

Dietary Advanced Glycation End Products: Their Role in the Insulin Resistance of Aging



Abstract
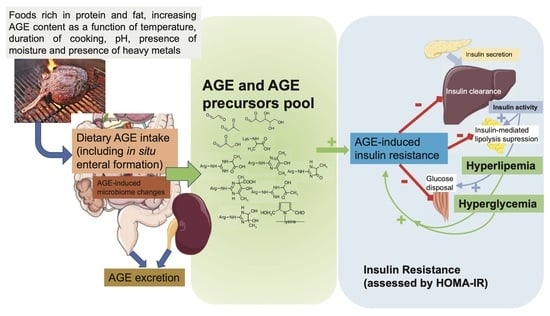
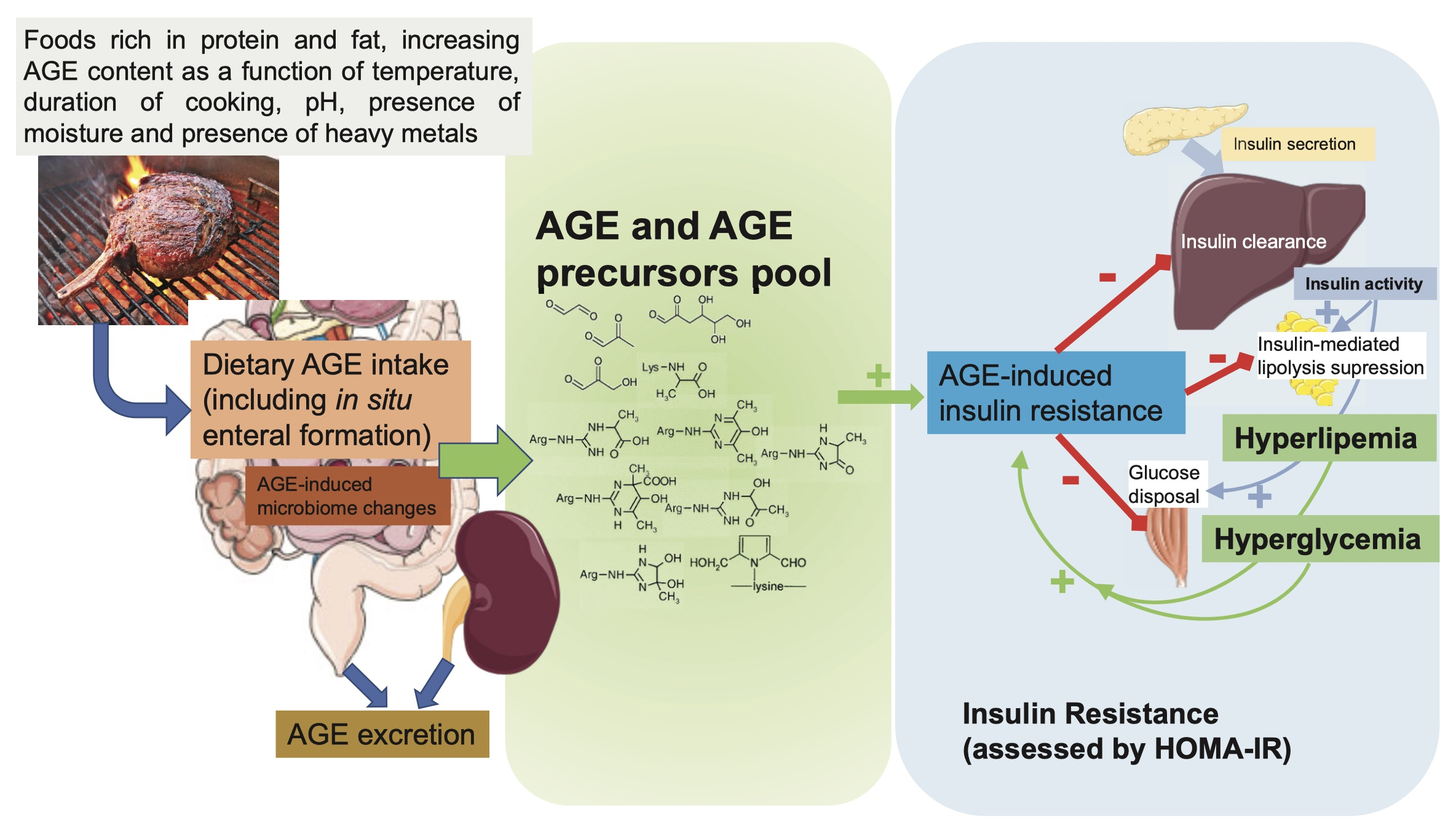
1. Introduction
2. What Advanced Glycation End Products (AGEs) and Their Biological Actions Are, and How They Can Produce Insulin Resistance
3. Evidence of an Association between Dietary AGEs and IR
3.1. Animal Data
3.2. Epidemiological Evidence Linking Dietary AGEs and IR in Humans
3.3. Randomized Controlled Interventional Studies Testing the Association between Dietary AGEs and IR in Humans
3.4. Clinical Trials with Mediterranean and Vegan Dietary Patterns Support the Association between Dietary AGEs and IR in Humans
4. Controversies about Documenting IR in Human Studies
5. Final Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, M.; Chi, X.; Wang, Y.; Setrerrahmane, S.; Xie, W.; Xu, H. Trends in insulin resistance: Insights into mechanisms and therapeutic strategy. Signal Transduct. Target. Ther. 2022, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Yeni-Komshian, H.; Carantoni, M.; Abbasi, F.; Reaven, G.M. Relationship between several surrogate estimates of insulin resistance and quantification of insulin-mediated glucose disposal in 490 healthy nondiabetic volunteers. Diabetes Care 2000, 23, 171–175. [Google Scholar] [CrossRef]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar] [PubMed]
- Gastaldelli, A. Measuring and estimating insulin resistance in clinical and research settings. Obesity 2022, 30, 1549–1563. [Google Scholar] [CrossRef]
- Costantino, S.; Paneni, F.; Cosentino, F. Ageing, metabolism and cardiovascular disease. J. Physiol. 2016, 594, 2061–2073. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.M.; Standley, R.A.; Anthony, S.J.; Grench, K.E.; Helbling, N.L.; DeLany, J.P.; Cornnell, H.H.; Yi, F.; Stefanovic-Racic, M.; Toledo, F.G.S.; et al. Weight Loss and Exercise Differentially Affect Insulin Sensitivity, Body Composition, Cardiorespiratory Fitness, and Muscle Strength in Older Adults With Obesity: A Randomized Controlled Trial. J. Gerontol. A Biol. Sci. Med. Sci. 2022, 77, 1088–1097. [Google Scholar] [CrossRef]
- Castro-Barquero, S.; Ruiz-León, A.M.; Sierra-Pérez, M.; Estruch, R.; Casas, R. Dietary Strategies for Metabolic Syndrome: A Comprehensive Review. Nutrients 2020, 12, 2983. [Google Scholar] [CrossRef] [PubMed]
- Lutsey, P.L.; Steffen, L.M.; Stevens, J. Dietary intake and the development of the metabolic syndrome: The Atherosclerosis Risk in Communities study. Circulation 2008, 117, 754–761. [Google Scholar] [CrossRef]
- Ottum, M.S.; Mistry, A.M. Advanced glycation end-products: Modifiable environmental factors profoundly mediate insulin resistance. J. Clin. Biochem. Nutr. 2015, 57, 1–12. [Google Scholar] [CrossRef]
- Uribarri, J.; del Castillo, M.D.; de la Maza, M.P.; Filip, R.; Gugliucci, A.; Luevano-Contreras, C.; Macías-Cervantes, M.H.; Markowicz Bastos, D.H.; Medrano, A.; Menini, T.; et al. Dietary advanced glycation end products and their role in health and disease. Adv. Nutr. 2015, 6, 461–473. [Google Scholar] [CrossRef]
- Fan, L.; Yu, W.; Zhang, B.; Cao, B.; Wang, M.; Hu, X. Distinctive effects of different types of advanced glycation end-products (AGEs) on liver glucose metabolism. Food Funct. 2022, 13, 11298–11306. [Google Scholar] [CrossRef] [PubMed]
