



Improved Media Formulations for Primary Cell Cultures Derived from a Colonial Urochordate


 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. B. schlosseri Husbandry
2.2. B. schlosseri Cell Extraction under Aseptic Conditions
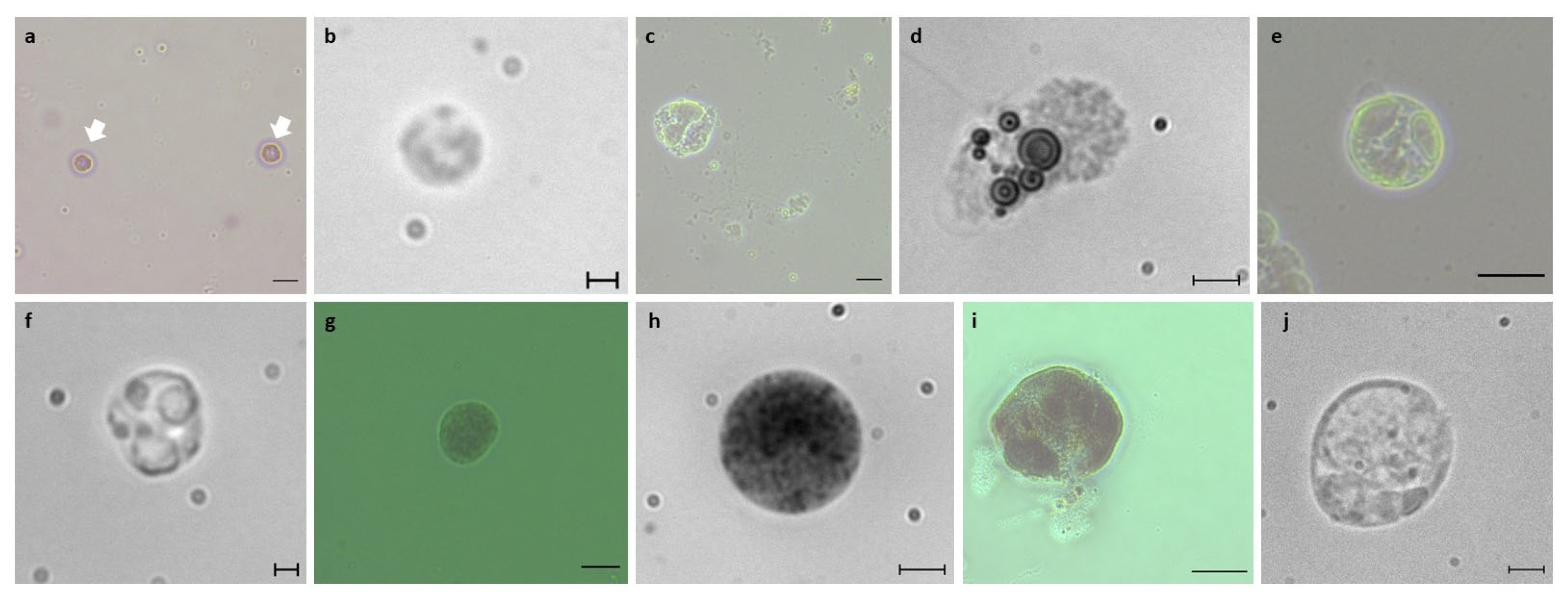
2.3. Blood Cell Observations
2.4. Media
2.5. General Culture Conditions
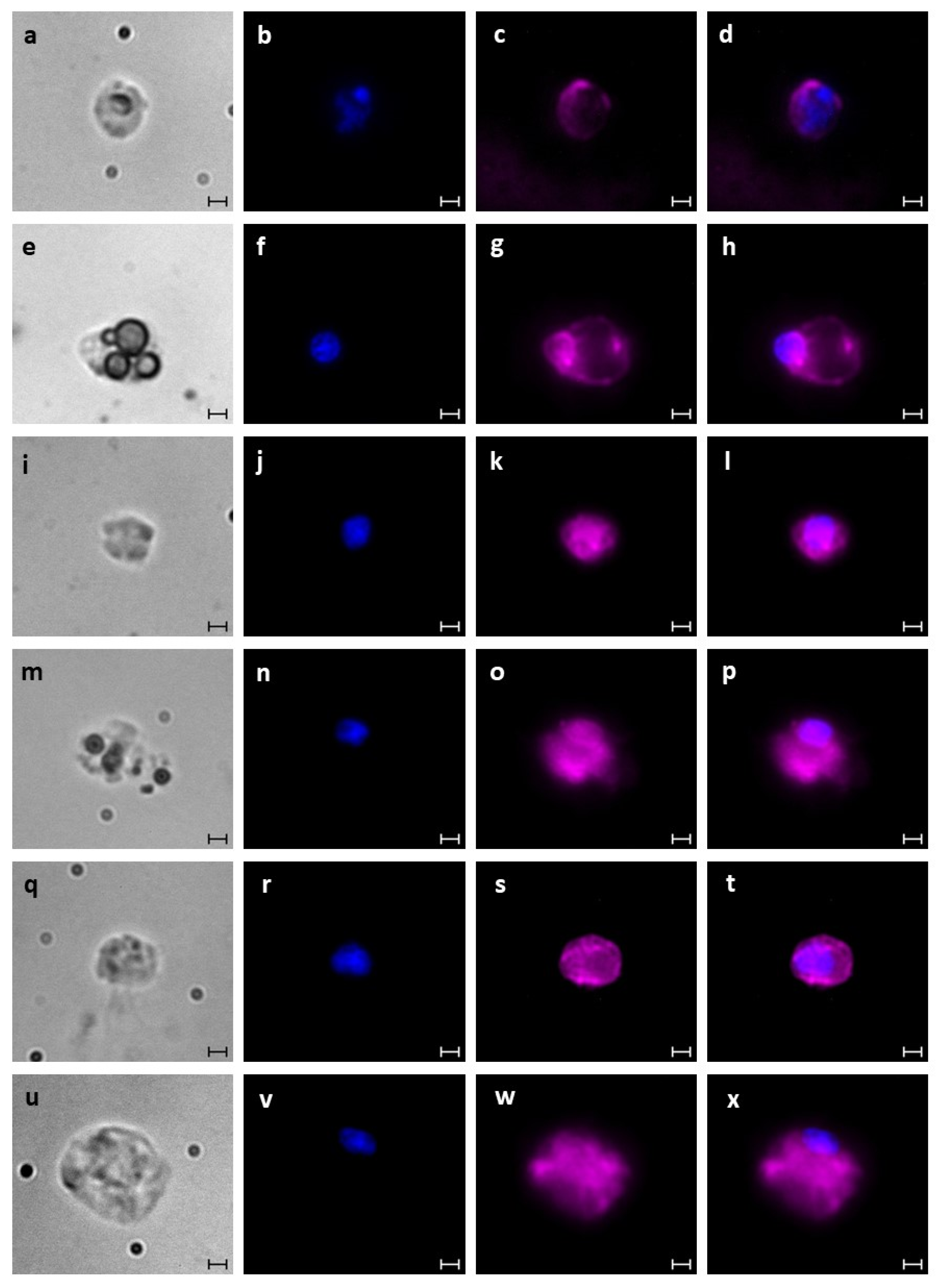
2.6. Blood Cell Characterization
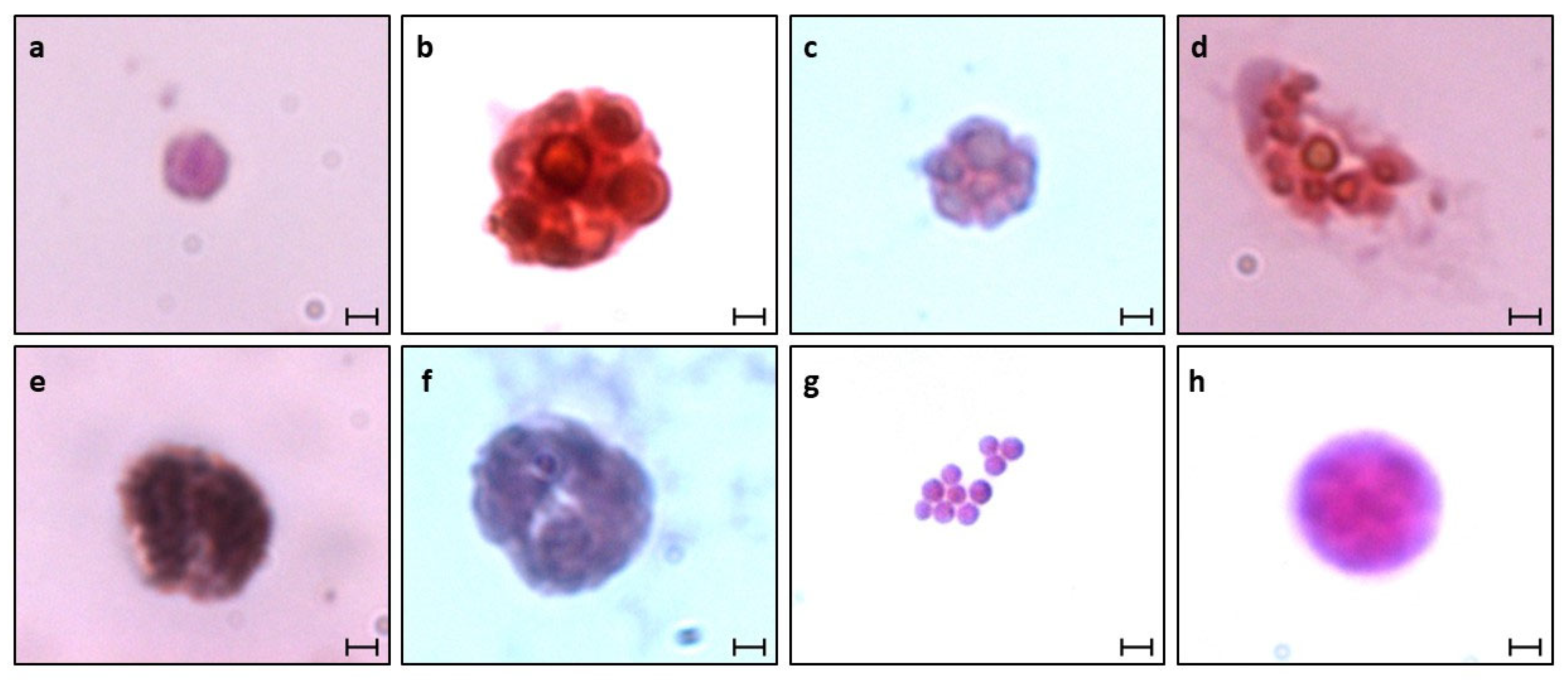
2.7. Hematoxylin and Eosin (H&E) Staining
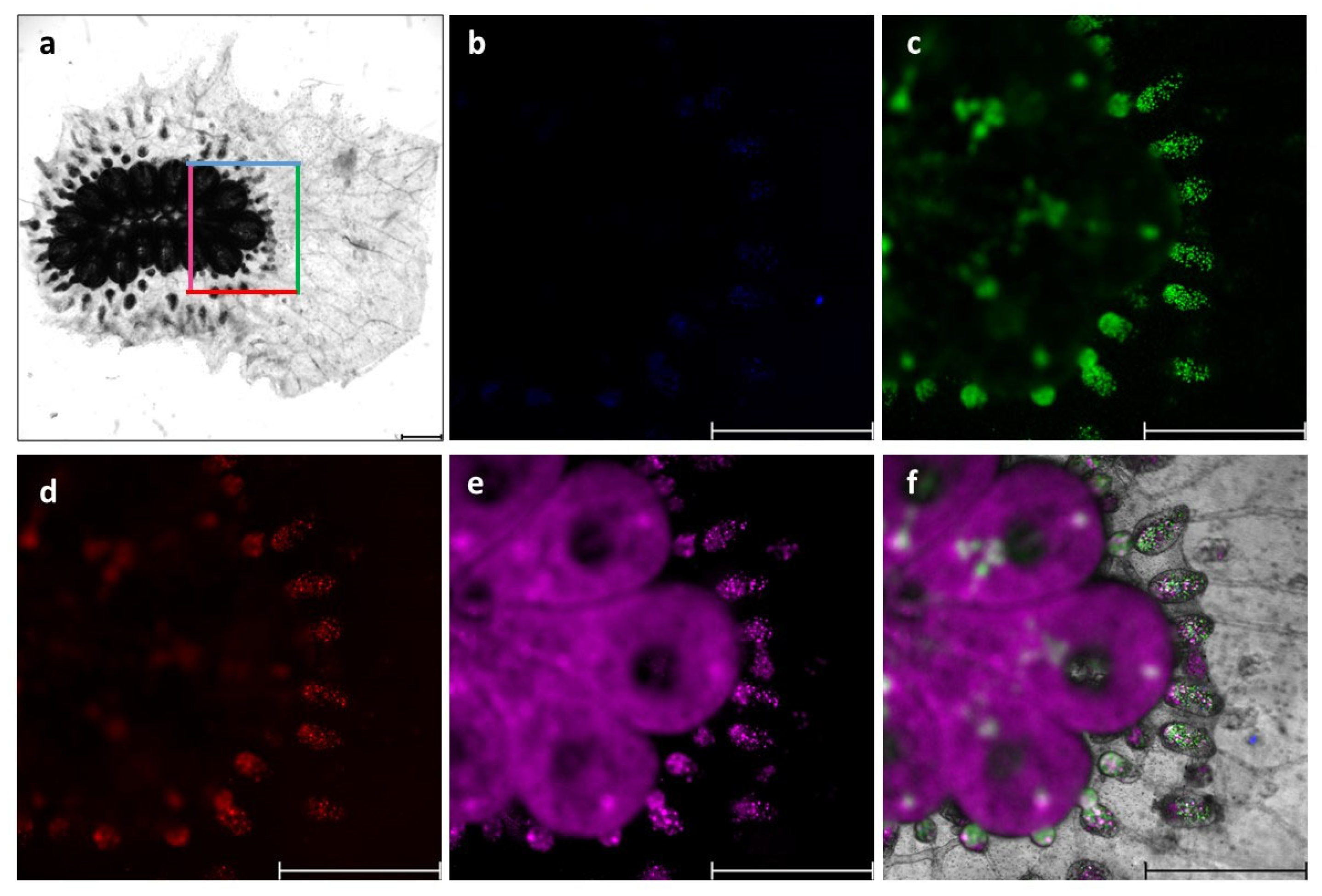
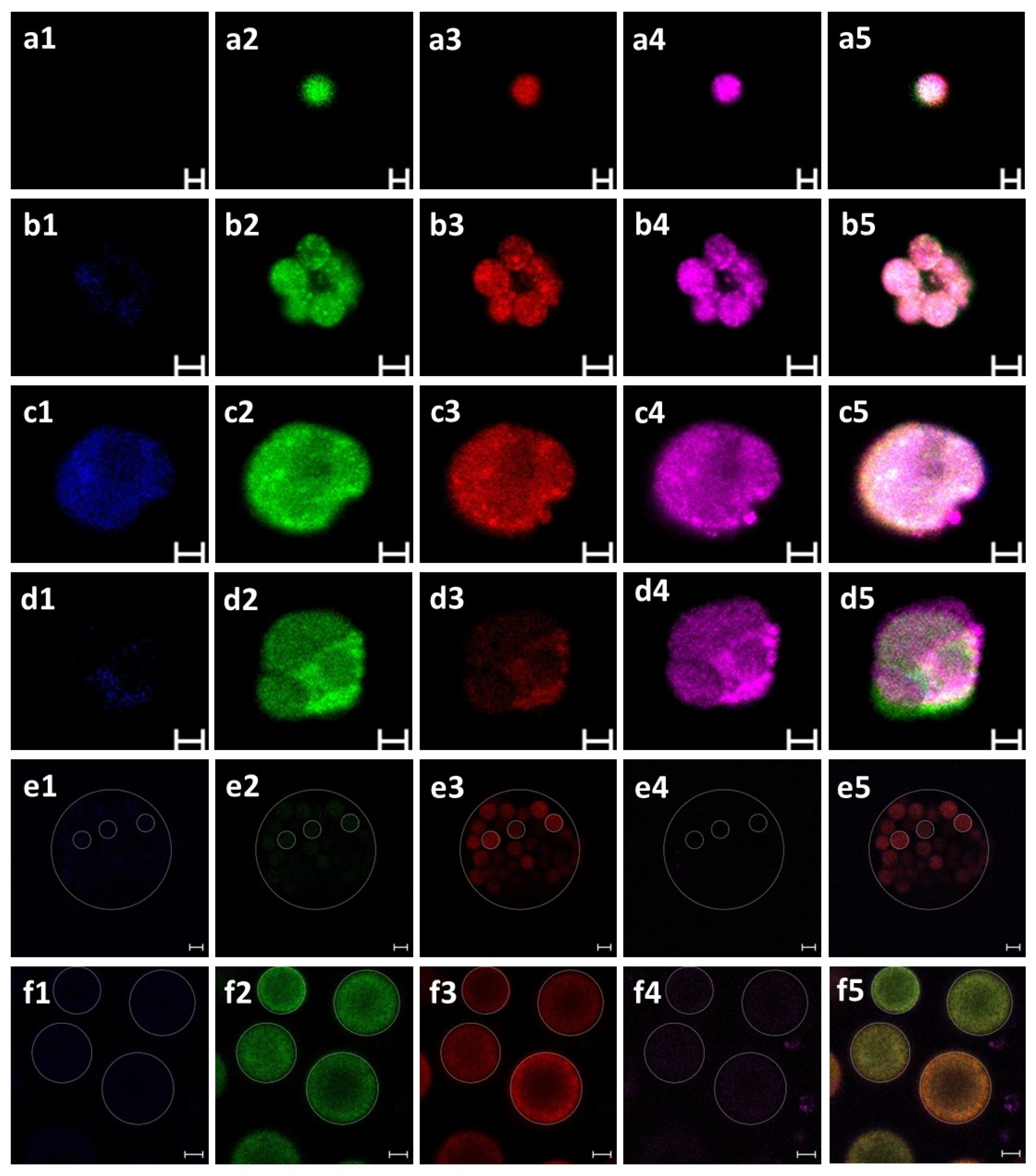
2.8. Immunofluorescence Staining
2.9. Statistics
3. Results
3.1. Identification of B. schlosseri Blood Cell Populations
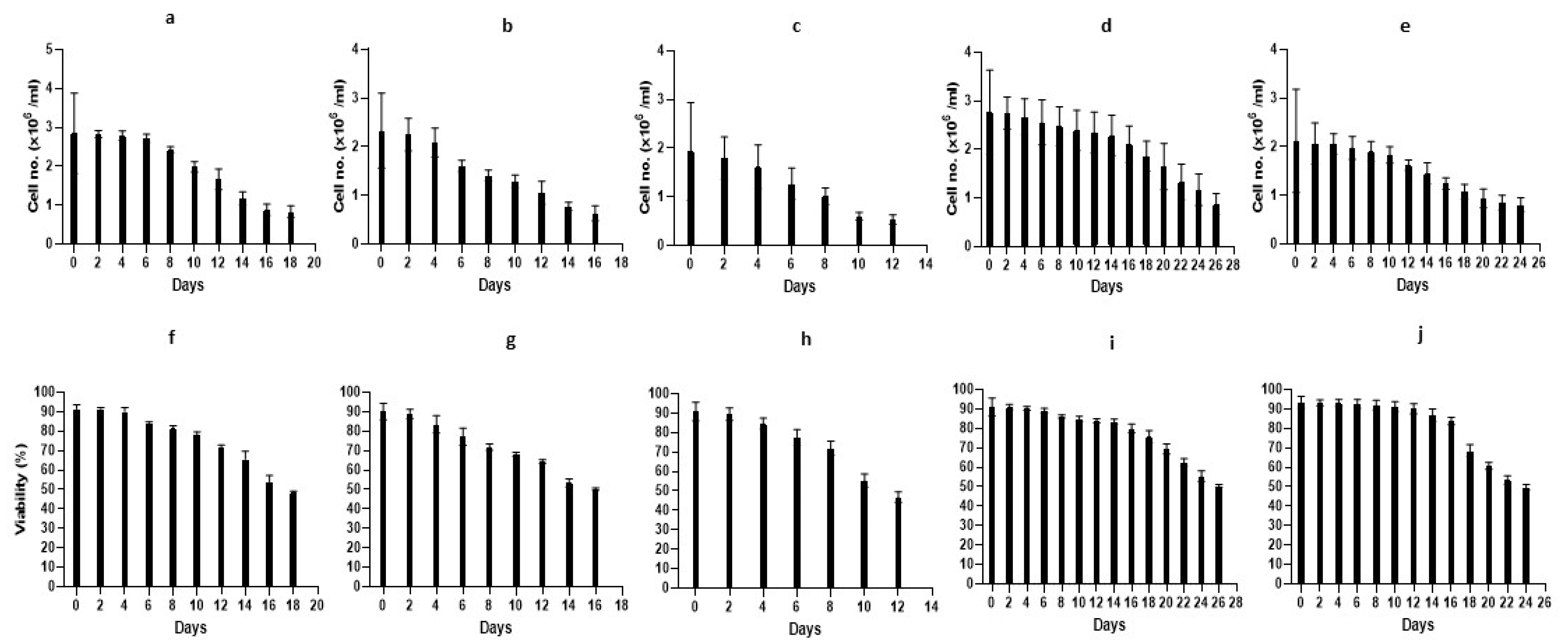
3.2. Cultivation of B. schlosseri Blood Cells
3.2.1. Primary Cultures: TGM1 Medium
3.2.2. Primary Cultures: TGM2 Medium
3.2.3. Primary Cultures: TGM3 Medium
3.2.4. Primary Cultures: TGM4 Medium
3.2.5. Primary Cultures: TGM5 Medium
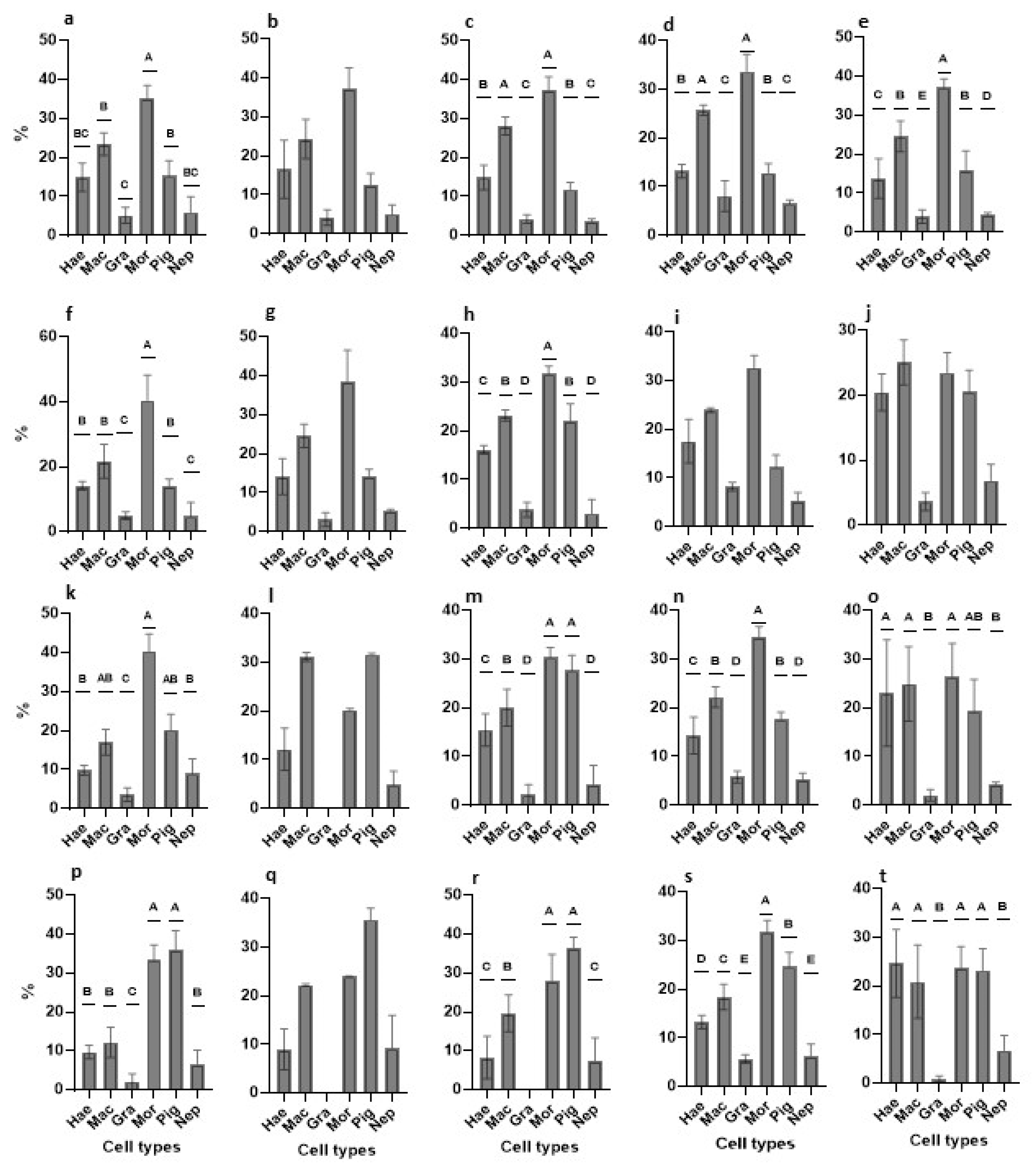
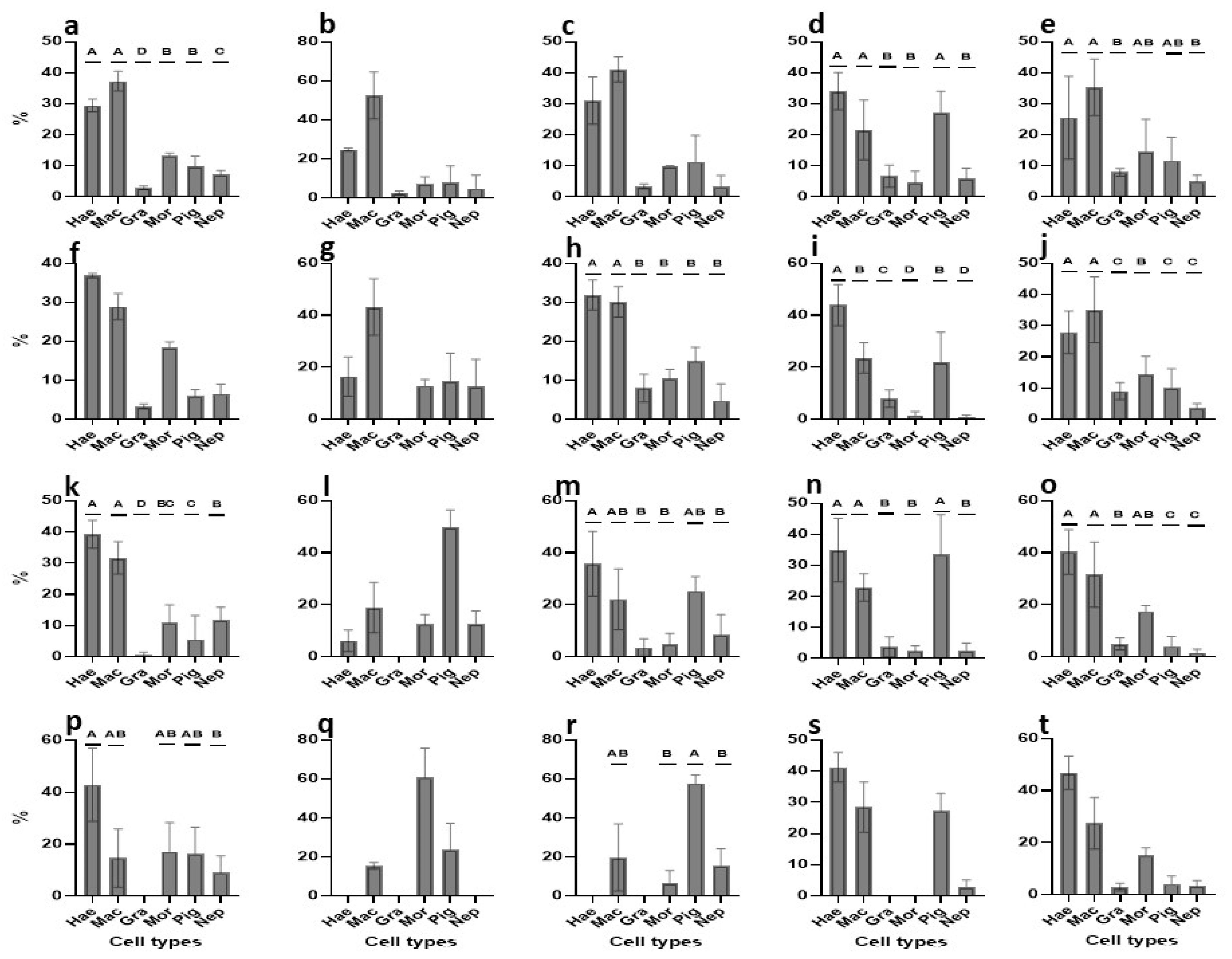
3.2.6. Cell Types Changes in Primary Cultures—An Overview
3.2.7. Proliferation of Primary Cultures—An Overview
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rinkevich, B. Cell cultures from marine invertebrates: Obstacles, new approaches and recent improvements. J. Biotechnol. 1999, 70, 133–153. [Google Scholar] [CrossRef]
- Rinkevich, B. Cell cultures from marine invertebrates: New insights for capturing endless stemness. J. Mar. Biotechnol. 2011, 13, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Mothersill, C.; Austin, B. Aquatic Invertebrate Cell Culture; Springer: Berlin/Heidelberg, Germany, 2000; pp. 133–153. [Google Scholar]
