

Lighting Up the Fire in the Microenvironment of Cold Tumors: A Major Challenge to Improve Cancer Immunotherapy



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Factors Involved in the Establishment of Immune Desert Tumors
2.1. Defects in the Cancer-Immunity Cycle
2.1.1. Low Expression of Tumor Antigens
2.1.2. Deficiency in APC Recruitment and Maturation/Activation
2.1.3. Immunosuppressive Tumor Microenvironment
2.1.4. Dysregulation of the Chemokines and Cytokines Network
3. Strategies to Turn Non-Inflamed Cold Tumors into Inflamed Hot Tumors
3.1. Neoadjuvant Chemotherapy
3.2. Radiotherapy
3.3. Targeting Autophagy
3.4. Oncolytic Viruses
3.5. Vaccines
3.6. The Use of Nanoparticles for Enhancing the Delivery of Therapies to the Tumor Site
3.7. Combining Adoptive T Cell Transfer with Other Therapies
- (i)
- TILs
- (ii)
- TCR-T cells
- (iii)
- CAR-T cells
3.8. Targeting Hypoxia
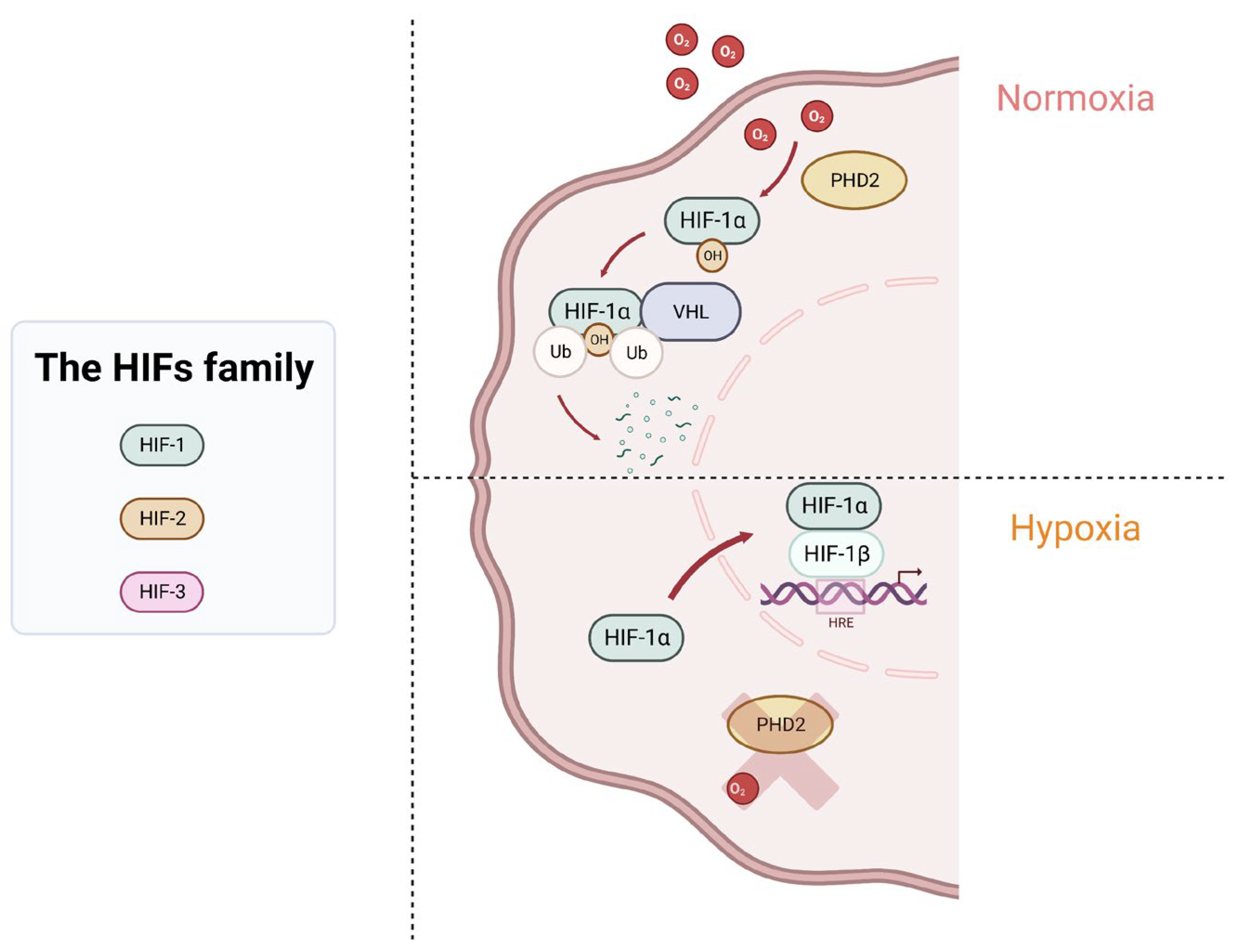
3.8.1. Hypoxia: Mechanisms and Pathways
3.8.2. Hypoxia Inducible Factors and Cancer
- (i)
- Regulation of signal transduction pathways
- (ii)
- Suppression of immune effector cells
- (iii)
- Recruitment of immunosuppressive cells
- (iv)
- Induction of autophagy
3.8.3. Targeting Hypoxia as a Strategy to Improve Patient’s Response to Immunotherapy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoober, J.K.; Eggink, L.L.; Cote, R. Stories From the Dendritic Cell Guardhouse. Front. Immunol. 2019, 10, 2880. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, L.; Zitvogel, L.; Eggermont, A.; Marabelle, A. PD-Loma: A cancer entity with a shared sensitivity to the PD-1/PD-L1 pathway blockade. Br. J. Cancer 2019, 120, 3–5. [Google Scholar] [CrossRef]
- Pol, J.; Kroemer, G. Anti-CTLA-4 immunotherapy: Uncoupling toxicity and efficacy. Cell Res. 2018, 28, 501–502. [Google Scholar] [CrossRef]
- Maleki Vareki, S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J. Immunother. Cancer 2018, 6, 157. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, X.; Zhang, F.; Zhang, X.; Tang, F.; Han, Z.; Li, Y. TCR-T Immunotherapy: The Challenges and Solutions. Front. Oncol. 2021, 11, 794183. [Google Scholar] [CrossRef]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising targets for cancer therapy. Signal. Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef]
- Leko, V.; Rosenberg, S.A. Identifying and Targeting Human Tumor Antigens for T Cell-Based Immunotherapy of Solid Tumors. Cancer Cell 2020, 38, 454–472. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Khatri, I.; de Wit, T.; de Rink, I.; Nieuwland, M.; Kerkhoven, R.; van Eenennaam, H.; Sun, C.; Garg, A.D.; Borst, J.; et al. CD4(+) helper T cells endow cDC1 with cancer-impeding functions in the human tumor micro-environment. Nat. Commun. 2023, 14, 217. [Google Scholar] [CrossRef] [PubMed]
- Von Euw, E.M.; Barrio, M.M.; Furman, D.; Bianchini, M.; Levy, E.M.; Yee, C.; Li, Y.; Wainstok, R.; Mordoh, J. Monocyte-derived dendritic cells loaded with a mixture of apoptotic/necrotic melanoma cells efficiently cross-present gp100 and MART-1 antigens to specific CD8(+) T lymphocytes. J. Transl. Med. 2007, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Borst, J.; Busselaar, J.; Bosma, D.M.T.; Ossendorp, F. Mechanism of action of PD-1 receptor/ligand targeted cancer immunotherapy. Eur. J. Immunol. 2021, 51, 1911–1920. [Google Scholar] [CrossRef] [PubMed]
- Melaiu, O.; Chierici, M.; Lucarini, V.; Jurman, G.; Conti, L.A.; De Vito, R.; Boldrini, R.; Cifaldi, L.; Castellano, A.; Furlanello, C.; et al. Cellular and gene signatures of tumor-infiltrating dendritic cells and natural-killer cells predict prognosis of neuroblastoma. Nat. Commun. 2020, 11, 5992. [Google Scholar] [CrossRef]