- Corica, D.; Pepe, G.; Currò, M.; Aversa, T.; Tropeano, A.; Ientile, R.; Wasniewska, M. Methods to investigate advanced glycation end-product and their application in clinical practice. Methods 2022, 203, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet. Assoc. 2010, 110, 911–916.e912. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jiang, Y.; Li, H.; Gao, Z.; Yu, G.; Xie, H.; Wang, Y.; Fu, L. Parallel generation of extra advanced glycation end-products during co-digestion of whey proteins and α-dicarbonyls in a simulated gastrointestinal model. Food Funct. 2023, 14, 5342–5354. [Google Scholar] [CrossRef]
- van der Lugt, T.; Vrolijk, M.F.; Bovee, T.F.H.; van Leeuwen, S.P.J.; Vonsovic, S.; Hamers, A.; Opperhuizen, A.; Bast, A. Gastrointestinal digestion of dietary advanced glycation endproducts increases their pro-inflammatory potential. Food Funct. 2021, 12, 6691–6696. [Google Scholar] [CrossRef] [PubMed]
- Garay-Sevilla, M.E.; Beeri, M.S.; de la Maza, M.P.; Rojas, A.; Salazar-Villanea, S.; Uribarri, J. The potential role of dietary advanced glycation endproducts in the development of chronic non-infectious diseases: A narrative review. Nutr. Res. Rev. 2020, 33, 298–311. [Google Scholar] [CrossRef]
- Snelson, M.; Coughlan, M.T. Dietary Advanced Glycation End Products: Digestion, Metabolism and Modulation of Gut Microbial Ecology. Nutrients 2019, 11, 215. [Google Scholar] [CrossRef]
- Garay-Sevilla, M.E.; Rojas, A.; Portero-Otin, M.; Uribarri, J. Dietary AGEs as Exogenous Boosters of Inflammation. Nutrients 2021, 13, 2802. [Google Scholar] [CrossRef]
- Poulsen, M.W.; Bak, M.J.; Andersen, J.M.; Monošík, R.; Giraudi-Futin, A.C.; Holst, J.J.; Nielsen, J.; Lauritzen, L.; Larsen, L.H.; Bügel, S.; et al. Effect of dietary advanced glycation end products on postprandial appetite, inflammation, and endothelial activation in healthy overweight individuals. Eur. J. Nutr. 2014, 53, 661–672. [Google Scholar] [CrossRef]
- Zhou, L.; Song, X.D.; Xu, H.; Liang, G.Q.; Wang, F.; Zhang, L.R.; Huang, F.; Cai, J.; Jiang, G.R. Exogenous 3-Deoxyglucosone-Induced Carbonyl and Oxidative Stress Causes β-Cells Dysfunction by Impairing Gut Permeability in Rats. Biochemistry 2018, 83, 1358–1368. [Google Scholar] [CrossRef]
- Treibmann, S.; Groß, J.; Pätzold, S.; Henle, T. Studies on the Reaction of Dietary Methylglyoxal and Creatine during Simulated Gastrointestinal Digestion and in Human Volunteers. Nutrients 2022, 14, 3598. [Google Scholar] [CrossRef] [PubMed]
- Sato, J.; Kanazawa, A.; Watada, H. Type 2 Diabetes and Bacteremia. Ann. Nutr. Metab. 2017, 71 (Suppl. S1), 17–22. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Litke, R.; Rianha, S.; Paul-Constant, C.; Lo Guidice, J.M.; Taront, S.; Tessier, F.J.; Boulanger, E.; Fradin, C. Exposure of Caenorhabditis elegans to Dietary Nε-Carboxymethyllysine Emphasizes Endocytosis as a New Route for Intestinal Absorption of Advanced Glycation End Products. Nutrients 2021, 13, 4398. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.J.; Santos, A.; Prada, P.O. Linking Gut Microbiota and Inflammation to Obesity and Insulin Resistance. Physiology 2016, 31, 283–293. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef]