- Munroe, S.; Sandoval, K.; Martens, D.E.; Sipkema, D.; Pomponi, S.A. Genetic algorithm as an optimization tool for the development of sponge cell culture media. In Vitro Cell. Dev. Biol. Anim. 2019, 55, 149–158. [Google Scholar] [CrossRef]
- Rosner, A.; Armengaud, J.; Ballarin, L.; Barnay-Verdier, S.; Cima, F.; Coelho, A.V.; Domart-Coulon, I.; Drobne, D.; Genevière, A.M.; Jemec, K.A.; et al. Stem cells of aquatic invertebrates as an advanced tool for assessing ecotoxicological impacts. Sci. Total Environ. 2021, 771, 144565. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, S.; Singh, I.B.; Puthumana, J. Status in molluscan cell line development in last one decade (2010–2020): Impediments and way forward. Cytotechnology 2022, 74, 433–457. [Google Scholar] [CrossRef]
- Domart-Coulon, I.; Blanchoud, S. From Primary Cell and Tissue Cultures to Aquatic Invertebrate Cell Lines: An Updated Overview. In Advances in Aquatic Invertebrate Stem Cell Research: From Basic Research to Innovative Applications, 3rd ed.; Ballarin., L., Rinkevich., B., Hobmayer, B., Eds.; MDPI: Basel, Switzerland, 2022; pp. 1–64. [Google Scholar] [CrossRef]
- Hesp, K.; van der Heijden, J.M.; Munroe, S.; Sipkema, D.; Martens, D.E.; Wijffels, R.H.; Pomponi, S.A. First continuous marine sponge cell line established. Sci. Rep. 2023, 13, 5766. [Google Scholar] [CrossRef]
- Rinkevich, B. Marine invertebrate cell cultures: New millennium trends. J. Mar. Biotechnol. 2005, 7, 429–439. [Google Scholar] [CrossRef]
- Rinkevich, B.; Frank, U.; Gateño, D.; Rabinowitz, C. The establishment of various cell lines from colonial marine invertebrates. In Use of Aquatic Invertebrates as Tools for Monitoring of Environmental Hazards; Mueller, W.E.G., Ed.; Gustav Fischer: Jena, Germany, 1994; pp. 253–263. [Google Scholar]