- Van Cruijsen, H.; van der Veldt, A.A.; Vroling, L.; Oosterhoff, D.; Broxterman, H.J.; Scheper, R.J.; Giaccone, G.; Haanen, J.B.; van den Eertwegh, A.J.; Boven, E.; et al. Sunitinib-induced myeloid lineage redistribution in renal cell cancer patients: CD1c+ dendritic cell frequency predicts progression-free survival. Clin. Cancer Res. 2008, 14, 5884–5892. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef]
- Ge, W.; Wu, W. Influencing Factors and Significance of Tumor-associated Macrophage Polarization in Tumor Microenvironment. Chin. J. Lung Cancer 2023, 26, 228–237. [Google Scholar] [CrossRef]
- Dallavalasa, S.; Beeraka, N.M.; Basavaraju, C.G.; Tulimilli, S.V.; Sadhu, S.P.; Rajesh, K.; Aliev, G.; Madhunapantula, S.V. The Role of Tumor Associated Macrophages (TAMs) in Cancer Progression, Chemoresistance, Angiogenesis and Metastasis—Current Status. Curr. Med. Chem. 2021, 28, 8203–8236. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Shimizu, J.; Yamazaki, S.; Sakihama, T.; Itoh, M.; Kuniyasu, Y.; Nomura, T.; Toda, M.; Takahashi, T. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: Their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol. Rev. 2001, 182, 18–32. [Google Scholar] [CrossRef]
- Alissafi, T.; Hatzioannou, A.; Legaki, A.I.; Varveri, A.; Verginis, P. Balancing cancer immunotherapy and immune-related adverse events: The emerging role of regulatory T cells. J. Autoimmun. 2019, 104, 102310. [Google Scholar] [CrossRef]
- Yin, Z.; Li, C.; Wang, J.; Xue, L. Myeloid-derived suppressor cells: Roles in the tumor microenvironment and tumor radiotherapy. Int. J. Cancer 2019, 144, 933–946. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Berchem, G.; Noman, M.Z.; Bosseler, M.; Paggetti, J.; Baconnais, S.; Le Cam, E.; Nanbakhsh, A.; Moussay, E.; Mami-Chouaib, F.; Janji, B.; et al. Hypoxic tumor-derived microvesicles negatively regulate NK cell function by a mechanism involving TGF-β and miR23a transfer. Oncoimmunology 2016, 5, e1062968. [Google Scholar] [CrossRef]
- Lequeux, A.; Noman, M.Z.; Xiao, M.; Sauvage, D.; Van Moer, K.; Viry, E.; Bocci, I.; Hasmim, M.; Bosseler, M.; Berchem, G.; et al. Impact of hypoxic tumor microenvironment and tumor cell plasticity on the expression of immune checkpoints. Cancer Lett. 2019, 458, 13–20. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef]
- Noman, M.Z.; Hasmim, M.; Lequeux, A.; Xiao, M.; Duhem, C.; Chouaib, S.; Berchem, G.; Janji, B. Improving Cancer Immunotherapy by Targeting the Hypoxic Tumor Microenvironment: New Opportunities and Challenges. Cells 2019, 8, 1083. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Hasmim, M.; Messai, Y.; Terry, S.; Kieda, C.; Janji, B.; Chouaib, S. Hypoxia: A key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015, 309, C569–C579. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lu, H.; Xiang, L.; Bullen, J.W.; Zhang, C.; Samanta, D.; Gilkes, D.M.; He, J.; Semenza, G.L. HIF-1 regulates CD47 expression in breast cancer cells to promote evasion of phagocytosis and maintenance of cancer stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E6215–E6223. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Patel, S.P.; Roszik, J.; Qin, Y. Hypoxia-Driven Immunosuppressive Metabolites in the Tumor Microenvironment: New Approaches for Combinational Immunotherapy. Front. Immunol. 2018, 9, 1591. [Google Scholar] [CrossRef]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef]
- Janji, B.; Chouaib, S. The Promise of Targeting Hypoxia to Improve Cancer Immunotherapy: Mirage or Reality? Front. Immunol. 2022, 13, 880810. [Google Scholar] [CrossRef]
- Gorbachev, A.V.; Fairchild, R.L. Regulation of chemokine expression in the tumor microenvironment. Crit. Rev. Immunol. 2014, 34, 103–120. [Google Scholar] [CrossRef]
- Hojo, S.; Koizumi, K.; Tsuneyama, K.; Arita, Y.; Cui, Z.; Shinohara, K.; Minami, T.; Hashimoto, I.; Nakayama, T.; Sakurai, H.; et al. High-level expression of chemokine CXCL16 by tumor cells correlates with a good prognosis and increased tumor-infiltrating lymphocytes in colorectal cancer. Cancer Res. 2007, 67, 4725–4731. [Google Scholar] [CrossRef]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef]
