi, P.; Shanabrough, M.; Borok, E.; Argente, J.; Chowen, J.A.; Perez-Tilve, D.; et al. Synaptic Input Organization of the Melanocortin System Predicts Diet-Induced Hypothalamic Reactive Gliosis and Obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 14875–14880. [Google Scholar] [CrossRef]
- Lee, D.A.; Bedont, J.L.; Pak, T.; Wang, H.; Song, J.; Miranda-Angulo, A.; Takiar, V.; Charubhumi, V.; Balordi, F.; Takebayashi, H.; et al. Tanycytes of the Hypothalamic Median Eminence Form a Diet-Responsive Neurogenic Niche. Nat. Neurosci. 2012, 15, 700–702. [Google Scholar] [CrossRef]
- Lee, D.A.; Yoo, S.; Pak, T.; Salvatierra, J.; Velarde, E.; Aja, S.; Blackshaw, S. Dietary and Sex-Specific Factors Regulate Hypothalamic Neurogenesis in Young Adult Mice. Front. Neurosci. 2014, 8, 157. [Google Scholar] [CrossRef]
- Jacobowitz, D.M.; O’Donohue, T.L. α-Melanocyte Stimulating Hormone: Immunohistochemical Identification and Mapping in Neurons of Rat Brain. Proc. Natl. Acad. Sci. USA 1978, 75, 6300–6304. [Google Scholar] [CrossRef] [PubMed]
- Mountjoy, K.G.; Mortrud, M.T.; Low, M.J.; Simerly, R.B.; Cone, R.D. Localization of the Melanocortin-4 Receptor (MC4-R) in Neuroendocrine and Autonomic Control Circuits in the Brain. Mol. Endocrinol. 1994, 8, 1298–1308. [Google Scholar] [CrossRef]
- Lu, D.; Willard, D.; Patel, I.R.; Kadwell, S.; Overton, L.; Kost, T.; Luther, M.; Chen, W.; Woychik, R.P.; Wilkison, W.O.; et al. Agouti Protein Is an Antagonist of the Melanocyte-Stimulating-Hormone Receptor. Nature 1994, 371, 799–802. [Google Scholar] [CrossRef]
- Ollmann, M.M.; Wilson, B.D.; Yang, Y.K.; Kerns, J.A.; Chen, Y.; Gantz, I.; Barsh, G.S. Antagonism of Central Melanocortin Receptors in Vitro and in Vivo by Agouti-Related Protein. Science 1997, 278, 135–138. [Google Scholar] [CrossRef]
- Michaud, J.L.; Rosenquist, T.; May, N.R.; Fan, C.M. Development of Neuroendocrine Lineages Requires the BHLH-PAS Transcription Factor SIM1. Genes Dev. 1998, 12, 3264–3275. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.L.; Boucher, F.; Melnyk, A.; Gauthier, F.; Goshu, E.; Lévy, E.; Mitchell, G.A.; Himms-Hagen, J.; Fan, C.M. Sim1 Haploinsufficiency Causes Hyperphagia, Obesity and Reduction of the Paraventricular Nucleus of the Hypothalamus. Hum. Mol. Genet. 2001, 10, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Tolson, K.P.; Gemelli, T.; Meyer, D.; Yazdani, U.; Kozlitina, J.; Zinn, A.R. Inducible Neuronal Inactivation of Sim1 in Adult Mice Causes Hyperphagic Obesity. Endocrinology 2014, 155, 2436–2444. [Google Scholar] [CrossRef]
- Hinney, A.; Schmidt, A.; Nottebom, K.; Heibült, O.; Becker, I.; Ziegler, A.; Gerber, G.; Sina, M.; Görg, T.; Mayer, H.; et al. Several Mutations in the Melanocortin-4 Receptor Gene Including a Nonsense and a Frameshift Mutation Associated with Dominantly Inherited Obesity in Humans. J. Clin. Endocrinol. Metab. 1999, 84, 1483–1486. [Google Scholar] [CrossRef] [PubMed]
- Vaisse, C.; Clement, K.; Durand, E.; Hercberg, S.; Guy-Grand, B.; Froguel, P. Melanocortin-4 Receptor Mutations Are a Frequent and Heterogeneous Cause of Morbid Obesity. J. Clin. Investig. 2000, 106, 253–262. [Google Scholar] [CrossRef]
- Farooqi, I.S.; Keogh, J.M.; Yeo, G.S.H.; Lank, E.J.; Cheetham, T.; O’Rahilly, S. Clinical Spectrum of Obesity and Mutations in the Melanocortin 4 Receptor Gene. N. Engl. J. Med. 2003, 348, 1085–1095. [Google Scholar] [CrossRef]
- Loos, R.J.F.; Lindgren, C.M.; Li, S.; Wheeler, E.; Hua Zhao, J.; Prokopenko, I.; Inouye, M.; Freathy, R.M.; Attwood, A.P.; Beckmann, J.S.; et al. Common Variants near MC4R Are Associated with Fat Mass, Weight and Risk of Obesity. Nat. Genet. 2008, 40, 768–775. [Google Scholar] [CrossRef]
- Li, Y.Q.; Shrestha, Y.; Pandey, M.; Chen, M.; Kablan, A.; Gavrilova, O.; Offermanns, S.; Weinstein, L.S. Gq/11α and Gsα Mediate Distinct Physiological Responses to Central Melanocortins. J. Clin. Investig. 2016, 126, 40–49. [Google Scholar] [CrossRef]
- Garfield, A.S.; Li, C.; Madara, J.C.; Shah, B.P.; Webber, E.; Steger, J.S.; Campbell, J.N.; Gavrilova, O.; Lee, C.E.; Olson, D.P.; et al. A Neural Basis for Melanocortin-4 Receptor Regulated Appetite. Nat. Neurosci. 2015, 18, 863. [Google Scholar] [CrossRef]
- Matsumura, S.; Miyakita, M.; Miyamori, H.; Kyo, S.; Shima, D.; Yokokawa, T.; Ishikawa, F.; Sasaki, T.; Jinno, T.; Tanaka, J.; et al. Stimulation of GSsignaling in MC4R Cells by DREADD Increases Energy Expenditure, Suppresses Food Intake, and Increases Locomotor Activity in Mice. Am. J. Physiol. Endocrinol. Metab. 2022, 322, E436–E445. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.Y.; Muniyappa, R.; Abel, B.S.; Mullins, K.P.; Staker, P.; Brychta, R.J.; Zhao, X.; Ring, M.; Psota, T.L.; Cone, R.D.; et al. RM-493, a Melanocortin-4 Receptor (MC4R) Agonist, Increases Resting Energy Expenditure in Obese Individuals. J. Clin. Endocrinol. Metab. 2015, 100, 1639–1645. [Google Scholar] [CrossRef]
- Wang, X.; Cui, X.; Li, Y.; Li, F.; Li, Y.; Dai, J.; Hu, H.; Wang, X.; Sun, J.; Yang, Y.; et al. MC4R Deficiency Causes Dysregulation of Postsynaptic Excitatory Synaptic Transmission as a Crucial Culprit for Obesity. Diabetes 2022, 71, 2331–2343. [Google Scholar] [CrossRef]
- Podyma, B.; Sun, H.; Wilson, E.A.; Carlson, B.; Pritikin, E.; Gavrilova, O.; Weinstein, L.S.; Chen, M. The Stimulatory G Protein Gsα Is Required in Melanocortin 4 Receptor–Expressing Cells for Normal Energy Balance, Thermogenesis, and Glucose Metabolism. J. Biol. Chem. 2018, 293, 10993. [Google Scholar] [CrossRef]
- Kuo, J.J.; Silva, A.A.; Hall, J.E. Hypothalamic Melanocortin Receptors and Chronic Regulation of Arterial Pressure and Renal Function. Hypertension 2003, 41, 768–774. [Google Scholar] [CrossRef]
- Sohn, J.W.; Harris, L.E.; Berglund, E.D.; Liu, T.; Vong, L.; Lowell, B.B.; Balthasar, N.; Williams, K.W.; Elmquist, J.K. Melanocortin 4 Receptors Reciprocally Regulate Sympathetic and Parasympathetic Preganglionic Neurons. Cell 2013, 152, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, M.; Kawabe, K.; Sapru, H.N. Activation of Melanocortin Receptors in the Intermediolateral Cell Column of the Upper Thoracic Cord Elicits Tachycardia in the Rat. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H885. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.D.; Liu, T.; Kong, X.; Sohn, J.W.; Vong, L.; Deng, Z.; Lee, C.E.; Lee, S.; Williams, K.W.; Olson, D.P.; et al. Melanocortin 4 Receptors in Autonomic Neurons Regulate Thermogenesis and Glycemia. Nat. Neurosci. 2014, 17, 911–913. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Madara, J.C.; Steger, J.S.; Krashes, M.J.; Balthasar, N.; Campbell, J.N.; Resch, J.M.; Conley, N.J.; Garfield, A.S.; Lowell, B.B. The Paraventricular Hypothalamus Regulates Satiety and Prevents Obesity via Two Genetically Distinct Circuits. Neuron 2019, 102, 653–667. [Google Scholar] [CrossRef]
- An, J.J.; Liao, G.Y.; Kinney, C.E.; Sahibzada, N.; Xu, B. Discrete BDNF Neurons in the Paraventricular Hypothalamus Control Feeding and Energy Expenditure. Cell Metab. 2015, 22, 175–188. [Google Scholar] [CrossRef]
- Li, C.; Navarrete, J.; Liang-Guallpa, J.; Lu, C.; Funderburk, S.C.; Chang, R.B.; Liberles, S.D.; Olson, D.P.; Krashes, M.J. Defined Paraventricular Hypothalamic Populations Exhibit Differential Responses to Food Contingent on Caloric State. Cell Metab. 2019, 29, 681–694.e5. [Google Scholar] [CrossRef]
- Varela, L.; Horvath, T.L. Parallel Paths in PVH Control of Feeding. Neuron 2019, 102, 514–516. [Google Scholar] [CrossRef] [PubMed]