- Bayne, C.J. Invertebrate cell culture considerations: Insects, ticks, shellfish, and worms. Methods Cell Biol. 1998, 57, 187–201. [Google Scholar] [CrossRef]
- Grasela, J.J.; Pomponi, S.A.; Rinkevich, B.; Grima, J. Efforts to develop a cultured sponge cell line: Revisiting an intractable problem. In Vitro Cell. Dev. Biol. Anim. 2012, 48, 12–20. [Google Scholar] [CrossRef]
- Conkling, M.; Hesp, K.; Munroe, S.; Sandoval, K.; Martens, D.E.; Sipkema, D.; Pomponi, S.A. Breakthrough in marine invertebrate cell culture: Sponge cells divide rapidly in improved nutrient medium. Sci. Rep. 2019, 9, 17321. [Google Scholar] [CrossRef]
- Manni, L.; Anselmi, C.; Cima, F.; Gasparini, F.; Voskoboynik, A.; Martini, M.; Peronato, A.; Burighel, P.; Zaniolo, G.; Ballarin, L. Sixty years of experimental studies on the blastogenesis of the colonial tunicate Botryllus schlosseri. Dev. Biol. 2019, 448, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Ben-Hamo, O.; Rinkevich, B. Botryllus schlosseri—A model colonial species in basic and applied studies. In Handbook of Marine Model Organisms in Experimental Biology. Established and Emerging, 2nd ed.; Boutet, A., Schierwater, B., Eds.; CRC Press: Boca Raton, FL, USA, 2021; pp. 385–402. [Google Scholar]
- Laird, D.J.; De Tomaso, A.W.; Weissman, I.L. Stem cells are units of natural selection in a colonial ascidian. Cell 2005, 123, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Rinkevich, B.; Rabinowitz, C. Acquiring embryo-derived cell cultures and aseptic metamorphosis of larvae from the colonial protochordate Botryllus schlosseri. Invertebr. Rep. Dev. 1994, 25, 59–72. [Google Scholar] [CrossRef]
- Rinkevich, B.; Rabinowitz, C. Initiation of epithelial cell cultures from palleal buds of Botryllus schlosseri, a colonial tunicate. In Vitro Cell. Dev. Biol. Anim. 1997, 33, 422–424. [Google Scholar] [CrossRef]