- Lequeux, A.; Noman, M.Z.; Xiao, M.; Van Moer, K.; Hasmim, M.; Benoit, A.; Bosseler, M.; Viry, E.; Arakelian, T.; Berchem, G.; et al. Targeting HIF-1 alpha transcriptional activity drives cytotoxic immune effector cells into melanoma and improves combination immunotherapy. Oncogene 2021, 40, 4725–4735. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef]
- Vinokurova, D.; Apetoh, L. The Emerging Role of IL-9 in the Anticancer Effects of Anti-PD-1 Therapy. Biomolecules 2023, 13, 670. [Google Scholar] [CrossRef]
- Pelekanou, V.; Carvajal-Hausdorf, D.E.; Altan, M.; Wasserman, B.; Carvajal-Hausdorf, C.; Wimberly, H.; Brown, J.; Lannin, D.; Pusztai, L.; Rimm, D.L. Effect of neoadjuvant chemotherapy on tumor-infiltrating lymphocytes and PD-L1 expression in breast cancer and its clinical significance. Breast Cancer Res. 2017, 19, 91. [Google Scholar] [CrossRef]
- Spring, L.M.; Fell, G.; Arfe, A.; Sharma, C.; Greenup, R.; Reynolds, K.L.; Smith, B.L.; Alexander, B.; Moy, B.; Isakoff, S.J.; et al. Pathologic Complete Response after Neoadjuvant Chemotherapy and Impact on Breast Cancer Recurrence and Survival: A Comprehensive Meta-analysis. Clin. Cancer Res. 2020, 26, 2838–2848. [Google Scholar] [CrossRef]
- Parra, E.R.; Villalobos, P.; Behrens, C.; Jiang, M.; Pataer, A.; Swisher, S.G.; William, W.N., Jr.; Zhang, J.; Lee, J.; Cascone, T.; et al. Effect of neoadjuvant chemotherapy on the immune microenvironment in non-small cell lung carcinomas as determined by multiplex immunofluorescence and image analysis approaches. J. Immunother. Cancer 2018, 6, 48. [Google Scholar] [CrossRef]
- Chen, J.; Li, C.; Cao, Y.; Zhu, L.; Zhang, B.; You, J.; Hou, H.; Wang, J.; Yuan, Z. Toripalimab combined with concurrent platinum-based Chemoradiotherapy in patients with locally advanced cervical Cancer: An open-label, single-arm, phase II trial. BMC Cancer 2022, 22, 793. [Google Scholar] [CrossRef]
- Valkema, M.J.; Mostert, B.; Lagarde, S.M.; Wijnhoven, B.P.L.; van Lanschot, J.J.B. The effectivity of targeted therapy and immunotherapy in patients with advanced metastatic and non-metastatic cancer of the esophagus and esophago-gastric junction. Updat. Surg. 2023, 75, 313–323. [Google Scholar] [CrossRef]
- Rébé, C.; Ghiringhelli, F. Cytotoxic effects of chemotherapy on cancer and immune cells: How can it be modulated to generate novel therapeutic strategies? Future Oncol. 2015, 11, 2645–2654. [Google Scholar] [CrossRef]
- Schaue, D. A Century of Radiation Therapy and Adaptive Immunity. Front. Immunol. 2017, 8, 431. [Google Scholar] [CrossRef]
- Sevenich, L. Turning “Cold” Into “Hot” Tumors-Opportunities and Challenges for Radio-Immunotherapy Against Primary and Metastatic Brain Cancers. Front. Oncol. 2019, 9, 163. [Google Scholar] [CrossRef] [PubMed]
- Lussier, D.M.; Alspach, E.; Ward, J.P.; Miceli, A.P.; Runci, D.; White, J.M.; Mpoy, C.; Arthur, C.D.; Kohlmiller, H.N.; Jacks, T.; et al. Radiation-induced neoantigens broaden the immunotherapeutic window of cancers with low mutational loads. Proc. Natl. Acad. Sci. USA 2021, 118, e2102611118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Pan, J.; Gan, L.; Xue, J. Ferroptosis, necroptosis, and pyroptosis in cancer: Crucial cell death types in radiotherapy and post-radiotherapy immune activation. Radiother. Oncol. 2023, 184, 109689. [Google Scholar] [CrossRef] [PubMed]
- Dewan, M.Z.; Galloway, A.E.; Kawashima, N.; Dewyngaert, J.K.; Babb, J.S.; Formenti, S.C.; Demaria, S. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 2009, 15, 5379–5388. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, G.; Fridman, M. Abscopal effect of radiation in papillary adenocarcinoma. Br. J. Radiol. 1973, 46, 220–222. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Wu, S.; Shen, J.; Huang, C.; Chen, Y.; Li, S. Abscopal effect of radiation therapy and nivolumab in a patient with combined small-cell lung cancer: A case report. Immunotherapy 2022, 14, 909–914. [Google Scholar] [CrossRef]