- Macia, L.; Thorburn, A.N.; Binge, L.C.; Marino, E.; Rogers, K.E.; Maslowski, K.M.; Vieira, A.T.; Kranich, J.; Mackay, C.R. Microbial influences on epithelial integrity and immune function as a basis for inflammatory diseases. Immunol. Rev. 2012, 245, 164–176. [Google Scholar] [CrossRef]
- Schilderink, R.; Verseijden, C.; de Jonge, W.J. Dietary inhibitors of histone deacetylases in intestinal immunity and homeostasis. Front. Immunol. 2013, 4, 226. [Google Scholar] [CrossRef]
- Wang, J.; Cai, W.; Yu, J.; Liu, H.; He, S.; Zhu, L.; Xu, J. Dietary Advanced Glycation End Products Shift the Gut Microbiota Composition and Induce Insulin Resistance in Mice. Diabetes Metab. Syndr. Obes. Targets Ther. 2022, 15, 427–437. [Google Scholar] [CrossRef]
- Yacoub, R.; Nugent, M.; Cai, W.; Nadkarni, G.N.; Chaves, L.D.; Abyad, S.; Honan, A.M.; Thomas, S.A.; Zheng, W.; Valiyaparambil, S.A.; et al. Advanced glycation end products dietary restriction effects on bacterial gut microbiota in peritoneal dialysis patients; a randomized open label controlled trial. PLoS ONE 2017, 12, e0184789. [Google Scholar] [CrossRef]
- Wang, X.; Liu, J.; Zhen, J.; Zhang, C.; Wan, Q.; Liu, G.; Wei, X.; Zhang, Y.; Wang, Z.; Han, H.; et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 2014, 86, 712–725. [Google Scholar] [CrossRef]
- Vlassara, H.; Striker, G.E. AGE restriction in diabetes mellitus: A paradigm shift. Nat. Rev. Endocrinol. 2011, 7, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef] [PubMed]
- Teissier, T.; Boulanger, É. The receptor for advanced glycation end-products (RAGE) is an important pattern recognition receptor (PRR) for inflammaging. Biogerontology 2019, 20, 279–301. [Google Scholar] [CrossRef]
- Khalid, M.; Petroianu, G.; Adem, A. Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives. Biomolecules 2022, 12, 542. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, J.P.; McKinley, B.; Eckel, R.H. The metabolic syndrome and inflammation. Metab. Syndr. Relat. Disord. 2004, 2, 82–104. [Google Scholar] [CrossRef]
- Riehl, A.; Németh, J.; Angel, P.; Hess, J. The receptor RAGE: Bridging inflammation and cancer. Cell Commun. Signal. 2009, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Yu, W.; Tao, M.; Zhao, Y.; Hu, X.; Wang, M. 4’-Methoxyresveratrol Alleviated AGE-Induced Inflammation via RAGE-Mediated NF-κB and NLRP3 Inflammasome Pathway. Molecules 2018, 23, 1447. [Google Scholar] [CrossRef]
- Finucane, O.M.; Lyons, C.L.; Murphy, A.M.; Reynolds, C.M.; Klinger, R.; Healy, N.P.; Cooke, A.A.; Coll, R.C.; McAllan, L.; Nilaweera, K.N.; et al. Monounsaturated fatty acid-enriched high-fat diets impede adipose NLRP3 inflammasome-mediated IL-1β secretion and insulin resistance despite obesity. Diabetes 2015, 64, 2116–2128. [Google Scholar] [CrossRef]
- Kierdorf, K.; Fritz, G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Pinto-Junior, D.C.; Silva, K.S.; Michalani, M.L.; Yonamine, C.Y.; Esteves, J.V.; Fabre, N.T.; Thieme, K.; Catanozi, S.; Okamoto, M.M.; Seraphim, P.M.; et al. Advanced glycation end products-induced insulin resistance involves repression of skeletal muscle GLUT4 expression. Sci. Rep. 2018, 8, 8109. [Google Scholar] [CrossRef]