- Rosner, A.; Moiseeva, E.; Rinkevich, Y.; Lapidot, Z.; Rinkevich, B. Vasa and the germ line lineage in a colonial urochordate. Dev. Biol. 2009, 331, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Rinkevich, Y.; Voskoboynik, A.; Rosner, A.; Rabinowitz, C.; Oren, M.; Paz, G.; Alfasi, G.; Moiseeva, E.; Ishizuka, K.J.; Palmeri, K.J.; et al. Repeated, long-term cycling of putative stem cells between niches in a basal chordate. Dev. Cell 2013, 24, 76–88. [Google Scholar] [CrossRef]
- Ballarin, L.; Karahan, A.; Salvetti, A.; Rossi, L.; Manni, L.; Rinkevich, B.; Rosner, A.; Voskoboynik, A.; Rosental, B.; Canesi, L.; et al. Stem cells and innate immunity in aquatic invertebrates: Bridging two seemingly disparate disciplines for new discoveries in biology. Front. Immunol. 2021, 12, 688106. [Google Scholar] [CrossRef]
- Vanni, V.; Anselmi, C.; Ballarin, L.; Drago, L.; Gasparini, F.; Gordon, T.; Peronato, A.; Rosental, B.; Rosner, A.; Rinkevich, B.; et al. Current knowledge on stem cells in ascidians. In Advances in Aquatic Invertebrate Stem Cell Research: From Basic Research to Innovative Applications, 3rd ed.; Ballarin., L., Rinkevich., B., Hobmayer, B., Eds.; MDPI: Basel, Switzerland, 2022; pp. 273–333. [Google Scholar]
- Berrill, N.J. The Tunicata with an Account of the British Species; Ray Society: London, UK, 1950. [Google Scholar]
- Voskoboynik, A.; Simon-Blecher, N.; Soen, Y.; Rinkevich, B.; De Tomaso, A.W.; Ishizuka, K.J.; Weissman, I.L. Striving for normality: Whole body regeneration through a series of abnormal generations. FASEB J. 2007, 21, 1335–1344. [Google Scholar] [CrossRef]
- Mukai, H.; Watanabe, H. Studies on the formation of germ cells in a compound ascidian Botryllus primigenus oka. J. Morphol. 1976, 148, 337–361. [Google Scholar] [CrossRef]