- Katsuki, S.; Takahashi, Y.; Tamari, K.; Minami, K.; Takenaka, W.; Ibuki, Y.; Yamamoto, J.; Tatekawa, S.; Hayashi, K.; Seo, Y.; et al. Radiation therapy enhances systemic antitumor efficacy in PD-L1 therapy regardless of sequence of radiation in murine osteosarcoma. PLoS ONE 2022, 17, e0271205. [Google Scholar] [CrossRef]
- Yoshimoto, Y.; Suzuki, Y.; Mimura, K.; Ando, K.; Oike, T.; Sato, H.; Okonogi, N.; Maruyama, T.; Izawa, S.; Noda, S.E.; et al. Radiotherapy-induced anti-tumor immunity contributes to the therapeutic efficacy of irradiation and can be augmented by CTLA-4 blockade in a mouse model. PLoS ONE 2014, 9, e92572. [Google Scholar] [CrossRef]
- Demaria, S.; Kawashima, N.; Yang, A.M.; Devitt, M.L.; Babb, J.S.; Allison, J.P.; Formenti, S.C. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 2005, 11, 728–734. [Google Scholar] [CrossRef]
- Koller, K.M.; Mackley, H.B.; Liu, J.; Wagner, H.; Talamo, G.; Schell, T.D.; Pameijer, C.; Neves, R.I.; Anderson, B.; Kokolus, K.M.; et al. Improved survival and complete response rates in patients with advanced melanoma treated with concurrent ipilimumab and radiotherapy versus ipilimumab alone. Cancer Biol. Ther. 2017, 18, 36–42. [Google Scholar] [CrossRef]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef]
- Ochoa-de-Olza, M.; Bourhis, J.; Coukos, G.; Herrera, F.G. Low-dose irradiation for reversing immunotherapy resistance: How to translate? J. Immunother. Cancer 2022, 10, e004939. [Google Scholar] [CrossRef]
- Mgrditchian, T.; Arakelian, T.; Paggetti, J.; Noman, M.Z.; Viry, E.; Moussay, E.; Van Moer, K.; Kreis, S.; Guerin, C.; Buart, S.; et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc. Natl. Acad. Sci. USA 2017, 114, E9271–E9279. [Google Scholar] [CrossRef]
- Noman, M.Z.; Paggetti, J.; Moussay, E.; Berchem, G.; Janji, B. Driving Natural Killer cells toward the melanoma tumor battlefield: Autophagy as a valuable therapeutic target. Oncoimmunology 2018, 7, e1452583. [Google Scholar] [CrossRef]
- Janji, B.; Hasmim, M.; Parpal, S.; Berchem, G.; Noman, M.Z. Firing up the cold tumors by targeting Vps34. Oncoimmunology 2020, 9, 1809936. [Google Scholar] [CrossRef]
- Janji, B.; Hasmim, M.; Parpal, S.; De Milito, A.; Berchem, G.; Noman, M.Z. Lighting up the fire in cold tumors to improve cancer immunotherapy by blocking the activity of the autophagy-related protein PIK3C3/VPS34. Autophagy 2020, 16, 2110–2111. [Google Scholar] [CrossRef]
- Noman, M.Z.; Parpal, S.; Van Moer, K.; Xiao, M.; Yu, Y.; Viklund, J.; De Milito, A.; Hasmim, M.; Andersson, M.; Amaravadi, R.K.; et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci. Adv. 2020, 6, eaax7881. [Google Scholar] [CrossRef]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef]
- Duffy, A.; Le, J.; Sausville, E.; Emadi, A. Autophagy modulation: A target for cancer treatment development. Cancer Chemother. Pharmacol. 2015, 75, 439–447. [Google Scholar] [CrossRef]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef]
- Bourgeois-Daigneault, M.C.; Roy, D.G.; Aitken, A.S.; El Sayes, N.; Martin, N.T.; Varette, O.; Falls, T.; St-Germain, L.E.; Pelin, A.; Lichty, B.D.; et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018, 10, eaao1641. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Chen, H.; Zhu, Z.; Liu, Z.; Ma, C.; Lee, Y.J.; Bartlett, D.L.; Guo, Z.S. Ferroptosis Inducer Improves the Efficacy of Oncolytic Virus-Mediated Cancer Immunotherapy. Biomedicines 2022, 10, 1425. [Google Scholar] [CrossRef] [PubMed]
- Kudling, T.V.; Clubb, J.H.A.; Quixabeira, D.C.A.; Santos, J.M.; Havunen, R.; Kononov, A.; Heiniö, C.; Cervera-Carrascon, V.; Pakola, S.; Basnet, S.; et al. Local delivery of interleukin 7 with an oncolytic adenovirus activates tumor-infiltrating lymphocytes and causes tumor regression. Oncoimmunology 2022, 11, 2096572. [Google Scholar] [CrossRef] [PubMed]