- Chikazawa, M.; Shibata, T.; Hatasa, Y.; Hirose, S.; Otaki, N.; Nakashima, F.; Ito, M.; Machida, S.; Maruyama, S.; Uchida, K. Identification of C1q as a Binding Protein for Advanced Glycation End Products. Biochemistry 2016, 55, 435–446. [Google Scholar] [CrossRef]
- Snelson, M.; Tan, S.M.; Clarke, R.E.; de Pasquale, C.; Thallas-Bonke, V.; Nguyen, T.V.; Penfold, S.A.; Harcourt, B.E.; Sourris, K.C.; Lindblom, R.S.; et al. Processed foods drive intestinal barrier permeability and microvascular diseases. Sci. Adv. 2021, 7, eabe4841. [Google Scholar] [CrossRef] [PubMed]
- Kellow, N.J.; Coughlan, M.T. Effect of diet-derived advanced glycation end products on inflammation. Nutr. Rev. 2015, 73, 737–759. [Google Scholar] [CrossRef] [PubMed]
- Walke, P.B.; Bansode, S.B.; More, N.P.; Chaurasiya, A.H.; Joshi, R.S.; Kulkarni, M.J. Molecular investigation of glycated insulin-induced insulin resistance via insulin signaling and AGE-RAGE axis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166029. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef]
- Riboulet-Chavey, A.; Pierron, A.; Durand, I.; Murdaca, J.; Giudicelli, J.; Van Obberghen, E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes 2006, 55, 1289–1299. [Google Scholar] [CrossRef]
- Gaens, K.H.; Goossens, G.H.; Niessen, P.M.; van Greevenbroek, M.M.; van der Kallen, C.J.; Niessen, H.W.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.; Blaak, E.E.; et al. Nε-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1199–1208. [Google Scholar] [CrossRef]
- Hofmann, S.M.; Dong, H.J.; Li, Z.; Cai, W.; Altomonte, J.; Thung, S.N.; Zeng, F.; Fisher, E.A.; Vlassara, H. Improved insulin sensitivity is associated with restricted intake of dietary glycoxidation products in the db/db mouse. Diabetes 2002, 51, 2082–2089. [Google Scholar] [CrossRef]
- Sandu, O.; Song, K.; Cai, W.; Zheng, F.; Uribarri, J.; Vlassara, H. Insulin resistance and type 2 diabetes in high-fat-fed mice are linked to high glycotoxin intake. Diabetes 2005, 54, 2314–2319. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Uribarri, J.; Zhu, L.; Chen, X.; Swamy, S.; Zhao, Z.; Grosjean, F.; Simonaro, C.; Kuchel, G.A.; Schnaider-Beeri, M.; et al. Oral glycotoxins are a modifiable cause of dementia and the metabolic syndrome in mice and humans. Proc. Natl. Acad. Sci. USA 2014, 111, 4940–4945. [Google Scholar] [CrossRef]
- Mastrocola, R.; Dal Bello, F.; Cento, A.S.; Gaens, K.; Collotta, D.; Aragno, M.; Medana, C.; Collino, M.; Wouters, K.; Schalkwijk, C.G. Altered hepatic sphingolipid metabolism in insulin resistant mice: Role of advanced glycation endproducts. Free Radic. Biol. Med. 2021, 169, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Pillai, J.A.; Bena, J.; Bekris, L.; Kodur, N.; Kasumov, T.; Leverenz, J.B.; Kashyap, S.R. Metabolic syndrome biomarkers relate to rate of cognitive decline in MCI and dementia stages of Alzheimer’s disease. Alzheimer’s Res. Ther. 2023, 15, 54. [Google Scholar] [CrossRef] [PubMed]