- An, J.J.; Kinney, C.E.; Tan, J.W.; Liao, G.Y.; Kremer, E.J.; Xu, B. TrkB-Expressing Paraventricular Hypothalamic Neurons Suppress Appetite through Multiple Neurocircuits. Nat. Commun. 2020, 11, 1729. [Google Scholar] [CrossRef]
- Krashes, M.J.; Lowell, B.B.; Garfield, A.S. Melanocortin-4 Receptor–Regulated Energy Homeostasis. Nat. Neurosci. 2016, 19, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Baldini, G.; Phelan, K.D. The Melanocortin Pathway and Control of Appetite-Progress and Therapeutic Implications. J. Endocrinol. 2019, 241, R1–R33. [Google Scholar] [CrossRef] [PubMed]
- King, B.M. The Rise, Fall, and Resurrection of the Ventromedial Hypothalamus in the Regulation of Feeding Behavior and Body Weight. Physiol. Behav. 2006, 87, 221–244. [Google Scholar] [CrossRef]
- Becker, E.E.; Kissileff, H.R. Inhibitory Controls of Feeding by the Ventromedial Hypothalamus. Am. J. Physiol. 1974, 226, 383–396. [Google Scholar] [CrossRef]
- Maes, H. Time Course of Feeding Induced by Pentobarbital-Injections into the Rat’s VMH. Physiol. Behav. 1980, 24, 1107–1114. [Google Scholar] [CrossRef]
- Gaur, A.; Pal, G.K.; Ananthanarayanan, P.H.; Pal, P. Role of Ventromedial Hypothalamus in High Fat Diet Induced Obesity in Male Rats: Association with Lipid Profile, Thyroid Profile and Insulin Resistance. Ann. Neurosci. 2014, 21, 104–107. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, B.; Stutz, B.; Liu, Z.W.; Horvath, T.L.; Yang, X. Ventromedial Hypothalamic OGT Drives Adipose Tissue Lipolysis and Curbs Obesity. Sci. Adv. 2022, 8, eabn8092. [Google Scholar] [CrossRef] [PubMed]
- Viskaitis, P.; Irvine, E.E.; Smith, M.A.; Choudhury, A.I.; Alvarez-Curto, E.; Glegola, J.A.; Hardy, D.G.; Pedroni, S.M.A.; Paiva Pessoa, M.R.; Fernando, A.B.P.; et al. Modulation of SF1 Neuron Activity Coordinately Regulates Both Feeding Behavior and Associated Emotional States. Cell Rep. 2017, 21, 3559–3572. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, E.A.; Okamoto, S.; Ishikawa, A.W.; Yokota, S.; Wada, N.; Hirabayashi, T.; Saito, K.; Sato, T.; Takagi, K.; Wang, C.C.; et al. Activation of SF1 Neurons in the Ventromedial Hypothalamus by DREADD Technology Increases Insulin Sensitivity in Peripheral Tissues. Diabetes 2017, 66, 2372–2386. [Google Scholar] [CrossRef] [PubMed]
- Sternson, S.M.; Shepherd, G.M.G.; Friedman, J.M. Topographic Mapping of VMH → Arcuate Nucleus Microcircuits and Their Reorganization by Fasting. Nat. Neurosci. 2005, 8, 1356–1363. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, D.; Sweeney, P.; Yang, Y. An Excitatory Ventromedial Hypothalamus to Paraventricular Thalamus Circuit That Suppresses Food Intake. Nat. Commun. 2020, 11, 6326. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, H.; Zigman, J.M.; Ye, C.; Lee, C.E.; McGovern, R.A.; Tang, V.; Kenny, C.D.; Christiansen, L.M.; White, R.D.; Edelstein, E.A.; et al. Leptin Directly Activates SF1 Neurons in the VMH, and This Action by Leptin Is Required for Normal Body-Weight Homeostasis. Neuron 2006, 49, 191–203. [Google Scholar] [CrossRef]
- Meek, T.H.; Nelson, J.T.; Matsen, M.E.; Dorfman, M.D.; Guyenet, S.J.; Damian, V.; Allison, M.B.; Scarlett, J.M.; Nguyen, H.T.; Thaler, J.P.; et al. Functional Identification of a Neurocircuit Regulating Blood Glucose. Proc. Natl. Acad. Sci. USA 2016, 113, E2073–E2082. [Google Scholar] [CrossRef]
- Fosch, A.; Zagmutt, S.; Casals, N.; Rodríguez-Rodríguez, R. New Insights of SF1 Neurons in Hypothalamic Regulation of Obesity and Diabetes. Int. J. Mol. Sci. 2021, 22, 22. [Google Scholar] [CrossRef]
- Bellinger, L.L.; Bernardis, L.L. The Dorsomedial Hypothalamic Nucleus and Its Role in Ingestive Behavior and Body Weight Regulation: Lessons Learned from Lesioning Studies. Physiol. Behav. 2002, 76, 431–442. [Google Scholar] [CrossRef]
- Kesterson, R.A.; Huszar, D.; Lynch, C.A.; Simerly, R.B.; Cone, R.D. Induction of Neuropeptide Y Gene Expression in the Dorsal Medial Hypothalamic Nucleus in Two Models of the Agouti Obesity Syndrome. Mol. Endocrinol. 1997, 11, 630–637. [Google Scholar] [CrossRef]
- Guan, X.M.; Yu, H.; Van Der Ploeg, L.H.T. Evidence of Altered Hypothalamic Pro-Opiomelanocortin/Neuropeptide Y MRNA Expression in Tubby Mice. Mol. Brain Res. 1998, 59, 273–279. [Google Scholar] [CrossRef]
- Bi, S.; Ladenheim, E.E.; Schwartz, G.J.; Moran, T.H. A Role for NPY Overexpression in the Dorsomedial Hypothalamus in Hyperphagia and Obesity of OLETF Rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R254–R260. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Scott, K.A.; Hyun, J.; Tamashiro, K.L.; Tray, N.; Moran, T.H.; Bi, S. Role of Dorsomedial Hypothalamic Neuropeptide Y in Modulating Food Intake and Energy Balance. J. Neurosci. 2009, 29, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Otgon-Uul, Z.; Suyama, S.; Onodera, H.; Yada, T. Optogenetic Activation of Leptin- and Glucose-Regulated GABAergic Neurons in Dorsomedial Hypothalamus Promotes Food Intake via Inhibitory Synaptic Transmission to Paraventricular Nucleus of Hypothalamus. Mol. Metab. 2016, 5, 709–715. [Google Scholar] [CrossRef]
- Chen, J.; Scott, K.A.; Zhao, Z.; Moran, T.H.; Bi, S. Characterization of the Feeding Inhibition and Neural Activation Produced by Dorsomedial Hypothalamic Cholecystokinin Administration. Neuroscience 2008, 152, 178–188. [Google Scholar] [CrossRef]
- Rust, V.A.; Crosby, K.M. Cholecystokinin Acts in the Dorsomedial Hypothalamus of Young Male Rats to Suppress Appetite in a Nitric Oxide-Dependent Manner. Neurosci. Lett. 2021, 764, 136295. [Google Scholar] [CrossRef] [PubMed]
- Imoto, D.; Yamamoto, I.; Matsunaga, H.; Yonekura, T.; Lee, M.L.; Kato, K.X.; Yamasaki, T.; Xu, S.; Ishimoto, T.; Yamagata, S.; et al. Refeeding Activates Neurons in the Dorsomedial Hypothalamus to Inhibit Food Intake and Promote Positive Valence. Mol. Metab. 2021, 54, 101366. [Google Scholar] [CrossRef]
- Han, Y.; He, Y.; Harris, L.; Xu, Y.; Wu, Q. Identification of a GABAergic Neural Circuit Governing Leptin Signaling Deficiency-Induced Obesity. Elife 2023, 12, e82649. [Google Scholar] [CrossRef]
- Haspula, D.; Clark, M.A. Neuroinflammation and Sympathetic Overactivity: Mechanisms and Implications in Hypertension. Auton. Neurosci. 2018, 210, 10–17. [Google Scholar] [CrossRef]
- Grill, H.J.; Norgren, R. Chronically Decerebrate Rats Demonstrate Satiation but Not Bait Shyness. Science 1978, 201, 267–269. [Google Scholar] [CrossRef]
- Dirocco, R.J.; Grill, H.J. The Forebrain Is Not Essential for Sympathoadrenal Hyperglycemic Response to Glucoprivation. Science 1979, 204, 1112–1114. [Google Scholar] [CrossRef]
- Jeong, J.K.; Dow, S.A.; Young, C.N. Sensory Circumventricular Organs, Neuroendocrine Control, and Metabolic Regulation. Metabolites 2021, 11, 494. [Google Scholar] [CrossRef] [PubMed]
- Watts, A.G.; Kanoski, S.E.; Sanchez-Watts, G.; Langhans, W. The Physiological Control of Eating: Signals, Neurons, and Networks. Physiol. Rev. 2022, 102, 689–813. [Google Scholar] [CrossRef] [PubMed]
- Grill, H.J.; Hayes, M.R. The Nucleus Tractus Solitarius: A Portal for Visceral Afferent Signal Processing, Energy Status Assessment and Integration of Their Combined Effects on Food Intake. Int. J. Obes. 2009, 33 (Suppl. 1), S11–S15. [Google Scholar] [CrossRef] [PubMed]
- Kandalam, U.; Sarmiento, N.; Haspula, D.; Clark, M.A. Angiotensin III Induces Signal Transducer and Activator of Transcription 3 and Interleukin-6 MRNA Levels in Cultured Rat Astrocytes. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 758–767. [Google Scholar] [CrossRef]