- Nisar, M.; Paracha, R.Z.; Adil, S.; Qureshi, S.N.; Janjua, H.A. An Extensive Review on Preclinical and Clinical Trials of Oncolytic Viruses Therapy for Pancreatic Cancer. Front. Oncol. 2022, 12, 875188. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Oduro, P.K.; Guo, R.; Li, R.; Leng, L.; Kong, X.; Wang, Q.; Yang, L. Oncolytic Viruses: Immunotherapy Drugs for Gastrointestinal Malignant Tumors. Front. Cell. Infect. Microbiol. 2022, 12, 921534. [Google Scholar] [CrossRef]
- Shoaf, M.L.; Desjardins, A. Oncolytic Viral Therapy for Malignant Glioma and Their Application in Clinical Practice. Neurother. J. Am. Soc. Exp. NeuroTher. 2022, 19, 1818–1831. [Google Scholar] [CrossRef]
- Ripp, J.; Hentzen, S.; Saeed, A. Oncolytic Viruses as an Adjunct to Immune Checkpoint Inhibition. Front. Biosci. (Landmark Ed.) 2022, 27, 151. [Google Scholar] [CrossRef]
- Newman, J.H.; Chesson, C.B.; Herzog, N.L.; Bommareddy, P.K.; Aspromonte, S.M.; Pepe, R.; Estupinian, R.; Aboelatta, M.M.; Buddhadev, S.; Tarabichi, S.; et al. Intratumoral injection of the seasonal flu shot converts immunologically cold tumors to hot and serves as an immunotherapy for cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 1119–1128. [Google Scholar] [CrossRef]
- Lin, Z.; Zhang, Y.; Cai, H.; Zhou, F.; Gao, H.; Deng, L.; Li, R. A PD-L1-Based Cancer Vaccine Elicits Antitumor Immunity in a Mouse Melanoma Model. Mol. Ther. Oncolytics 2019, 14, 222–232. [Google Scholar] [CrossRef]
- Bevers, S.; Kooijmans, S.A.A.; Van de Velde, E.; Evers, M.J.W.; Seghers, S.; Gitz-Francois, J.; van Kronenburg, N.C.H.; Fens, M.; Mastrobattista, E.; Hassler, L.; et al. mRNA-LNP vaccines tuned for systemic immunization induce strong antitumor immunity by engaging splenic immune cells. Mol. Ther. J. Am. Soc. Gene Ther. 2022, 30, 3078–3094. [Google Scholar] [CrossRef]
- Sun, H.; Zhang, Y.; Wang, G.; Yang, W.; Xu, Y. mRNA-Based Therapeutics in Cancer Treatment. Pharmaceutics 2023, 15, 622. [Google Scholar] [CrossRef]
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Türeci, Ö.; Vormehr, M.; Kranz, L.M. mRNA therapeutics in cancer immunotherapy. Mol. Cancer 2021, 20, 69. [Google Scholar] [CrossRef]
- Jiang, X.T.; Liu, Q. mRNA vaccination in breast cancer: Current progress and future direction. J. Cancer Res. Clin. Oncol. 2023. [Google Scholar] [CrossRef]
- Li, Q.; Teng, Z.; Tao, J.; Shi, W.; Yang, G.; Zhang, Y.; Su, X.; Chen, L.; Xiu, W.; Yuwen, L.; et al. Elastic Nanovaccine Enhances Dendritic Cell-Mediated Tumor Immunotherapy. Small 2022, 18, e2201108. [Google Scholar] [CrossRef]
- Ju, F.; Luo, Y.; Lin, C.; Jia, X.; Xu, Z.; Tian, R.; Lin, Y.; Zhao, M.; Chang, Y.; Huang, X.; et al. Oncolytic virus expressing PD-1 inhibitors activates a collaborative intratumoral immune response to control tumor and synergizes with CTLA-4 or TIM-3 blockade. J. Immunother. Cancer 2022, 10, e004762. [Google Scholar] [CrossRef]
- Hagihara, K.; Chan, S.; Zhang, L.; Oh, D.Y.; Wei, X.X.; Simko, J.; Fong, L. Neoadjuvant sipuleucel-T induces both Th1 activation and immune regulation in localized prostate cancer. Oncoimmunology 2019, 8, e1486953. [Google Scholar] [CrossRef]
- Rodallec, A.; Sicard, G.; Fanciullino, R.; Benzekry, S.; Lacarelle, B.; Milano, G.; Ciccolini, J. Turning cold tumors into hot tumors: Harnessing the potential of tumor immunity using nanoparticles. Expert. Opin. Drug Metab. Toxicol. 2018, 14, 1139–1147. [Google Scholar] [CrossRef]
- Duan, X.; Chan, C.; Lin, W. Nanoparticle-Mediated Immunogenic Cell Death Enables and Potentiates Cancer Immunotherapy. Angew. Chem. (Int. Ed. Engl.) 2019, 58, 670–680. [Google Scholar] [CrossRef]