- Beeri, M.S.; Moshier, E.; Schmeidler, J.; Godbold, J.; Uribarri, J.; Reddy, S.; Sano, M.; Grossman, H.T.; Cai, W.; Vlassara, H.; et al. Serum concentration of an inflammatory glycotoxin, methylglyoxal, is associated with increased cognitive decline in elderly individuals. Mech. Ageing Dev. 2011, 132, 583–587. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Chow, H.M.; Shi, M.; Cheng, A.; Gao, Y.; Chen, G.; Song, X.; So, R.W.L.; Zhang, J.; Herrup, K. Age-related hyperinsulinemia leads to insulin resistance in neurons and cell-cycle-induced senescence. Nat. Neurosci. 2019, 22, 1806–1819. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Kootte, R.S.; Levin, E.; Salojärvi, J.; Smits, L.P.; Hartstra, A.V.; Udayappan, S.D.; Hermes, G.; Bouter, K.E.; Koopen, A.M.; Holst, J.J.; et al. Improvement of Insulin Sensitivity after Lean Donor Feces in Metabolic Syndrome Is Driven by Baseline Intestinal Microbiota Composition. Cell Metab. 2017, 26, 611–619.e616. [Google Scholar] [CrossRef]
- Csongová, M.; Scheijen, J.; van de Waarenburg, M.P.H.; Gurecká, R.; Koborová, I.; Tábi, T.; Szökö, É.; Schalkwijk, C.G.; Šebeková, K. Association of α-Dicarbonyls and Advanced Glycation End Products with Insulin Resistance in Non-Diabetic Young Subjects: A Case-Control Study. Nutrients 2022, 14, 4929. [Google Scholar] [CrossRef]
- Saha, A.; Poojary, P.; Chan, L.; Chauhan, K.; Nadkarni, G.; Coca, S.; Uribarri, J. Increased odds of metabolic syndrome with consumption of high dietary advanced glycation end products in adolescents. Diabetes Metab. 2017, 43, 469–471. [Google Scholar] [CrossRef] [PubMed]
- Baye, E.; Kiriakova, V.; Uribarri, J.; Moran, L.J.; de Courten, B. Consumption of diets with low advanced glycation end products improves cardiometabolic parameters: Meta-analysis of randomised controlled trials. Sci. Rep. 2017, 7, 2266. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.R.; Matta, S.T.; Haymond, M.W.; Chung, S.T. Measuring Insulin Resistance in Humans. Horm. Res. Paediatr. 2020, 93, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Birlouez-Aragon, I.; Saavedra, G.; Tessier, F.J.; Galinier, A.; Ait-Ameur, L.; Lacoste, F.; Niamba, C.N.; Alt, N.; Somoza, V.; Lecerf, J.M. A diet based on high-heat-treated foods promotes risk factors for diabetes mellitus and cardiovascular diseases. Am. J. Clin. Nutr. 2010, 91, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Mark, A.B.; Poulsen, M.W.; Andersen, S.; Andersen, J.M.; Bak, M.J.; Ritz, C.; Holst, J.J.; Nielsen, J.; de Courten, B.; Dragsted, L.O.; et al. Consumption of a diet low in advanced glycation end products for 4 weeks improves insulin sensitivity in overweight women. Diabetes Care 2014, 37, 88–95. [Google Scholar] [CrossRef]
- de Courten, B.; de Courten, M.P.; Soldatos, G.; Dougherty, S.L.; Straznicky, N.; Schlaich, M.; Sourris, K.C.; Chand, V.; Scheijen, J.L.; Kingwell, B.A.; et al. Diet low in advanced glycation end products increases insulin sensitivity in healthy overweight individuals: A double-blind, randomized, crossover trial. Am. J. Clin. Nutr. 2016, 103, 1426–1433. [Google Scholar] [CrossRef]
- Vlassara, H.; Cai, W.; Tripp, E.; Pyzik, R.; Yee, K.; Goldberg, L.; Tansman, L.; Chen, X.; Mani, V.; Fayad, Z.A.; et al. Oral AGE restriction ameliorates insulin resistance in obese individuals with the metabolic syndrome: A randomised controlled trial. Diabetologia 2016, 59, 2181–2192. [Google Scholar] [CrossRef]