- Haspula, D.; Clark, M.A. MAPK Activation Patterns of AT1R and CB1R in SHR versus Wistar Astrocytes: Evidence of CB1R Hypofunction and Crosstalk between AT1R and CB1R. Cell. Signal. 2017, 40, 81–90. [Google Scholar] [CrossRef]
- Haspula, D.; Clark, M.A. Molecular Basis of the Brain Renin Angiotensin System in Cardiovascular and Neurologic Disorders: Uncovering a Key Role for the Astroglial Angiotensin Type 1 Receptor AT1R. J. Pharmacol. Exp. Ther. 2018, 366, 251–264. [Google Scholar] [CrossRef]
- O’Connor, A.T.; Haspula, D.; Alanazi, A.Z.; Clark, M.A. Roles of Angiotensin III in the Brain and Periphery. Peptides 2022, 153, 170802. [Google Scholar] [CrossRef]
- Moura-Assis, A.; Friedman, J.M.; Velloso, L.A. Gut-to-Brain Signals in Feeding Control. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E326. [Google Scholar] [CrossRef]
- Kaelberer, M.M.; Buchanan, K.L.; Klein, M.E.; Barth, B.B.; Montoya, M.M.; Shen, X.; Bohórquez, D.V. A Gut-Brain Neural Circuit for Nutrient Sensory Transduction. Science 2018, 361, eaat5236. [Google Scholar] [CrossRef]
- Owyang, C.; Heldsinger, A. Vagal Control of Satiety and Hormonal Regulation of Appetite. J. Neurogastroenterol. Motil. 2011, 17, 338–348. [Google Scholar] [CrossRef]
- Berthoud, H.R.; Neuhuber, W.L. Vagal Mechanisms as Neuromodulatory Targets for the Treatment of Metabolic Disease. Ann. N. Y. Acad. Sci. 2019, 1454, 42–55. [Google Scholar] [CrossRef]
- Gibbs, J.; Young, R.C.; Smith, G.P. Cholecystokinin Decreases Food Intake in Rats. J. Comp. Physiol. Psychol. 1973, 84, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Widdop, R.E.; Krstew, E.; Mercer, L.D.; Carlsberg, M.; Beart, P.M.; Jarrott, B. Electrophysiological and Autoradiographical Evidence for Cholecystokinin A Receptors on Rat Isolated Nodose Ganglia. J. Auton. Nerv. Syst. 1994, 46, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.J.; Moran, T.H. CCK Elicits and Modulates Vagal Afferent Activity Arising from Gastric and Duodenal Sites. Ann. N. Y. Acad. Sci. 1994, 713, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Leon Mercado, L.; Caron, A.; Wang, Y.; Burton, M.; Gautron, L. Identification of Leptin Receptor–Expressing Cells in the Nodose Ganglion of Male Mice. Endocrinology 2019, 160, 1307–1322. [Google Scholar] [CrossRef] [PubMed]
- Barrachina, M.D.; Martínez, V.; Wang, L.; Wei, J.Y.; Taché, Y. Synergistic Interaction between Leptin and Cholecystokinin to Reduce Short-Term Food Intake in Lean Mice. Proc. Natl. Acad. Sci. USA 1997, 94, 10455. [Google Scholar] [CrossRef]
- Brierley, D.I.; de Lartigue, G. Reappraising the Role of the Vagus Nerve in GLP-1-Mediated Regulation of Eating. Br. J. Pharmacol. 2022, 179, 584–599. [Google Scholar] [CrossRef]
- Campbell, J.E.; Drucker, D.J. Pharmacology, Physiology, and Mechanisms of Incretin Hormone Action. Cell Metab. 2013, 17, 819–837. [Google Scholar] [CrossRef]
- Krieger, J.P.; Arnold, M.; Pettersen, K.G.; Lossel, P.; Langhans, W.; Lee, S.J. Knockdown of GLP-1 Receptors in Vagal Afferents Affects Normal Food Intake and Glycemia. Diabetes 2016, 65, 34–43. [Google Scholar] [CrossRef]
- Bai, L.; Mesgarzadeh, S.; Ramesh, K.S.; Huey, E.L.; Liu, Y.; Gray, L.A.; Aitken, T.J.; Chen, Y.; Beutler, L.R.; Ahn, J.S.; et al. Genetic Identification of Vagal Sensory Neurons That Control Feeding. Cell 2019, 179, 1129–1143.e23. [Google Scholar] [CrossRef]
- Egerod, K.L.; Petersen, N.; Timshel, P.N.; Rekling, J.C.; Wang, Y.; Liu, Q.; Schwartz, T.W.; Gautron, L. Profiling of G Protein-Coupled Receptors in Vagal Afferents Reveals Novel Gut-to-Brain Sensing Mechanisms. Mol. Metab. 2018, 12, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, I.; Pacheco-López, G.; Rüttimann, E.B.; Arnold, M.; Asarian, L.; Langhans, W.; Geary, N.; Hillebrand, J.J.G. Hepatic-Portal Vein Infusions of Glucagon-like Peptide-1 Reduce Meal Size and Increase c-Fos Expression in the Nucleus Tractus Solitarii, Area Postrema and Central Nucleus of the Amygdala in Rats. J. Neuroendocrinol. 2010, 22, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Campos, C.A.; Wright, J.S.; Czaja, K.; Ritter, R.C. CCK-Induced Reduction of Food Intake and Hindbrain MAPK Signaling Are Mediated by NMDA Receptor Activation. Endocrinology 2012, 153, 2633–2646. [Google Scholar] [CrossRef] [PubMed]
- Punjabi, M.; Arnold, M.; Rüttimann, E.; Graber, M.; Geary, N.; Pacheco-López, G.; Langhans, W. Circulating Glucagon-like Peptide-1 (GLP-1) Inhibits Eating in Male Rats by Acting in the Hindbrain and Without Inducing Avoidance. Endocrinology 2014, 155, 1690–1699. [Google Scholar] [CrossRef]
- Barrera, J.G.; Jones, K.R.; Herman, J.P.; D’Alessio, D.A.; Woods, S.C.; Seeley, R.J. Hyperphagia and Increased Fat Accumulation in Two Models of Chronic CNS Glucagon-Like Peptide-1 Loss of Function. J. Neurosci. 2011, 31, 3904–3913. [Google Scholar] [CrossRef]
- Finan, B.; Ma, T.; Ottaway, N.; Müller, T.D.; Habegger, K.M.; Heppner, K.M.; Kirchner, H.; Holland, J.; Hembree, J.; Raver, C.; et al. Unimolecular Dual Incretins Maximize Metabolic Benefits in Rodents, Monkeys, and Humans. Sci. Transl. Med. 2013, 5, 209ra151. [Google Scholar] [CrossRef]
- Zhang, C.; Vincelette, L.K.; Reimann, F.; Liberles, S.D. A Brainstem Circuit for Nausea Suppression. Cell Rep. 2022, 39, 110953. [Google Scholar] [CrossRef]
- Samms, R.J.; Cosgrove, R.; Snider, B.M.; Furber, E.C.; Droz, B.A.; Briere, D.A.; Dunbar, J.; Dogra, M.; Alsina-Fernandez, J.; Borner, T.; et al. GIPR Agonism Inhibits PYY-Induced Nausea-Like Behavior. Diabetes 2022, 71, 1410–1423. [Google Scholar] [CrossRef]
- Lutz, T.A.; Mollet, A.; Rushing, P.A.; Riediger, T.; Scharrer, E. The Anorectic Effect of a Chronic Peripheral Infusion of Amylin Is Abolished in Area Postrema/Nucleus of the Solitary Tract (AP/NTS) Lesioned Rats. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 1005–1011. [Google Scholar] [CrossRef]
- Halatchev, I.G.; Cone, R.D. Peripheral Administration of PYY(3-36) Produces Conditioned Taste Aversion in Mice. Cell Metab. 2005, 1, 159–168. [Google Scholar] [CrossRef]
- Woods, S.C.; Lutz, T.A.; Geary, N.; Langhans, W. Pancreatic Signals Controlling Food Intake; Insulin, Glucagon and Amylin. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1219. [Google Scholar] [CrossRef] [PubMed]
- Braegger, F.E.; Asarian, L.; Dahl, K.; Lutz, T.A.; Boyle, C.N. The Role of the Area Postrema in the Anorectic Effects of Amylin and Salmon Calcitonin: Behavioral and Neuronal Phenotyping. Eur. J. Neurosci. 2014, 40, 3055–3066. [Google Scholar] [CrossRef] [PubMed]
- Coester, B.; Le Foll, C.; Lutz, T.A. Viral Depletion of Calcitonin Receptors in the Area Postrema: A Proof-of-Concept Study. Physiol. Behav. 2020, 223, 112992. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.M.; Williams, K.W.; Rossi, J.; Lee, C.E.; Elmquist, J.K. Leptin Receptor Expression in Hindbrain Glp-1 Neurons Regulates Food Intake and Energy Balance in Mice. J. Clin. Investig. 2011, 121, 2413–2421. [Google Scholar] [CrossRef]
- Kanoski, S.E.; Zhao, S.; Guarnieri, D.J.; DiLeone, R.J.; Yan, J.; De Jonghe, B.C.; Bence, K.K.; Hayes, M.R.; Grill, H.J. Endogenous Leptin Receptor Signaling in the Medial Nucleus Tractus Solitarius Affects Meal Size and Potentiates Intestinal Satiation Signals. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E496–E503. [Google Scholar] [CrossRef]
- Cheng, W.; Ndoka, E.; Hutch, C.; Roelofs, K.; MacKinnon, A.; Khoury, B.; Magrisso, J.; Kim, K.S.; Rhodes, C.J.; Olson, D.P.; et al. Leptin Receptor-Expressing Nucleus Tractus Solitarius Neurons Suppress Food Intake Independently of GLP1 in Mice. JCI Insight. 2020, 5, e134359. [Google Scholar] [CrossRef]