- Navarro-Ocón, A.; Blaya-Cánovas, J.L.; López-Tejada, A.; Blancas, I.; Sánchez-Martín, R.M.; Garrido, M.J.; Griñán-Lisón, C.; Calahorra, J.; Cara, F.E.; Ruiz-Cabello, F.; et al. Nanomedicine as a Promising Tool to Overcome Immune Escape in Breast Cancer. Pharmaceutics 2022, 14, 505. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, C. Regulation of cancer-immunity cycle and tumor microenvironment by nanobiomaterials to enhance tumor immunotherapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, e1612. [Google Scholar] [CrossRef]
- Zhang, D.; Li, Q.; Chen, X.; Nie, X.; Xue, F.; Xu, W.; Luan, Y. An Injectable Hydrogel to Modulate T Cells for Cancer Immunotherapy. Small 2022, 18, e2202663. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, D.; Bertelli, G.; Pronzato, P.; Vidili, M.G.; Gulisano, M.; Lionetto, R.; Rosso, R. Continuous venous infusion of vindesine in metastatic breast cancer: Experience with a subcutaneously implanted system and portable pump. Anticancer Res. 1989, 9, 141–143. [Google Scholar] [PubMed]
- Song, P.; Han, X.; Zheng, R.; Yan, J.; Wu, X.; Wang, Y.; Zhang, H. Upregulation of MHC-I and downregulation of PD-L1 expression by doxorubicin and deferasirox codelivered liposomal nanoparticles for chemoimmunotherapy of melanoma. Int. J. Pharm. 2022, 624, 122002. [Google Scholar] [CrossRef] [PubMed]
- Merino, M.; Zalba, S.; Garrido, M.J. Immunoliposomes in clinical oncology: State of the art and future perspectives. J. Control. Release 2018, 275, 162–176. [Google Scholar] [CrossRef]
- Merino, M.; Lozano, T.; Casares, N.; Lana, H.; Troconiz, I.F.; Ten Hagen, T.L.M.; Kochan, G.; Berraondo, P.; Zalba, S.; Garrido, M.J. Dual activity of PD-L1 targeted Doxorubicin immunoliposomes promoted an enhanced efficacy of the antitumor immune response in melanoma murine model. J. Nanobiotechnol. 2021, 19, 102. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, J.; Wang, Y.; Han, Y.; Zhao, X.; Hu, H.; Qiao, M.; Chen, D. Blockage of the IDO1 pathway by charge-switchable nanoparticles amplifies immunogenic cell death for enhanced cancer immunotherapy. Acta Biomater. 2022, 150, 353–366. [Google Scholar] [CrossRef]
- Abadi, B.; Yazdanpanah, N.; Nokhodchi, A.; Rezaei, N. Smart biomaterials to enhance the efficiency of immunotherapy in glioblastoma: State of the art and future perspectives. Adv. Drug Deliv. Rev. 2021, 179, 114035. [Google Scholar] [CrossRef]
- Shabani, L.; Abbasi, M.; Amini, M.; Amani, A.M.; Vaez, A. The brilliance of nanoscience over cancer therapy: Novel promising nanotechnology-based methods for eradicating glioblastoma. J. Neurol. Sci. 2022, 440, 120316. [Google Scholar] [CrossRef]
- Liu, S.; Liu, J.; Li, H.; Mao, K.; Wang, H.; Meng, X.; Wang, J.; Wu, C.; Chen, H.; Wang, X.; et al. An optimized ionizable cationic lipid for brain tumor-targeted siRNA delivery and glioblastoma immunotherapy. Biomaterials 2022, 287, 121645. [Google Scholar] [CrossRef]
- Alghamri, M.S.; Banerjee, K.; Mujeeb, A.A.; Mauser, A.; Taher, A.; Thalla, R.; McClellan, B.L.; Varela, M.L.; Stamatovic, S.M.; Martinez-Revollar, G.; et al. Systemic Delivery of an Adjuvant CXCR4-CXCL12 Signaling Inhibitor Encapsulated in Synthetic Protein Nanoparticles for Glioma Immunotherapy. ACS Nano 2022, 16, 8729–8750. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, H.; Qiu, W.; Han, Y.; Liu, H.; Li, Z. Biomimetic nanoparticles directly remodel immunosuppressive microenvironment for boosting glioblastoma immunotherapy. Bioact. Mater. 2022, 16, 418–432. [Google Scholar] [CrossRef]
- Qian, Y.; Mao, J.; Leng, X.; Zhu, L.; Rui, X.; Jin, Z.; Jiang, H.; Liu, H.; Zhang, F.; Bi, X.; et al. Co-delivery of proanthocyanidin and mitoxantrone induces synergistic immunogenic cell death to potentiate cancer immunotherapy. Biomater. Sci. 2022, 10, 4549–4560. [Google Scholar] [CrossRef]