- Goudarzi, R.; Sedaghat, M.; Hedayati, M.; Hekmatdoost, A.; Sohrab, G. Low advanced Glycation end product diet improves the central obesity, insulin resistance and inflammatory profiles in Iranian patients with metabolic syndrome: A randomized clinical trial. J. Diabetes Metab. Disord. 2020, 19, 1129–1138. [Google Scholar] [CrossRef]
- Linkens, A.M.; Houben, A.J.; Niessen, P.M.; Wijckmans, N.E.; de Goei, E.E.; Van den Eynde, M.D.; Scheijen, J.L.; van den Waarenburg, M.P.; Mari, A.; Berendschot, T.T.; et al. A 4-week high-AGE diet does not impair glucose metabolism and vascular function in obese individuals. JCI Insight 2022, 7, e156950. [Google Scholar] [CrossRef]
- Uribarri, J.; Cai, W.; Ramdas, M.; Goodman, S.; Pyzik, R.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: Potential role of AGER1 and SIRT1. Diabetes Care 2011, 34, 1610–1616. [Google Scholar] [CrossRef]
- Luévano-Contreras, C.; Garay-Sevilla, M.E.; Wrobel, K.; Malacara, J.M.; Wrobel, K. Dietary advanced glycation end products restriction diminishes inflammation markers and oxidative stress in patients with type 2 diabetes mellitus. J. Clin. Biochem. Nutr. 2013, 52, 22–26. [Google Scholar] [CrossRef]
- Scheijen, J.; Clevers, E.; Engelen, L.; Dagnelie, P.C.; Brouns, F.; Stehouwer, C.D.A.; Schalkwijk, C.G. Analysis of advanced glycation endproducts in selected food items by ultra-performance liquid chromatography tandem mass spectrometry: Presentation of a dietary AGE database. Food Chem. 2016, 190, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Kodate, A.; Otoki, Y.; Shimizu, N.; Ito, J.; Kato, S.; Umetsu, N.; Miyazawa, T.; Nakagawa, K. Development of quantitation method for glycated aminophospholipids at the molecular species level in powdered milk and powdered buttermilk. Sci. Rep. 2018, 8, 8729. [Google Scholar] [CrossRef] [PubMed]
- Chiu, K.C.; Cohan, P.; Lee, N.P.; Chuang, L.M. Insulin sensitivity differs among ethnic groups with a compensatory response in beta-cell function. Diabetes Care 2000, 23, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Herpich, C.; Kochlik, B.; Weber, D.; Ott, C.; Grune, T.; Norman, K.; Raupbach, J. Fasting Concentrations and Postprandial Response of 1,2-Dicarbonyl Compounds 3-Deoxyglucosone, Glyoxal, and Methylglyoxal Are Not Increased in Healthy Older Adults. J. Gerontol. A Biol. Sci. Med. Sci. 2022, 77, 934–940. [Google Scholar] [CrossRef]
- Mahmoudinezhad, M.; Farhangi, M.A.; Kahroba, H.; Dehghan, P. Personalized diet study of dietary advanced glycation end products (AGEs) and fatty acid desaturase 2 (FADS(2)) genotypes in obesity. Sci. Rep. 2021, 11, 19725. [Google Scholar] [CrossRef]
- Gutierrez-Mariscal, F.M.; Cardelo, M.P.; de la Cruz, S.; Alcala-Diaz, J.F.; Roncero-Ramos, I.; Guler, I.; Vals-Delgado, C.; López-Moreno, A.; Luque, R.M.; Delgado-Lista, J.; et al. Reduction in Circulating Advanced Glycation End Products by Mediterranean Diet Is Associated with Increased Likelihood of Type 2 Diabetes Remission in Patients with Coronary Heart Disease: From the Cordioprev Study. Mol. Nutr. Food Res. 2021, 65, e1901290. [Google Scholar] [CrossRef]