- Venkatraman, A.; Edlow, B.L.; Immordino-Yang, M.H. The Brainstem in Emotion: A Review. Front. Neuroanat. 2017, 11, 15. [Google Scholar] [CrossRef]
- Vander Weele, C.M.; Siciliano, C.A.; Matthews, G.A.; Namburi, P.; Izadmehr, E.M.; Espinel, I.C.; Nieh, E.H.; Schut, E.H.S.; Padilla-Coreano, N.; Burgos-Robles, A.; et al. Dopamine Enhances Signal-to-Noise Ratio in Cortical-Brainstem Encoding of Aversive Stimuli. Nature 2018, 563, 397–401. [Google Scholar] [CrossRef]
- Wu, Q.; Clark, M.S.; Palmiter, R.D. Deciphering a Neuronal Circuit That Mediates Appetite. Nature 2012, 483, 594–597. [Google Scholar] [CrossRef]
- Roman, C.W.; Derkach, V.A.; Palmiter, R.D. Genetically and Functionally Defined NTS to PBN Brain Circuits Mediating Anorexia. Nat. Commun. 2016, 7, 11905. [Google Scholar] [CrossRef]
- D’Agostino, G.; Lyons, D.J.; Cristiano, C.; Burke, L.K.; Madara, J.C.; Campbell, J.N.; Garcia, A.P.; Land, B.B.; Lowell, B.B.; Dileone, R.J.; et al. Appetite Controlled by a Cholecystokinin Nucleus of the Solitary Tract to Hypothalamus Neurocircuit. eLife 2016, 5, e12225. [Google Scholar] [CrossRef]
- Roman, C.W.; Sloat, S.R.; Palmiter, R.D. A Tale of Two Circuits: CCKNTS Neuron Stimulation Controls Appetite and Induces Opposing Motivational States by Projections to Distinct Brain Regions. Neuroscience 2017, 358, 316–324. [Google Scholar] [CrossRef]
- Cheng, W.; Gonzalez, I.; Pan, W.; Tsang, A.H.; Adams, J.; Ndoka, E.; Gordian, D.; Khoury, B.; Roelofs, K.; Evers, S.S.; et al. Calcitonin Receptor Neurons in the Mouse Nucleus Tractus Solitarius Control Energy Balance via the Non-Aversive Suppression of Feeding. Cell Metab. 2020, 31, 301–312.e5. [Google Scholar] [CrossRef]
- Larsen, P.J.; Tang-Christensen, M.; Holst, J.J.; Ørskov, C. Distribution of Glucagon-like Peptide-1 and Other Preproglucagon-Derived Peptides in the Rat Hypothalamus and Brainstem. Neuroscience 1997, 77, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Alhadeff, A.L.; Rupprecht, L.E.; Hayes, M.R. GLP-1 Neurons in the Nucleus of the Solitary Tract Project Directly to the Ventral Tegmental Area and Nucleus Accumbens to Control for Food Intake. Endocrinology 2012, 153, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Palkovits, M.; Eskay, R.L. Distribution and Possible Origin of β-Endorphin and ACTH in Discrete Brainstem Nuclei of Rats. Neuropeptides 1987, 9, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Bronstein, D.M.; Schafer, M.K.H.; Watson, S.J.; Akil, H. Evidence That Beta-Endorphin Is Synthesized in Cells in the Nucleus Tractus Solitarius: Detection of POMC MRNA. Brain Res 1992, 587, 269–275. [Google Scholar] [CrossRef]
- Appleyard, S.M.; Bailey, T.W.; Doyle, M.W.; Jin, Y.H.; Smart, J.L.; Low, M.J.; Andresen, M.C. Proopiomelanocortin Neurons in Nucleus Tractus Solitarius Are Activated by Visceral Afferents: Regulation by Cholecystokinin and Opioids. J. Neurosci. 2005, 25, 3578–3585. [Google Scholar] [CrossRef]
- Georgescu, T.; Lyons, D.; Doslikova, B.; Garcia, A.P.; Marston, O.; Burke, L.K.; Chianese, R.; Lam, B.Y.H.; Yeo, G.S.H.; Rochford, J.J.; et al. Neurochemical Characterization of Brainstem Pro-Opiomelanocortin Cells. Endocrinology 2020, 161, bqaa032. [Google Scholar] [CrossRef]
- D’Agostino, G.; Lyons, D.; Cristiano, C.; Lettieri, M.; Olarte-Sanchez, C.; Burke, L.K.; Greenwald-Yarnell, M.; Cansell, C.; Doslikova, B.; Georgescu, T.; et al. Nucleus of the Solitary Tract Serotonin 5-HT2C Receptors Modulate Food Intake. Cell Metab. 2018, 28, 619–630.e5. [Google Scholar] [CrossRef]
- Wagner, S.; Brierley, D.I.; Leeson-Payne, A.; Jiang, W.; Chianese, R.; Lam, B.Y.H.; Dowsett, G.K.C.; Cristiano, C.; Lyons, D.; Reimann, F.; et al. Obesity Medication Lorcaserin Activates Brainstem GLP-1 Neurons to Reduce Food Intake and Augments GLP-1 Receptor Agonist Induced Appetite Suppression. Mol. Metab. 2023, 68, 101665. [Google Scholar] [CrossRef]
- Ritter, S.; Dinh, T.T.; Zhang, Y. Localization of Hindbrain Glucoreceptive Sites Controlling Food Intake and Blood Glucose. Brain Res. 2000, 856, 37–47. [Google Scholar] [CrossRef]
- Boychuk, C.R.; Smith, K.C.; Peterson, L.E.; Boychuk, J.A.; Butler, C.R.; Derera, I.D.; McCarthy, J.J.; Smith, B.N. A Hindbrain Inhibitory Microcircuit Mediates Vagally-Coordinated Glucose Regulation. Sci. Rep. 2019, 9, 2722. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Koller, J.; Ip, C.K.; Gopalasingam, G.; Bajaj, N.; Lee, N.J.; Enriquez, R.F.; Herzog, H. Lack of Neuropeptide FF Signalling in Mice Leads to Reduced Repetitive Behavior, Altered Drinking Behavior, and Fuel Type Selection. FASEB J. 2021, 35, e21980. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Koller, J.; Gopalasingam, G.; Qi, Y.; Herzog, H. Central NPFF Signalling Is Critical in the Regulation of Glucose Homeostasis. Mol. Metab. 2022, 62, 101525. [Google Scholar] [CrossRef]
- Wulsin, L.R.; Horn, P.S.; Perry, J.L.; Massaro, J.M.; D’Agostino, R.B. Autonomic Imbalance as a Predictor of Metabolic Risks, Cardiovascular Disease, Diabetes, and Mortality. J. Clin. Endocrinol. Metab. 2015, 100, 2443–2448. [Google Scholar] [CrossRef]
- Licht, C.M.M.; Vreeburg, S.A.; Van Reedt Dortland, A.K.B.; Giltay, E.J.; Hoogendijk, W.J.G.; DeRijk, R.H.; Vogelzangs, N.; Zitman, F.G.; De Geus, E.J.C.; Penninx, B.W.J.H. Increased Sympathetic and Decreased Parasympathetic Activity Rather than Changes in Hypothalamic-Pituitary-Adrenal Axis Activity Is Associated with Metabolic Abnormalities. J. Clin. Endocrinol. Metab. 2010, 95, 2458–2466. [Google Scholar] [CrossRef]
- Aronson, D.; Rayfield, E.J. How Hyperglycemia Promotes Atherosclerosis: Molecular Mechanisms. Cardiovasc. Diabetol. 2002, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- Haspula, D.; Vallejos, A.K.; Moore, T.M.; Tomar, N.; Dash, R.K.; Hoffmann, B.R. Influence of a Hyperglycemic Microenvironment on a Diabetic versus Healthy Rat Vascular Endothelium Reveals Distinguishable Mechanistic and Phenotypic Responses. Front. Physiol. 2019, 10, 558. [Google Scholar] [CrossRef]
- Jansen, A.S.P.; Hoffman, J.L.; Loewy, A.D. CNS Sites Involved in Sympathetic and Parasympathetic Control of the Pancreas: A Viral Tracing Study. Brain Res. 1997, 766, 29–38. [Google Scholar] [CrossRef]
- Buijs, R.M.; La Fleur, S.E.; Wortel, J.; Van Heyningen, C.; Zuiddam, L.; Mettenleiter, T.C.; Kalsbeek, A.; Nagai, K.; Niijima, A. The Suprachiasmatic Nucleus Balances Sympathetic and Parasympathetic Output to Peripheral Organs through Separate Preautonomic Neurons. J. Comp. Neurol. 2003, 464, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Rosario, W.; Singh, I.; Wautlet, A.; Patterson, C.; Flak, J.; Becker, T.C.; Ali, A.; Tamarina, N.; Philipson, L.H.; Enquist, L.W.; et al. The Brain-to-Pancreatic Islet Neuronal Map Reveals Differential Glucose Regulation from Distinct Hypothalamic Regions. Diabetes 2016, 65, 2711–2723. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ratnasabapathy, R.; Izzi-Engbeaya, C.; Nguyen-Tu, M.S.; Richardson, E.; Hussain, S.; De Backer, I.; Holton, C.; Norton, M.; Carrat, G.; et al. Hypothalamic Arcuate Nucleus Glucokinase Regulates Insulin Secretion and Glucose Homeostasis. Diabetes Obes. Metab. 2018, 20, 2246–2254. [Google Scholar] [CrossRef] [PubMed]
- Papazoglou, I.; Lee, J.-H.; Cui, Z.; Enquist, L.W.; Krashes, M.J.; Rane, S.G. A Distinct Hypothalamus-to-β Cell Circuit Modulates Insulin Secretion. Cell Metab. 2022, 34, 285–298.e7. [Google Scholar] [CrossRef]