- Wu, X.; Wei, Y.; Lin, R.; Chen, P.; Hong, Z.; Zeng, R.; Xu, Q.; Li, T. Multi-responsive mesoporous polydopamine composite nanorods cooperate with nano-enzyme and photosensitiser for intensive immunotherapy of bladder cancer. Immunology 2022, 167, 247–262. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Foley, K.C.; Nishimura, M.I.; Moore, T.V. Combination immunotherapies implementing adoptive T-cell transfer for advanced-stage melanoma. Melanoma Res. 2018, 28, 171–184. [Google Scholar] [CrossRef]
- Yin, H.; Guo, W.; Sun, X.; Li, R.; Feng, C.; Tan, Y. TILs and Anti-PD1 Therapy: An Alternative Combination Therapy for PDL1 Negative Metastatic Cervical Cancer. J. Immunol. Res. 2020, 2020, 8345235. [Google Scholar] [CrossRef]
- Shafer, P.; Kelly, L.M.; Hoyos, V. Cancer Therapy With TCR-Engineered T Cells: Current Strategies, Challenges, and Prospects. Front. Immunol. 2022, 13, 835762. [Google Scholar] [CrossRef]
- Yarza, R.; Bover, M.; Herrera-Juarez, M.; Rey-Cardenas, M.; Paz-Ares, L.; Lopez-Martin, J.A.; Haanen, J. Efficacy of T-Cell Receptor-Based Adoptive Cell Therapy in Cutaneous Melanoma: A Meta-Analysis. Oncol. 2023, 28, e406–e415. [Google Scholar] [CrossRef]
- Biernacki, M.A.; Brault, M.; Bleakley, M. T-Cell Receptor-Based Immunotherapy for Hematologic Malignancies. Cancer J. 2019, 25, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Hu, W. Advances in adoptive cellular therapy for colorectal cancer: A narrative review. Ann. Transl. Med. 2022, 10, 1404. [Google Scholar] [CrossRef] [PubMed]
- Salter, A.I.; Rajan, A.; Kennedy, J.J.; Ivey, R.G.; Shelby, S.A.; Leung, I.; Templeton, M.L.; Muhunthan, V.; Voillet, V.; Sommermeyer, D.; et al. Comparative analysis of TCR and CAR signaling informs CAR designs with superior antigen sensitivity and in vivo function. Sci. Signal. 2021, 14, eabe2606. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef]
- Fergusson, N.J.; Adeel, K.; Kekre, N.; Atkins, H.; Hay, K.A. A systematic review and meta-analysis of CD22 CAR T-cells alone or in combination with CD19 CAR T-cells. Front. Immunol. 2023, 14, 1178403. [Google Scholar] [CrossRef]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D., Jr.; June, C.H. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef]
- Yang, S.L.; Wu, C.; Xiong, Z.F.; Fang, X. Progress on hypoxia-inducible factor-3: Its structure, gene regulation and biological function (Review). Mol. Med. Rep. 2015, 12, 2411–2416. [Google Scholar] [CrossRef]
- Palazon, A.; Tyrakis, P.A.; Macias, D.; Veliça, P.; Rundqvist, H.; Fitzpatrick, S.; Vojnovic, N.; Phan, A.T.; Loman, N.; Hedenfalk, I.; et al. An HIF-1α/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell 2017, 32, 669–683.e5. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci. STKE 2007, 2007, cm8. [Google Scholar] [CrossRef]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef]
- Carmeliet, P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69 (Suppl. S3), 4–10. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef]
- Mohl, D.A.; Lagies, S.; Zodel, K.; Zumkeller, M.; Peighambari, A.; Ganner, A.; Plattner, D.A.; Neumann-Haefelin, E.; Adlesic, M.; Frew, I.J.; et al. Integrated Metabolomic and Transcriptomic Analysis of Modified Nucleosides for Biomarker Discovery in Clear Cell Renal Cell Carcinoma. Cells 2023, 12, 1102. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim. Biophys. Acta 2011, 1813, 1263–1268. [Google Scholar] [CrossRef]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef]
- Joseph, J.P.; Harishankar, M.K.; Pillai, A.A.; Devi, A. Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 2018, 80, 23–32. [Google Scholar] [CrossRef]
- Piñeiro Fernández, J.; Luddy, K.A.; Harmon, C.; O’Farrelly, C. Hepatic Tumor Microenvironments and Effects on NK Cell Phenotype and Function. Int. J. Mol. Sci. 2019, 20, 4131. [Google Scholar] [CrossRef]