- Kahleova, H.; Znayenko-Miller, T.; Uribarri, J.; Holubkov, R.; Branard, N.D. Dietary advanced glycation products and their associations with insulin sensitivity and body weight: A 16-week randomized clinical trial. Obes. Sci. Pract. 2022, 9, 235–242. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Tobin, J.D.; Andres, R. Glucose clamp technique: A method for quantifying insulin secretion and resistance. Am. J. Physiol. 1979, 237, E214–E223. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef]
- Chen, H.; Sullivan, G.; Quon, M.J. Assessing the predictive accuracy of QUICKI as a surrogate index for insulin sensitivity using a calibration model. Diabetes 2005, 54, 1914–1925. [Google Scholar] [CrossRef] [PubMed]
- Henríquez, S.; Jara, N.; Bunout, D.; Hirsch, S.; de la Maza, M.P.; Leiva, L.; Barrera, G. Variability of formulas to assess insulin sensitivity and their association with the Matsuda index. Nutr. Hosp. 2013, 28, 1594–1598. [Google Scholar] [CrossRef] [PubMed]
- Placzkowska, S.; Pawlik-Sobecka, L.; Kokot, I.; Piwowar, A. Indirect insulin resistance detection: Current clinical trends and laboratory limitations. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2019, 163, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.M.; Levy, J.C.; Matthews, D.R. Use and abuse of HOMA modeling. Diabetes Care 2004, 27, 1487–1495. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Author, Reference | Animal Model | Study Design | Intervention | Findings |
|---|---|---|---|---|
| Hofmann [50] | Db/Db mice (5 week old) | Dietary intervention with random assignment into two parallel groups for 20 weeks (n = 20) | High versus low AGE diets | Lower body weight, lower serum AGEs, better response to both glucose and insulin tolerance tests and better preservation of pancreatic islets than with the high AGE diet |
| Sandu [51] | C57/BL6 female mice (6 week old) | Dietary intervention with random assignment into two parallel groups for 6 months | High fat (35% fat) high AGE diet (HAGE-HF) versus High fat low AGE diet (LAGE-HF) | None of the LAGE-HF mice became diabetic, while 75% of HAGE-HF did. |
| Cai [52] | C57/BL6 | Dietary intervention with random assignment into three parallel groups for 18 months | Pair-fed three diets throughout life: (1) low AGE (MG−) *, (2) MG supplemented low AGE chow (MG+) and regular chow (Reg) | Older MG+ and Reg fed mice developed IR (higher fasting insulin levels and abnormal intraperitoneally glucose tolerance test) and dementia, which did not happen in MG− mice. |
| Wang [28] | C57/BL6 male mice (12 week old) | Dietary intervention with random assignment into three parallel groups for 24 weeks | Three parallel diets: (1) regular chow (n = 10), (2) regular chow + MG (n = 15) or (3) heat-treated chow (n = 15) | IR (high fasting insulin, HOMA and abnormal intraperitoneal glucose tolerance test) developed in groups 2 and 3, but not 1. Microbiota was also altered in groups 2 and 3 (not in group 1) leading to loss of butyrate-producing bacteria |
| Mastrocola [53] | Db/Db and C57/BL6 mice | Dietary intervention followed by pharmacological intervention in the C57/BL6 mice | C57/BL6 mice were randomly assigned to 4 groups for 12 weeks: (1) standard diet, (2) high fat diet (60%trans-fat), (3) standard diet + pyridoxamine for last 8 weeks, (4) high fat diet + pyridoxamine for last 8 weeks | High levels of AGEs and RAGE and abnormal enzymes of sphingolipid metabolism were found in the liver of Db/Db and group 2 C57/BL6, but not in groups 1, 3 and C57/BL6 |