- Wan, S.; Coleman, F.H.; Travagli, R.A. Glucagon-like Peptide-1 Excites Pancreas-Projecting Preganglionic Vagal Motoneurons. Am. J. Physiol. Gastrointest Liver Physiol. 2007, 292, G1474–G1482. [Google Scholar] [CrossRef][Green Version]
- Wan, S.; Browning, K.N.; Travagli, R.A. Glucagon-like Peptide-1 Modulates Synaptic Transmission to Identified Pancreas-Projecting Vagal Motoneurons. Peptides 2007, 28, 2184–2191. [Google Scholar] [CrossRef]
- Bruning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Muller-Wieland, D.; Kahn, C.R. Role of Brain Insulin Receptor in Control of Body Weight and Reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef]
- Dampney, R.A.L. Arcuate Nucleus—A Gateway for Insulin’s Action on Sympathetic Activity. J. Physiol. 2011, 589, 2109–2110. [Google Scholar] [CrossRef]
- Gelling, R.W.; Morton, G.J.; Morrison, C.D.; Niswender, K.D.; Myers, M.G.; Rhodes, C.J.; Schwartz, M.W. Insulin Action in the Brain Contributes to Glucose Lowering during Insulin Treatment of Diabetes. Cell Metab. 2006, 3, 67–73. [Google Scholar] [CrossRef]
- Kishore, P.; Boucai, L.; Zhang, K.; Li, W.; Koppaka, S.; Kehlenbrink, S.; Schiwek, A.; Esterson, Y.B.; Mehta, D.; Bursheh, S.; et al. Activation of K(ATP) Channels Suppresses Glucose Production in Humans. J. Clin. Investig. 2011, 121, 4916–4920. [Google Scholar] [CrossRef]
- Obici, S.; Zhang, B.B.; Karkanias, G.; Rossetti, L. Hypothalamic Insulin Signaling Is Required for Inhibition of Glucose Production. Nat. Med. 2002, 8, 1376–1382. [Google Scholar] [CrossRef]
- Shin, A.C.; Filatova, N.; Lindtner, C.; Chi, T.; Degann, S.; Oberlin, D.; Buettner, C. Insulin Receptor Signaling in POMC, but Not AgRP, Neurons Controls Adipose Tissue Insulin Action. Diabetes 2017, 66, 1560–1571. [Google Scholar] [CrossRef]
- Dodd, G.T.; Michael, N.J.; Lee-Young, R.S.; Mangiafico, S.P.; Pryor, J.T.; Munder, A.C.; Simonds, S.E.; Brüning, J.C.; Zhang, Z.Y.; Cowley, M.A.; et al. Insulin Regulates POMC Neuronal Plasticity to Control Glucose Metabolism. Elife 2018, 7, e38704. [Google Scholar] [CrossRef] [PubMed]
- El Mehdi, M.; Takhlidjt, S.; Devère, M.; Arabo, A.; Le Solliec, M.A.; Maucotel, J.; Bénani, A.; Nedelec, E.; Duparc, C.; Lefranc, B.; et al. The 26RFa (QRFP)/GPR103 Neuropeptidergic System in Mice Relays Insulin Signalling into the Brain to Regulate Glucose Homeostasis. Diabetologia 2022, 65, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Niu, S.N.; Huang, Z.B.; Wang, H.; Rao, X.R.; Kong, H.; Xu, J.; Li, X.J.; Yang, C.; Sheng, G.Q. Brainstem Hap1-Ahi1 Is Involved in Insulin-Mediated Feeding Control. FEBS Lett. 2011, 585, 85–91. [Google Scholar] [CrossRef]
- Filippi, B.M.; Yang, C.S.; Tang, C.; Lam, T.K.T. Insulin Activates Erk1/2 Signaling in the Dorsal Vagal Complex to Inhibit Glucose Production. Cell Metab. 2012, 16, 500–510. [Google Scholar] [CrossRef]
- Krowicki, Z.K.; Nathan, N.A.; Hornby, P.J. Gastric Motor and Cardiovascular Effects of Insulin in Dorsal Vagal Complex of the Rat. Am. J. Physiol. 1998, 275, G964–G972. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.B.; Smith, B.N. Insulin Reduces Excitation in Gastric-Related Neurons of the Dorsal Motor Nucleus of the Vagus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R807–R814. [Google Scholar] [CrossRef]
- Blake, C.B.; Smith, B.N. CAMP-Dependent Insulin Modulation of Synaptic Inhibition in Neurons of the Dorsal Motor Nucleus of the Vagus Is Altered in Diabetic Mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R711–R720. [Google Scholar] [CrossRef]
- Schechter, R.; Holtzclaw, L.; Sadiq, F.; Kahn, A.; Devaskar, S. Insulin Synthesis by Isolated Rabbit Neurons. Endocrinology 1988, 123, 505–513. [Google Scholar] [CrossRef]
- Devaskar, S.U.; Singh, B.S.; Carnaghi, L.R.; Rajakumar, P.A.; Giddings, S.J. Insulin II Gene Expression in Rat Central Nervous System. Regul. Pept. 1993, 48, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Tong, Q.; Ye, C.; Koda, S.; Fuller, P.M.; Krashes, M.J.; Vong, L.; Ray, R.S.; Olson, D.P.; Lowell, B.B. GABAergic RIP-Cre Neurons in the Arcuate Nucleus Selectively Regulate Energy Expenditure. Cell 2012, 151, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Rother, E.; Belgardt, B.F.; Tsaousidou, E.; Hampel, B.; Waisman, A.; Myers, M.G.; Brüning, J.C. Acute Selective Ablation of Rat Insulin Promoter-Expressing (RIP HER) Neurons Defines Their Orexigenic Nature. Proc. Natl. Acad. Sci. USA 2012, 109, 18132–18137. [Google Scholar] [CrossRef]
- Eerola, K.; Longo, F.; Reinbothe, T.M.; Richard, J.E.; Shevchouk, O.T.; López-Ferreras, L.; Mishra, D.; Asker, M.; Tolö, J.; Miranda, C.; et al. Hindbrain Insulin Controls Feeding Behavior. Mol. Metab. 2022, 66, 101614. [Google Scholar] [CrossRef]
- Püschel, G.P.; Püschel, P.; Pü, G.P. Control of Hepatocyte Metabolism by Sympathetic and Parasympathetic Hepatic Nerves. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 2004, 280A, 854–867. [Google Scholar] [CrossRef]
- Yi, C.X.; la Fleur, S.E.; Fliers, E.; Kalsbeek, A. The Role of the Autonomic Nervous Liver Innervation in the Control of Energy Metabolism. Biochim. Biophys Acta. 2010, 1802, 416–431. [Google Scholar] [CrossRef] [PubMed]
- Pocal, A.; Lam, T.K.T.; Gutierrez-Juarez, R.; Obici, S.; Schwartz, G.J.; Bryan, J.; Aguilar-Bryan, L.; Rossetti, L. Hypothalamic K(ATP) Channels Control Hepatic Glucose Production. Nature 2005, 434, 1026–1031. [Google Scholar] [CrossRef]
- Liss, B.; Roeper, J. Molecular Physiology of Neuronal K-ATP Channels. Mol. Membr. Biol. 2001, 18, 117–127. [Google Scholar] [CrossRef]
- Van Den Hoek, A.M.; Van Heijningen, C.; Schröder-van Der Elst, J.P.; Ouwens, D.M.; Havekes, L.M.; Romijn, J.A.; Kalsbeek, A.; Pijl, H. Intracerebroventricular Administration of Neuropeptide Y Induces Hepatic Insulin Resistance via Sympathetic Innervation. Diabetes 2008, 57, 2304–2310. [Google Scholar] [CrossRef]
- Kimura, K.; Tanida, M.; Nagata, N.; Inaba, Y.; Watanabe, H.; Nagashimada, M.; Ota, T.; Asahara, S.I.; Kido, Y.; Matsumoto, M.; et al. Central Insulin Action Activates Kupffer Cells by Suppressing Hepatic Vagal Activation via the Nicotinic Alpha 7 Acetylcholine Receptor. Cell Rep. 2016, 14, 2362–2374. [Google Scholar] [CrossRef]
- Williams, K.W.; Margatho, L.O.; Lee, C.E.; Choi, M.; Lee, S.; Scott, M.M.; Elias, C.F.; Elmquist, J.K. Segregation of Acute Leptin and Insulin Effects in Distinct Populations of Arcuate Proopiomelanocortin Neurons. J. Neurosci. 2010, 30, 2472–2479. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Zhang, C.; Borgquist, A.; Nestor, C.C.; Smith, A.W.; Bosch, M.A.; Ku, S.; Wagner, E.J.; Rønnekleiv, O.K.; Kelly, M.J. Insulin Excites Anorexigenic Proopiomelanocortin Neurons via Activation of Canonical Transient Receptor Potential Channels. Cell Metab. 2014, 19, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Wagner, E.J.; Rønnekleiv, O.K.; Kelly, M.J. Insulin and Leptin Excite Anorexigenic Pro-Opiomelanocortin Neurones via Activation of TRPC5 Channels. J. Neuroendocrinol. 2018, 30, e12501. [Google Scholar] [CrossRef] [PubMed]
- Huo, L.; Gamber, K.; Greeley, S.; Silva, J.; Huntoon, N.; Leng, X.H.; Bjørbæk, C. Leptin-Dependent Control of Glucose Balance and Locomotor Activity by POMC Neurons. Cell Metab. 2009, 9, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.D.; Vianna, C.R.; Donato, J.; Kim, M.H.; Chuang, J.C.; Lee, C.E.; Lauzon, D.A.; Lin, P.; Brule, L.J.; Scott, M.M.; et al. Direct Leptin Action on POMC Neurons Regulates Glucose Homeostasis and Hepatic Insulin Sensitivity in Mice. J. Clin. Investig. 2012, 122, 1000–1009. [Google Scholar] [CrossRef]