- Lauzon, R.J.; Patton, C.W.; Weissman, I.L. A morphological and immunohistochemical study of programmed cell death in Botryllus schlosseri (Tunicata, Ascidiacea). Cell Tissue Res. 1993, 272, 115–127. [Google Scholar] [CrossRef]
- Qarri, A.; Rosner, A.; Rabinowitz, C.; Rinkevich, B. UV-B radiation bearings on ephemeral soma in the shallow water tunicate Botryllus schlosseri. Ecotoxicol. Environ. Saf. 2020, 196, 110489. [Google Scholar] [CrossRef] [PubMed]
- Rinkevich, B. The tail of the underwater phoenix. Dev. Biol. 2019, 448, 291–292. [Google Scholar] [CrossRef] [PubMed]
- Rinkevich, B.; Rabinowitz, C. In vitro culture of blood cells from the colonial protochordate Botryllus schlosseri. In Vitro Cell. Dev. Biol. Anim. 1993, 29, 79–85. [Google Scholar] [CrossRef]
- Rabinowitz, C.; Rinkevich, B. In vitro delayed senescence of extirpated buds from zooids of the colonial tunicate, Botryllus schlosseri. J. Exp. Biol. 2004, 207, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Qarri, A.; Rinkevich, Y.; Rinkevich, B. Employing marine invertebrate cell culture media for isolation and cultivation of thraustochytrids. Bot. Mar. 2021, 64, 447–454. [Google Scholar] [CrossRef]
- Qarri, A.; Rinkevich, Y.; Rinkevich, B. Improving the Yields of Blood Cell Extractions from Botryllus schlosseri Vasculature. In Advances in Aquatic Invertebrate Stem Cell Research: From Basic Research to Innovative Applications, 3rd ed.; Ballarin, L., Rinkevich, B., Hobmayer, B., Eds.; MDPI: Basel, Switzerland, 2022; pp. 335–349. [Google Scholar] [CrossRef]
- Rabinowitz, C.; Rinkevich, B. Epithelial cell cultures from Botryllus schlosseri palleal buds: Accomplishments and challenges. Methods Cell Biol. 2005, 25, 137–148. [Google Scholar] [CrossRef]