- Hayes, C.; Donohoe, C.L.; Davern, M.; Donlon, N.E. The oncogenic and clinical implications of lactate induced immunosuppression in the tumour microenvironment. Cancer Lett. 2021, 500, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Murthy, A.; Gerber, S.A.; Koch, C.J.; Lord, E.M. Intratumoral Hypoxia Reduces IFN-γ-Mediated Immunity and MHC Class I Induction in a Preclinical Tumor Model. ImmunoHorizons 2019, 3, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Sethumadhavan, S.; Silva, M.; Philbrook, P.; Nguyen, T.; Hatfield, S.M.; Ohta, A.; Sitkovsky, M.V. Hypoxia and hypoxia-inducible factor (HIF) downregulate antigen-presenting MHC class I molecules limiting tumor cell recognition by T cells. PLoS ONE 2017, 12, e0187314. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, Y.; Lu, Y.; Wang, M.; Li, Y.; Wang, C.; Li, Q.; Zhao, H. The regulation of immune checkpoints by the hypoxic tumor microenvironment. PeerJ 2021, 9, e11306. [Google Scholar] [CrossRef]
- Labiano, S.; Palazon, A.; Melero, I. Immune response regulation in the tumor microenvironment by hypoxia. Semin. Oncol. 2015, 42, 378–386. [Google Scholar] [CrossRef]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Xiao, M.; Benoit, A.; Hasmim, M.; Duhem, C.; Vogin, G.; Berchem, G.; Noman, M.Z.; Janji, B. Targeting Cytoprotective Autophagy to Enhance Anticancer Therapies. Front. Oncol. 2021, 11, 626309. [Google Scholar] [CrossRef]
- Noman, M.Z.; Janji, B.; Kaminska, B.; Van Moer, K.; Pierson, S.; Przanowski, P.; Buart, S.; Berchem, G.; Romero, P.; Mami-Chouaib, F.; et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011, 71, 5976–5986. [Google Scholar] [CrossRef]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, X.; Li, E.; Zhang, G.; Wang, X.; Tang, T.; Bai, X.; Liang, T. VISTA: An immune regulatory protein checking tumor and immune cells in cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 83. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Li, X.F. The Hypoxia-Activated Prodrug TH-302: Exploiting Hypoxia in Cancer Therapy. Front. Pharmacol. 2021, 12, 636892. [Google Scholar] [CrossRef]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Sun, Y.; et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef]
- Phillips, R.M. Targeting the hypoxic fraction of tumours using hypoxia-activated prodrugs. Cancer Chemother. Pharmacol. 2016, 77, 441–457. [Google Scholar] [CrossRef]
- Wang, Y.; Shang, W.; Niu, M.; Tian, J.; Xu, K. Hypoxia-active nanoparticles used in tumor theranostic. Int. J. Nanomed. 2019, 14, 3705–3722. [Google Scholar] [CrossRef]
- Zhou, T.; Liang, X.; Wang, P.; Hu, Y.; Qi, Y.; Jin, Y.; Du, Y.; Fang, C.; Tian, J. A Hepatocellular Carcinoma Targeting Nanostrategy with Hypoxia-Ameliorating and Photothermal Abilities that, Combined with Immunotherapy, Inhibits Metastasis and Recurrence. ACS Nano 2020, 14, 12679–12696. [Google Scholar] [CrossRef]
- Lee, K.; Zhang, H.; Qian, D.Z.; Rey, S.; Liu, J.O.; Semenza, G.L. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci. USA 2009, 106, 17910–17915. [Google Scholar] [CrossRef]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benoit, A.; Vogin, G.; Duhem, C.; Berchem, G.; Janji, B. Lighting Up the Fire in the Microenvironment of Cold Tumors: A Major Challenge to Improve Cancer Immunotherapy. Cells 2023, 12, 1787. https://doi.org/10.3390/cells12131787
Benoit A, Vogin G, Duhem C, Berchem G, Janji B. Lighting Up the Fire in the Microenvironment of Cold Tumors: A Major Challenge to Improve Cancer Immunotherapy. Cells. 2023; 12(13):1787. https://doi.org/10.3390/cells12131787
Chicago/Turabian StyleBenoit, Alice, Guillaume Vogin, Caroline Duhem, Guy Berchem, and Bassam Janji. 2023. "Lighting Up the Fire in the Microenvironment of Cold Tumors: A Major Challenge to Improve Cancer Immunotherapy" Cells 12, no. 13: 1787. https://doi.org/10.3390/cells12131787
APA StyleBenoit, A., Vogin, G., Duhem, C., Berchem, G., & Janji, B. (2023). Lighting Up the Fire in the Microenvironment of Cold Tumors: A Major Challenge to Improve Cancer Immunotherapy. Cells, 12(13), 1787. https://doi.org/10.3390/cells12131787