- Liu, L.; Karkanias, G.B.; Morales, J.C.; Hawkins, M.; Barzilai, N.; Wang, J.; Rossetti, L. Intracerebroventricular Leptin Regulates Hepatic but Not Peripheral Glucose Fluxes. J. Biol. Chem. 1998, 273, 31160–31167. [Google Scholar] [CrossRef]
- Gutiérrez-Juárez, R.; Obici, S.; Rossetti, L. Melanocortin-Independent Effects of Leptin on Hepatic Glucose Fluxes. J. Biol. Chem. 2004, 279, 49704–49715. [Google Scholar] [CrossRef]
- Tong, Q.; Ye, C.P.; McCrimmon, R.J.; Dhillon, H.; Choi, B.; Kramer, M.D.; Yu, J.; Yang, Z.; Christiansen, L.M.; Lee, C.E.; et al. Synaptic Glutamate Release by Ventromedial Hypothalamic Neurons Is Part of the Neurocircuitry That Prevents Hypoglycemia. Cell Metab. 2007, 5, 383–393. [Google Scholar] [CrossRef]
- Shimazu, T.; Ogasawara, S. Effects of Hypothalamic Stimulation on Gluconeogenesis and Glycolysis in Rat Liver. Am. J. Physiol. 1975, 228, 1787–1793. [Google Scholar] [CrossRef][Green Version]
- Tu, L.; Fukuda, M.; Tong, Q.; Xu, Y. The Ventromedial Hypothalamic Nucleus: Watchdog of Whole-Body Glucose Homeostasis. Cell Biosci. 2022, 12, 71. [Google Scholar] [CrossRef]
- Gao, H.; Molinas, A.J.R.; Miyata, K.; Qiao, X.; Zsombok, A. Overactivity of Liver-Related Neurons in the Paraventricular Nucleus of the Hypothalamus: Electrophysiological Findings in Db/Db Mice. J. Neurosci. 2017, 37, 11140. [Google Scholar] [CrossRef]
- Pocai, A.; Obici, S.; Schwartz, G.J.; Rossetti, L. A Brain-Liver Circuit Regulates Glucose Homeostasis. Cell Metab. 2005, 1, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.; Joung, H.Y.; Liu, S.M.; Chua, S.C.; Schwartz, G.J.; Jo, Y.H. Optogenetic Stimulation of the Liver-Projecting Melanocortinergic Pathway Promotes Hepatic Glucose Production. Nat. Commun. 2020, 11, 6295. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.K.L.; Chari, M.; Su, B.B.; Cheung, G.W.C.; Kokorovic, A.; Yang, C.S.; Wang, P.Y.T.; Lai, T.Y.Y.; Lam, T.K.T. Activation of N-Methyl-d-Aspartate (NMDA) Receptors in the Dorsal Vagal Complex Lowers Glucose Production. J. Biol. Chem. 2010, 285, 21913. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.Y.T.; Caspi, L.; Lam, C.K.L.; Chari, M.; Li, X.; Light, P.E.; Gutierrez-Juarez, R.; Ang, M.; Schwartz, G.J.; Lam, T.K.T. Upper Intestinal Lipids Trigger a Gut-Brain-Liver Axis to Regulate Glucose Production. Nature 2008, 452, 1012–1016. [Google Scholar] [CrossRef]
- Wang, L.; Pydi, S.P.; Zhu, L.; Barella, L.F.; Cui, Y.; Gavrilova, O.; Bence, K.K.; Vernochet, C.; Wess, J. Adipocyte Gi Signaling Is Essential for Maintaining Whole-Body Glucose Homeostasis and Insulin Sensitivity. Nat. Commun. 2020, 11, 2995. [Google Scholar] [CrossRef]
- Tajima, K.; Ikeda, K.; Tanabe, Y.; Thomson, E.A.; Yoneshiro, T.; Oguri, Y.; Ferro, M.D.; Poon, A.S.Y.; Kajimura, S. Wireless Optogenetics Protects against Obesity via Stimulation of Non-Canonical Fat Thermogenesis. Nat. Commun. 2020, 11, 1730. [Google Scholar] [CrossRef]
- Lyons, C.E.; Razzoli, M.; Larson, E.; Svedberg, D.; Frontini, A.; Cinti, S.; Vulchanova, L.; Sanders, M.; Thomas, M.; Bartolomucci, A. Optogenetic-Induced Sympathetic Neuromodulation of Brown Adipose Tissue Thermogenesis. FASEB J. 2020, 34, 2765–2773. [Google Scholar] [CrossRef]
- Kimura, T.; Pydi, S.P.; Wang, L.; Haspula, D.; Cui, Y.; Lu, H.; König, G.M.; Kostenis, E.; Steinberg, G.R.; Gavrilova, O.; et al. Adipocyte Gq Signaling Is a Regulator of Glucose and Lipid Homeostasis in Mice. Nat. Commun. 2022, 13, 1652. [Google Scholar] [CrossRef]
- Bamshad, M.; Aoki, V.T.; Adkison, M.G.; Warren, W.S.; Bartness, T.J. Central Nervous System Origins of the Sympathetic Nervous System Outflow to White Adipose Tissue. Am. J. Physiol. 1998, 275, R291–R299. [Google Scholar] [CrossRef]
- Bartness, T.J.; Song, C.K. Brain-Adipose Tissue Neural Crosstalk. Physiol. Behav. 2007, 91, 343–351. [Google Scholar] [CrossRef]
- Stanley, S.; Pinto, S.; Segal, J.; Pérez, C.A.; Viale, A.; DeFalco, J.; Cai, X.; Heisler, L.K.; Friedman, J.M. Identification of Neuronal Subpopulations That Project from Hypothalamus to Both Liver and Adipose Tissue Polysynaptically. Proc. Natl. Acad. Sci. USA 2010, 107, 7024–7029. [Google Scholar] [CrossRef] [PubMed]
- Ryu, V.; Bartness, T.J. Short and Long Sympathetic-Sensory Feedback Loops in White Fat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R886–R900. [Google Scholar] [CrossRef] [PubMed]
- Steculorum, S.M.; Ruud, J.; Karakasilioti, I.; Backes, H.; Engström Ruud, L.; Timper, K.; Hess, M.E.; Tsaousidou, E.; Mauer, J.; Vogt, M.C.; et al. AgRP Neurons Control Systemic Insulin Sensitivity via Myostatin Expression in Brown Adipose Tissue. Cell 2016, 165, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti-de-Albuquerque, J.P.; Bober, J.; Zimmer, M.R.; Dietrich, M.O. Regulation of Substrate Utilization and Adiposity by Agrp Neurons. Nat. Commun. 2019, 10, 311. [Google Scholar] [CrossRef]
- Chao, P.T.; Yang, L.; Aja, S.; Moran, T.H.; Bi, S. Knockdown of NPY Expression in the Dorsomedial Hypothalamus Promotes Development of Brown Adipocytes and Prevents Diet-Induced Obesity. Cell Metab. 2011, 13, 573–583. [Google Scholar] [CrossRef]
- Gómez-Valadés, A.G.; Pozo, M.; Varela, L.; Boudjadja, M.B.; Ramírez, S.; Chivite, I.; Eyre, E.; Haddad-Tóvolli, R.; Obri, A.; Milà-Guasch, M.; et al. Mitochondrial Cristae-Remodeling Protein OPA1 in POMC Neurons Couples Ca2+ Homeostasis with Adipose Tissue Lipolysis. Cell Metab. 2021, 33, 1820–1835.e9. [Google Scholar] [CrossRef]
- López, M. Hypothalamic AMPK and Energy Balance. Eur J. Clin. Investig. 2018, 48, e12996. [Google Scholar] [CrossRef]
- Martínez De Morentin, P.B.; González-García, I.; Martins, L.; Lage, R.; Fernández-Mallo, D.; Martínez-Sánchez, N.; Ruíz-Pino, F.; Liu, J.; Morgan, D.A.; Pinilla, L.; et al. Estradiol Regulates Brown Adipose Tissue Thermogenesis via Hypothalamic AMPK. Cell Metab. 2014, 20, 41–53. [Google Scholar] [CrossRef]
- Seoane-Collazo, P.; Roa, J.; Rial-Pensado, E.; Liñares-Pose, L.; Beiroa, D.; Ruíz-Pino, F.; López-González, T.; Morgan, D.A.; Pardavila, J.Á.; Sánchez-Tapia, M.J.; et al. SF1-Specific AMPKa1 Deletion Protects against Diet-Induced Obesity. Diabetes 2018, 67, 2213–2226. [Google Scholar] [CrossRef] [PubMed]
- Buettner, C.; Muse, E.D.; Cheng, A.; Chen, L.; Scherer, T.; Pocai, A.; Su, K.; Cheng, B.; Li, X.; Harvey-White, J.; et al. Leptin Controls Adipose Tissue Lipogenesis via Central, STAT3-Independent Mechanisms. Nat. Med. 2008, 14, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Pirzgalska, R.M.; Pereira, M.M.A.; Kubasova, N.; Barateiro, A.; Seixas, E.; Lu, Y.H.; Kozlova, A.; Voss, H.; Martins, G.G.; et al. Sympathetic Neuro-Adipose Connections Mediate Leptin-Driven Lipolysis. Cell 2015, 163, 84–94. [Google Scholar] [CrossRef]
- Dodd, G.T.; Decherf, S.; Loh, K.; Simonds, S.E.; Wiede, F.; Balland, E.; Merry, T.L.; Münzberg, H.; Zhang, Z.Y.; Kahn, B.B.; et al. Leptin and Insulin Act on POMC Neurons to Promote the Browning of White Fat. Cell 2015, 160, 88–104. [Google Scholar] [CrossRef] [PubMed]
- Mark, A.L.; Agassandian, K.; Morgan, D.A.; Liu, X.; Cassell, M.D.; Rahmouni, K. Leptin Signaling in the Nucleus Tractus Solitarii Increases Sympathetic Nerve Activity to the Kidney. Hypertension 2009, 53, 375–380. [Google Scholar] [CrossRef]