- Rabinowitz, C.; Rinkevich, B. De novo emerged stemness signatures in epithelial monolayers developed from extirpated palleal buds. In Vitro Cell. Dev. Biol. Anim. 2011, 47, 26–31. [Google Scholar] [CrossRef]
- Rabinowitz, C.; Alfassi, G.; Rinkevich, B. Further portrayal of epithelial monolayers emergent de novo from extirpated ascidians palleal buds. In Vitro Cell. Dev. Biol. Anim. 2009, 45, 334–342. [Google Scholar] [CrossRef]
- Rinkevich, B.; Rabinowitz, C. Urochordate Cell Cultures: From in vivo to in vitro approaches. In Aquatic Invertebrate Cell Cultures; Mothersill, C., Austin, B., Eds.; Springer-Praxis: Chichester, UK, 2000; pp. 225–244. [Google Scholar]
- Rabinowitz, C.; Douek, J.; Weisz, R.; Shabtay, A.; Rinkevich, B. Isolation and characterization of four novel thraustochytrid strains from a colonial tunicate. Indian J. Med. Sci. 2006, 35, 341–350. [Google Scholar]
- Rinkevich, B.; Ballarin, L.; Martinez, P.; Somorjai, I.; Ben-Hamo, O.; Borisenko, I.; Berezikov, E.; Ereskovsky, A.; Gazave, E.; Khnykin, D.; et al. A pan-metazoan concept for adult stem cells: The wobbling Penrose landscape. Biol. Rev. 2022, 97, 299–325. [Google Scholar] [CrossRef]
- Rinkevich, B. Rejection patterns in botryllid ascidian immunity: The first tier of allorecognition. Can. J. Zool. 2005, 83, 101–121. [Google Scholar] [CrossRef]
- Rinkevich, B. Natural chimerism in colonial urochordates. J. Exp. Mar. Biol. Ecol. 2005, 322, 93–109. [Google Scholar] [CrossRef]
- Rinkevich, B. Senescence in Modular Animals: Botryllid Ascidians as a unique ageing system. In The Evolution of Senescence in the Tree of Life, 2nd ed.; Shefferson, R., Jones, O., Salguero-Gomez, R., Eds.; Cambridge University Press: Cambridge, UK, 2017; pp. 220–237. [Google Scholar] [CrossRef]