| Author, Year, Reference | Study Design | Intervention | Number of Participants | Randomized | Participant Characteristics | Duration and Allocation | Specified Outcomes | Findings |
|---|---|---|---|---|---|---|---|---|
| Birlouez, 2010 [64] | Crossover | High- and low-AGE diets | 62 | Yes | Healthy individuals | 1 month, France | Changes in serum AGEs and HOMA | Decreased serum AGEs and HOMA |
| Uribarri, 2011 [70] | Parallel | High- and low-AGE diets | 18 | Yes | Patients with diabetes | 3 months, USA | Changes in serum AGEs, markers of OS and inflammation and HOMA | Decreased levels of serum AGEs, markers of OS *, inflammation and HOMA |
| Luevano-Contreras 2013 [71] | Parallel | High- and low-AGE diets | 26 | Yes | Patients with diabetes | 1.5 months, Mexico | Changes in serum AGEs, markers of OS and inflammation and HOMA | Decreased markers of OS and inflammation, but no changes in serum AGEs or HOMA |
| Mark, 2014 [65] | Parallel | High- and low-AGE diets | 74 | Yes | Overweight women | 1 month, Denmark | Changes in urinary AGEs and HOMA | Decreased urinary AGEs and HOMA |
| De courten 2016 [66] | Crossover | High- and low-AGE diets | 20 | Yes | Overweight individuals | 0.5 months, Denmark | Changes in serum AGEs and insulin resistance (hyperinsulinemic–euglycemic clamp and intravenous glucose tolerance test) | Decreased serum AGEs and insulin resistance |
| Vlassara, 2016 [67] | Parallel | High- and low-AGE diets | 138 | Yes | Patients with metabolic syndrome | 12 months, USA | Changes in serum AGEs, markers of OS and inflammation and HOMA | Decreased levels of serum AGEs, markers of OS, inflammation and HOMA |
| Goudarzi 2020 [68] | Parallel | High- and low-AGE diets | 40 | Yes | Patients with metabolic syndrome | 2 months, Iran | Changes in serum AGEs, markers of OS and inflammation and HOMA | Decreased serum AGEs, markers of OS, inflammation, HOMA and weight (were also calorie restricted) |
| Linkens 2022 [69] | Parallel | High- and low-AGE diets | 82 | Yes | Patients with obesity | 1.5 months, Netherlands | Changes in serum AGEs, markers of OS and inflammation and insulin resistance (hyperinsulinemic–euglycemic and hyperglycemic clamp) | Decreased circulating AGEs but not markers of OS/inflammation or insulin sensitivity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Portero-Otin, M.; de la Maza, M.P.; Uribarri, J. Dietary Advanced Glycation End Products: Their Role in the Insulin Resistance of Aging. Cells 2023, 12, 1684. https://doi.org/10.3390/cells12131684
Portero-Otin M, de la Maza MP, Uribarri J. Dietary Advanced Glycation End Products: Their Role in the Insulin Resistance of Aging. Cells. 2023; 12(13):1684. https://doi.org/10.3390/cells12131684
Chicago/Turabian StylePortero-Otin, Manuel, M. Pia de la Maza, and Jaime Uribarri. 2023. "Dietary Advanced Glycation End Products: Their Role in the Insulin Resistance of Aging" Cells 12, no. 13: 1684. https://doi.org/10.3390/cells12131684
APA StylePortero-Otin, M., de la Maza, M. P., & Uribarri, J. (2023). Dietary Advanced Glycation End Products: Their Role in the Insulin Resistance of Aging. Cells, 12(13), 1684. https://doi.org/10.3390/cells12131684