- Barnes, M.J.; McDougal, D.H. Leptin into the Rostral Ventral Lateral Medulla (RVLM) Augments Renal Sympathetic Nerve Activity and Blood Pressure. Front. Neurosci. 2014, 8, 232. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Pelletier, N.E.; Wong, J.; Li, B.; Sdrulla, A.D.; Madden, C.J.; Marks, D.L.; Brooks, V.L. Leptin Increases Sympathetic Nerve Activity via Induction of Its Own Receptor in the Paraventricular Nucleus. Elife 2020, 9, e55357. [Google Scholar] [CrossRef] [PubMed]
- Coester, B.; Koester-Hegmann, C.; Lutz, T.A.; Foll, C. Le Amylin/Calcitonin Receptor–Mediated Signaling in POMC Neurons Influences Energy Balance and Locomotor Activity in Chow-Fed Male Mice. Diabetes 2020, 69, 1110–1125. [Google Scholar] [CrossRef]
- Psichas, A.; Reimann, F.; Gribble, F.M. Gut Chemosensing Mechanisms. J. Clin. Investig. 2015, 125, 908–917. [Google Scholar] [CrossRef]
- Kim, W.; Egan, J.M. The Role of Incretins in Glucose Homeostasis and Diabetes Treatment. Pharmacol. Rev. 2008, 60, 470–512. [Google Scholar] [CrossRef]
- Ørskov, C.; Poulsen, S.S.; Møller, M.; Holst, J.J. Glucagon-like Peptide I Receptors in the Subfornical Organ and the Area Postrema Are Accessible to Circulating Glucagon-like Peptide I. Diabetes 1996, 45, 832–835. [Google Scholar] [CrossRef]
- Gu, G.; Roland, B.; Tomaselli, K.; Dolman, C.S.; Lowe, C.; Heilig, J.S. Glucagon-like Peptide-1 in the Rat Brain: Distribution of Expression and Functional Implication. J. Comp. Neurol. 2013, 521, 2235–2261. [Google Scholar] [CrossRef] [PubMed]
- Secher, A.; Jelsing, J.; Baquero, A.F.; Hecksher-Sørensen, J.; Cowley, M.A.; Dalbøge, L.S.; Hansen, G.; Grove, K.L.; Pyke, C.; Raun, K.; et al. The Arcuate Nucleus Mediates GLP-1 Receptor Agonist Liraglutide-Dependent Loss. J. Clin. Investig. 2014, 124, 4473–4488. [Google Scholar] [CrossRef]
- Adriaenssens, A.E.; Biggs, E.K.; Darwish, T.; Tadross, J.; Sukthankar, T.; Girish, M.; Polex-Wolf, J.; Lam, B.Y.; Zvetkova, I.; Pan, W.; et al. Glucose-Dependent Insulinotropic Polypeptide Receptor-Expressing Cells in the Hypothalamus Regulate Food Intake. Cell Metab. 2019, 30, 987–996.e6. [Google Scholar] [CrossRef]
- Zhang, Q.; Delessa, C.T.; Augustin, R.; Bakhti, M.; Colldén, G.; Drucker, D.J.; Feuchtinger, A.; Caceres, C.G.; Grandl, G.; Harger, A.; et al. The Glucose-Dependent Insulinotropic Polypeptide (GIP) Regulates Body Weight and Food Intake via CNS-GIPR Signaling. Cell Metab. 2021, 33, 833–844.e5. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M. The Role of GIP Receptor in the CNS for the Pathogenesis of Obesity. Diabetes 2021, 70, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Turton, M.D.; O’Shea, D.; Gunn, I.; Beak, S.A.; Edwards, C.M.B.; Meeran, K.; Choi, S.J.; Taylor, G.M.; Heath, M.M.; Lambert, P.D.; et al. A Role for Glucagon-like Peptide-1 in the Central Regulation of Feeding. Nature 1996, 379, 69–72. [Google Scholar] [CrossRef]
- Meeran, K.; O’Shea, D.; Edwards, C.M.B.; Turton, M.D.; Heath, M.M.; Gunn, I.; Abusnana, S.; Rossi, M.; Small, C.J.; Goldstone, A.P.; et al. Repeated Intracerebroventricular Administration of Glucagon-Like Peptide-1-(7–36) Amide or Exendin-(9–39) Alters Body Weight in the Rat*This Work Was Supported by the United Kingdom Medical Research Council. Endocrinology 1999, 140, 244–250. [Google Scholar] [CrossRef]
- Heppner, K.M.; Kirigiti, M.; Secher, A.; Paulsen, S.J.; Buckingham, R.; Pyke, C.; Knudsen, L.B.; Vrang, N.; Grove, K.L. Expression and Distribution of Glucagon-like Peptide-1 Receptor MRNA, Protein and Binding in the Male Nonhuman Primate (Macaca Mulatta) Brain. Endocrinology 2015, 156, 255–267. [Google Scholar] [CrossRef]
- NamKoong, C.; Kim, M.S.; Jang, B.T.; Lee, Y.H.; Cho, Y.M.; Choi, H.J. Central Administration of GLP-1 and GIP Decreases Feeding in Mice. Biochem. Biophys. Res. Commun. 2017, 490, 247–252. [Google Scholar] [CrossRef]
- Costa, A.; Ai, M.; Nunn, N.; Culotta, I.; Hunter, J.; Boudjadja, M.B.; Valencia-Torres, L.; Aviello, G.; Hodson, D.J.; Snider, B.M.; et al. Anorectic and Aversive Effects of GLP-1 Receptor Agonism Are Mediated by Brainstem Cholecystokinin Neurons, and Modulated by GIP Receptor Activation. Mol. Metab. 2022, 55, 101407. [Google Scholar] [CrossRef]
- Samms, R.J.; Sloop, K.W.; Gribble, F.M.; Reimann, F.; Adriaenssens, A.E. GIPR Function in the Central Nervous System: Implications and Novel Perspectives for GIP-Basedz Therapies in Treating Metabolic Disorders. Diabetes 2021, 70, 1938–1944. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Nian, C.; Karunakaran, S.; Clee, S.M.; Isales, C.M.; McIntosh, C.H.S. GIP-Overexpressing Mice Demonstrate Reduced Diet-Induced Obesity and Steatosis, and Improved Glucose Homeostasis. PLoS ONE 2012, 7, e40156. [Google Scholar] [CrossRef] [PubMed]
- Mroz, P.A.; Finan, B.; Gelfanov, V.; Yang, B.; Tschöp, M.H.; DiMarchi, R.D.; Perez-Tilve, D. Optimized GIP Analogs Promote Body Weight Lowering in Mice through GIPR Agonism Not Antagonism. Mol. Metab. 2019, 20, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Wang, L.; Ohbayashi, K.; Takeuchi, M.; O’Farrell, L.; Coskun, T.; Rakhat, Y.; Yabe, D.; Iwasaki, Y.; Seino, Y.; et al. Glucose-Dependent Insulinotropic Polypeptide Counteracts Diet-Induced Obesity along with Reduced Feeding, Elevated Plasma Leptin and Activation of Leptin-Responsive and Proopiomelanocortin Neurons in the Arcuate Nucleus. Diabetes Obes. Metab. 2023, 25, 1534–1546.e6. [Google Scholar] [CrossRef]
- Falko, J.M.; Crockett, S.E.; Cataland, S.; Mazzaferri, E.L. Gastric Inhibitory Polypeptide (GIP) Stimulated by Fat Ingestion in Man. J. Clin. Endocrinol. Metab. 1975, 41, 260–265.e5. [Google Scholar] [CrossRef]
- Brøns, C.; Jensen, C.B.; Storgaard, H.; Hiscock, N.J.; White, A.; Appel, J.S.; Jacobsen, S.; Nilsson, E.; Larsen, C.M.; Astrup, A.; et al. Impact of Short-Term High-Fat Feeding on Glucose and Insulin Metabolism in Young Healthy Men. J. Physiol. 2009, 587, 2387–2397. [Google Scholar] [CrossRef]
- Miyawaki, K.; Yamada, Y.; Ban, N.; Ihara, Y.; Tsukiyama, K.; Zhou, H.; Fujimoto, S.; Oku, A.; Tsuda, K.; Toyokuni, S.; et al. Inhibition of Gastric Inhibitory Polypeptide Signaling Prevents Obesity. Nat. Med. 2002, 8, 738–742. [Google Scholar] [CrossRef]
- Szecowka, J.; Grill, V.; Sandberg, E.; Efendic, S. Effect of GIP on the Secretion of Insulin and Somatostatin and the Accumulation of Cyclic AMP in Vitro in the Rat. Acta. Endocrinol. 1982, 99, 416–421. [Google Scholar] [CrossRef]
- Drucker, D.J.; Philippe, J.; Mojsov, S.; Chick, W.L.; Habener, J.F. Glucagon-like Peptide I Stimulates Insulin Gene Expression and Increases Cyclic AMP Levels in a Rat Islet Cell Line. Proc. Natl. Acad. Sci. USA 1987, 84, 3434–3438. [Google Scholar] [CrossRef]
- Ahrén, B. Sensory Nerves Contribute to Insulin Secretion by Glucagon-like Peptide-1 in Mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R269–R272. [Google Scholar] [CrossRef]
- Baggio, L.L.; Drucker, D.J. Biology of Incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.W.C.; Kokorovic, A.; Lam, C.K.L.; Chari, M.; Lam, T.K.T. Intestinal Cholecystokinin Controls Glucose Production through a Neuronal Network. Cell Metab. 2009, 10, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Blouet, C.; Schwartz, G.J. Duodenal Lipid Sensing Activates Vagal Afferents to Regulate Non-Shivering Brown Fat Thermogenesis in Rats. PLoS ONE 2012, 7, e51898. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, D.A.; Bagnol, D.; Woods, S.C.; D’Alessio, D.A.; Seeley, R.J. Arcuate Glucagon-like Peptide 1 Receptors Regulate Glucose Homeostasis but Not Food Intake. Diabetes 2008, 57, 2046–2054. [Google Scholar] [CrossRef]