- Cima, F. Searching for the Origin and the Differentiation of Haemocytes before and after Larval Settlement of the Colonial Ascidian Botryllus schlosseri: An Ultrastructural Viewpoint. J. Mar. Sci. Eng. 2022, 10, 987. [Google Scholar] [CrossRef]
- Ballarin, L.; Del Favero, M.; Manni, L. Relationships among hemocytes, tunic cells, germ cells, and accessory cells in the colonial ascidian Botryllus schlosseri. J. Exp. Zool. B. Mol. Dev. Evol. 2011, 316, 284–295. [Google Scholar] [CrossRef]
- Domart-Coulon, I.; Tambutté, S.; Tambutté, E.; Allemand, D. Short term viability of soft tissue detached from the skeleton of reef-building corals. J. Exp. Mar. Biol. Ecol. 2004, 309, 199–217. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medium | Exp. No. | No. of Colonies | Blastogenic Stage | No. of Zooids | WS Type | Cell Numbers/Exp n ± STD (×106) | Viability ± STD (%) |
|---|---|---|---|---|---|---|---|
| TGM1 | 1 | 2 | A, D | 7, 11 | WS1 | 3.1 ± 1.1 | 92.1 ± 1.3 |
| 2 | 3 | B, C, D | 12, 8, 8 | 2.9 ± 1.4 | 92.3 ± 2.7 | ||
| 3 | 4 | A, C, C, D | 15, 10, 8, 12 | 2.7 ± 0.7 | 90.4 ± 1.7 | ||
| TGM2 | 1 | 3 | B, C, D | 10, 11, 9 | WS1 | 2.6 ± 0.6 | 88.6 ± 2.9 |
| 2 | 3 | C, C, D | 8, 8, 12 | 2.1 ± 0.9 | 91.6 ± 4.9 | ||
| TGM3 | 1 | 2 | A, D | 14, 11 | WS2 | 1.3 ± 0.33 | 94.6 ± 2.0 |
| 2 | 2 | B, D | 8, 10 | 2.4 ± 0.9 | 87.1 ± 6.4 | ||
| 3 | 3 | B, C, D | 7, 8, 12 | 2.1 ± 1.2 | 90.8 ± 2.9 | ||
| TGM4 | 1 | 2 | A, C | 11, 12 | WS3 | 2.8 ± 0.9 | 91.1 ± 1.4 |
| 2 | 2 | B, C | 10, 7 | 2.3 ± 0.5 | 92.8 ± 1.3 | ||
| 3 | 2 | C, D | 15, 10 | 3.2 ± 0.9 | 90.7 ± 6.7 | ||
| 4 | 3 | C, C, D | 11, 8, 12 | 2.8 ± 1.0 | 89.9 ± 6.2 | ||
| TGM5 | 1 | 3 | C, C, D | 7, 8, 10 | WS1 | 1.6 ± 0.6 | 94 ± 3.4 |
| 2 | 3 | B, C, D | 10, 12, 15 | 2.6 ± 1.4 | 91.9 ± 4.0 | ||
| 3 | 3 | A, D, D | 11, 8, 7 | 2.2 ± 1.0 | 94.3 ± 1.6 |
| Medium | Major Outcomes | Onset | 24-h | Day-3 | Day-8 |