- Burmeister, M.A.; Ferre, T.; Ayala, J.E.; King, E.M.; Holt, R.M.; Ayala, J.E. Acute Activation of Central GLP-1 Receptors Enhances Hepatic Insulin Action and Insulin Secretion in High-Fat-Fed, Insulin Resistant Mice. Am. J. Physiol. Endocrinol. Metab. 2012, 302, 334–343. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, L.; Zhang, J.; Conde, K.; Phansalkar, J.; Li, Z.; Yao, L.; Xu, Z.; Wang, W.; Zhou, J.; et al. Glucose-Sensing Glucagon-like Peptide-1 Receptor Neurons in the Dorsomedial Hypothalamus Regulate Glucose Metabolism. Sci. Adv. 2022, 8, eabn5345. [Google Scholar] [CrossRef]
- Pérez-Tilve, D.; González-Matías, L.; Aulinger, B.A.; Alvarez-Crespo, M.; Gil-Lozano, M.; Alvarez, E.; Andrade-Olivie, A.M.; Tschöp, M.H.; D’Alessio, D.A.; Mallo, F. Exendin-4 Increases Blood Glucose Levels Acutely in Rats by Activation of the Sympathetic Nervous System. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1088–E1096. [Google Scholar] [CrossRef] [PubMed]
- Jessen, L.; Smith, E.P.; Ulrich-Lai, Y.; Herman, J.P.; Seeley, R.J.; Sandoval, D.; D’Alessio, D. Central Nervous System GLP-1 Receptors Regulate Islet Hormone Secretion and Glucose Homeostasis in Male Rats. Endocrinology 2017, 158, 2124–2133. [Google Scholar] [CrossRef]
- Marcelin, G.; Jo, Y.H.; Li, X.; Schwartz, G.J.; Zhang, Y.; Dun, N.J.; Lyu, R.M.; Blouet, C.; Chang, J.K.; Chua, S. Central Action of FGF19 Reduces Hypothalamic AGRP/NPY Neuron Activity and Improves Glucose Metabolism. Mol. Metab. 2013, 3, 19–28. [Google Scholar] [CrossRef]
- Liu, S.; Marcelin, G.; Blouet, C.; Jeong, J.H.; Jo, Y.H.; Schwartz, G.J.; Chua, S. A Gut-Brain Axis Regulating Glucose Metabolism Mediated by Bile Acids and Competitive Fibroblast Growth Factor Actions at the Hypothalamus. Mol. Metab. 2018, 8, 37–50. [Google Scholar] [CrossRef]
- Liu, S.-M.; Ifebi, B.; Johnson, F.; Xu, A.; Ho, J.; Yang, Y.; Schwartz, G.J.; Jo, Y.-H.; Chua, S. The Gut Signals to AGRP-Expressing Cells of the Pituitary to Control Glucose Homeostasis. J. Clin. Investig. 2023, 133, e164185. [Google Scholar] [CrossRef]
- Wean, J.B.; Smith, B.N. FGF19 in the Hindbrain Lowers Blood Glucose and Alters Excitability of Vagal Motor Neurons in Hyperglycemic Mice. Endocrinology 2021, 162, bqab021. [Google Scholar] [CrossRef]
- Fan, K.; Li, Q.; Pan, D.; Liu, H.; Li, P.; Hai, R.; Du, C. Effects of Amylin on Food Intake and Body Weight via Sympathetic Innervation of the Interscapular Brown Adipose Tissue. Nutr. Neurosci. 2020, 25, 343–355. [Google Scholar] [CrossRef]
- Batterham, R.L.; Ffytche, D.H.; Rosenthal, J.M.; Zelaya, F.O.; Barker, G.J.; Withers, D.J.; Williams, S.C.R. PYY Modulation of Cortical and Hypothalamic Brain Areas Predicts Feeding Behaviour in Humans. Nature 2007, 450, 106–109. [Google Scholar] [CrossRef] [PubMed]
- De Silva, A.; Salem, V.; Long, C.J.; Makwana, A.; Newbould, R.D.; Rabiner, E.A.; Ghatei, M.A.; Bloom, S.R.; Matthews, P.M.; Beaver, J.D.; et al. The Gut Hormones PYY 3-36 and GLP-1 7-36 Amide Reduce Food Intake and Modulate Brain Activity in Appetite Centers in Humans. Cell Metab. 2011, 14, 700–706. [Google Scholar] [CrossRef]
- Tak, Y.J.; Lee, S.Y. Long-Term Efficacy and Safety of Anti-Obesity Treatment: Where Do We Stand? Curr. Obes. Rep. 2021, 10, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Haspula, D.; Clark, M.A. Cannabinoid Receptors: An Update on Cell Signaling, Pathophysiological Roles and Therapeutic Opportunities in Neurological, Cardiovascular, and Inflammatory Diseases. Int. J. Mol. Sci. 2020, 21, 7693. [Google Scholar] [CrossRef] [PubMed]
- Coulter, A.A.; Rebello, C.J.; Greenway, F.L. Centrally Acting Agents for Obesity: Past, Present, and Future. Drugs 2018, 78, 1113–1132. [Google Scholar] [CrossRef]
- Greenway, F.L.; Fujioka, K.; Plodkowski, R.A.; Mudaliar, S.; Guttadauria, M.; Erickson, J.; Kim, D.D.; Dunayevich, E. Effect of Naltrexone plus Bupropion on Weight Loss in Overweight and Obese Adults (COR-I): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2010, 376, 595–605. [Google Scholar] [CrossRef]
- Padwal, R.S.; Majumdar, S.R. Drug Treatments for Obesity: Orlistat, Sibutramine, and Rimonabant. Lancet 2007, 369, 71–77. [Google Scholar] [CrossRef]
- Blundell, J.; Finlayson, G.; Axelsen, M.; Flint, A.; Gibbons, C.; Kvist, T.; Hjerpsted, J.B. Effects of Once-Weekly Semaglutide on Appetite, Energy Intake, Control of Eating, Food Preference and Body Weight in Subjects with Obesity. Diabetes Obes. Metab. 2017, 19, 1242–1251. [Google Scholar] [CrossRef]
- Hjerpsted, J.B.; Flint, A.; Brooks, A.; Axelsen, M.B.; Kvist, T.; Blundell, J. Semaglutide Improves Postprandial Glucose and Lipid Metabolism, and Delays First-Hour Gastric Emptying in Subjects with Obesity. Diabetes Obes. Metab. 2018, 20, 610–619. [Google Scholar] [CrossRef]
- Chao, A.M.; Wadden, T.A.; Berkowitz, R.I.; Quigley, K.; Silvestry, F. The Risk of Cardiovascular Complications with Current Obesity Drugs. Expert Opin. Drug. Saf. 2020, 19, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Josipovic, M.; Staricoff, E.O.; Riches, C.H.; Sheppard, L.; Evans, M. 1311-P: Glucokinase in AGRP Neurons Is Required for Normal Insulin Secretion and Action in Female Mice. Diabetes 2022, 71, 1311. [Google Scholar] [CrossRef]
- Caughey, S.D.; Wilson, P.W.; Mukhtar, N.; Brocklehurst, S.; Reid, A.; D’Eath, R.B.; Boswell, T.; Dunn, I.C. Sex Differences in Basal Hypothalamic Anorectic and Orexigenic Gene Expression and the Effect of Quantitative and Qualitative Food Restriction. Biol Sex Differ 2018, 9, 20. [Google Scholar] [CrossRef]
- Kroll, D.S.; Feldman, D.E.; Biesecker, C.L.; McPherson, K.L.; Manza, P.; Joseph, P.V.; Volkow, N.D.; Wang, G.J. Neuroimaging of Sex/Gender Differences in Obesity: A Review of Structure, Function, and Neurotransmission. Nutrients 2020, 12, 1942. [Google Scholar] [CrossRef]
- Pi-Sunyer, X. The Medical Risks of Obesity. Postgrad. Med. 2009, 121, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Haththotuwa, R.N.; Wijeyaratne, C.N.; Senarath, U. Worldwide Epidemic of Obesity. Obes. Obstet. 2020, 3–8. [Google Scholar] [CrossRef]
- Karalliedde, J.; Gnudi, L. Diabetes Mellitus, a Complex and Heterogeneous Disease, and the Role of Insulin Resistance as a Determinant of Diabetic Kidney Disease. Nephrol. Dial. Transplant. 2016, 31, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Neeland, I.J.; Poirier, P.; Després, J.P. The Cardiovascular and Metabolic Heterogeneity of Obesity: Clinical Challenges and Implications for Management. Circulation 2018, 137, 1391–1406. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, R.J.; Tschöp, M.H.; Wilding, J.P.H. Anti-Obesity Drugs: Past, Present and Future. Dis. Model Mech. 2012, 5, 621–626. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haspula, D.; Cui, Z. Neurochemical Basis of Inter-Organ Crosstalk in Health and Obesity: Focus on the Hypothalamus and the Brainstem. Cells 2023, 12, 1801. https://doi.org/10.3390/cells12131801
Haspula D, Cui Z. Neurochemical Basis of Inter-Organ Crosstalk in Health and Obesity: Focus on the Hypothalamus and the Brainstem. Cells. 2023; 12(13):1801. https://doi.org/10.3390/cells12131801
Chicago/Turabian StyleHaspula, Dhanush, and Zhenzhong Cui. 2023. "Neurochemical Basis of Inter-Organ Crosstalk in Health and Obesity: Focus on the Hypothalamus and the Brainstem" Cells 12, no. 13: 1801. https://doi.org/10.3390/cells12131801
APA StyleHaspula, D., & Cui, Z. (2023). Neurochemical Basis of Inter-Organ Crosstalk in Health and Obesity: Focus on the Hypothalamus and the Brainstem. Cells, 12(13), 1801. https://doi.org/10.3390/cells12131801