|---|---|---|---|---|---|
| TGM1 | abundant cell types | mac, mor | mac, mor | mor | mor, pig |
| PCNA+ cells | hae, mac | hae, mac, mor | hae, mac | hae | |
| TGM2 | abundant cell types | mac, mor | mac, mor | mac, mor, pig | mac, mor, pig |
| PCNA+ cells | hae, mac | mac | pig | mor | |
| TGM3 | abundant cell types | mac, mor | mac, mor, pig | hae, mac, mor, pig | mac, mor, pig |
| PCNA+ cells | hae, mac | hae, mac | hae, mac, pig | pig | |
| TGM4 | abundant cell types | mac, mor | hae, mac, mor | mac, mor, pig | hae, mac, mor, pig |
| PCNA+ cells | hae, mac, pig | mac, mor, pig | hae, mac, pig | hae, mac, pig | |
| TGM5 | abundant cell types | mac, mor | hae, mac, mor, pig | hae, mac, mor, pig | hae, mac, mor, pig |
| PCNA+ cells | hae, mac | hae, mac | hae, mac, mor | hae, mac, mor |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qarri, A.; Kültz, D.; Gardell, A.M.; Rinkevich, B.; Rinkevich, Y. Improved Media Formulations for Primary Cell Cultures Derived from a Colonial Urochordate. Cells 2023, 12, 1709. https://doi.org/10.3390/cells12131709
Qarri A, Kültz D, Gardell AM, Rinkevich B, Rinkevich Y. Improved Media Formulations for Primary Cell Cultures Derived from a Colonial Urochordate. Cells. 2023; 12(13):1709. https://doi.org/10.3390/cells12131709
Chicago/Turabian StyleQarri, Andy, Dietmar Kültz, Alison M. Gardell, Baruch Rinkevich, and Yuval Rinkevich. 2023. "Improved Media Formulations for Primary Cell Cultures Derived from a Colonial Urochordate" Cells 12, no. 13: 1709. https://doi.org/10.3390/cells12131709
APA StyleQarri, A., Kültz, D., Gardell, A. M., Rinkevich, B., & Rinkevich, Y. (2023). Improved Media Formulations for Primary Cell Cultures Derived from a Colonial Urochordate. Cells, 12(13), 1709. https://doi.org/10.3390/cells12131709