Abstract
The transient receptor potential vanilloid 4 (TRPV4) channel is a non-selective cation channel that is mostly permeable to calcium (Ca2+), which participates in intracellular Ca2+ handling in cardiac cells. It is widely expressed through the body and is activated by a large spectrum of physicochemical stimuli, conferring it a role in a variety of sensorial and physiological functions. Within the cardiovascular system, TRPV4 expression is reported in cardiomyocytes, endothelial cells (ECs) and smooth muscle cells (SMCs), where it modulates mitochondrial activity, Ca2+ homeostasis, cardiomyocytes electrical activity and contractility, cardiac embryonic development and fibroblast proliferation, as well as vascular permeability, dilatation and constriction. On the other hand, TRPV4 channels participate in several cardiac pathological processes such as the development of cardiac fibrosis, hypertrophy, ischemia–reperfusion injuries, heart failure, myocardial infarction and arrhythmia. In this manuscript, we provide an overview of TRPV4 channel implications in cardiac physiology and discuss the potential of the TRPV4 channel as a therapeutic target against cardiovascular diseases.
1. Introduction
The mammalian TRP channels are widely expressed in the heart and can be considered as real “cellular switches” able to respond to numerous physical and chemical stimuli related to sensory physiology [,,]. In mammals, the TRP family is composed of 28 members and classified according to amino acid sequence homology into six families: TRPA (ankyrin; TRPA1), TRPC (canonical; TRPC1–TRPC7), TRPM (melastatin; TRPM1–TRPM8), TRPML (mucolipin; TRPML1–TRPML3), TRPP (polycystin; TRPP1–TRPP3) and TRPV (vanilloid; TRPV1–TRPV6) [,]. With some exceptions, most of the TRP channels are able to conduct Ca2+, which plays an important role in stimulus–response reactions of cardiac cells. Interestingly, the altered expression, localization and regulation of TRP channels have already been related to cardiovascular disorders due to Ca2+ handling dysregulation []. Among the TRPV channel family [], the TRPV4 channel has emerged as a key modulator in cardiac cell structure and activity [,]. The recent identification of pharmacological modulators and construction of TRPV4 knockout (KO) mice unmasked several physiopathological roles of this channel [,,]. However, the mechanisms that regulate cardiac TRPV4 channels still remain poorly understood. In this context, this review will discuss the latest findings on the pathophysiological role of the TRPV4 channel in the cardiovascular system and why an understanding of TRPV4 regulation may lead to novel therapeutical strategies related to cardiac diseases.
2. Gene, Structure, Function and Electrical Properties
The TRPV4 channel has received particular attention due to its large expression in the cardiovascular system [,,,]. The TRPV4 protein is encoded by the trpv4 gene, present on the long (q) arm of chromosome 12 at position 24.1 in humans []. The corresponding locus is found between bases 109,783,087 and 109,783,406 of the genome and is composed of 15 exons. Interestingly, five channel isoforms (TRPV4-A to -E) were found and produced by alternate exon splicing in a human bronchial epithelial cell line. Only the TRPV4-A and TRPV4-D isoforms are localized in the plasma membrane and display identical biophysical properties [], while the other splice variants (N-terminal deletions) remain in the endoplasmic reticulum (ER) and do not form functional ion channels []. Because the functional differences between these two isoforms have not been explored in the cardiovascular system, we will refer in the following section to TRPV4 as the TRPV4-A and TRPV4-D isoforms without distinction. TRPV4 has a tetrameric structure, with each subunit being composed of six transmembrane segments (S1 to S6), a pattern shared with other TRP channels and voltage-gated ion channel subunits (VGIC). The S1 to S4 segments constitute the peripheral structure, while the central S5-S6 loops border the pore of the channel [,,,]. The N- and C-terminal extremities are intracellular and contain a variety of functional domains []. A recent crystallographic study combined with an X-ray approach gives more details about the structure of Xenopus tropicalis TRPV4, with a resolution of 3.8 Å [].
The N-terminal region represents more than half of the protein and plays a critical role in channel assembly, trafficking and regulation [,,,,,]. It harbours six Ankyrin repeated domains (ARD1–6) that participate in channel oligomerization, protein–protein interaction and trafficking []. Note that the absence of these specific domains blocks the TRPV4 trafficking at the ER level []. This region also presents a Ca2+-calmodulin kinase type II (CaMKII) regulation site conferring channel sensitivity to intracellular Ca2+. In HEK-293 cells overexpressing TRPV4, a proline-rich sequence in the TRPV4 N-terminal can interact with the cytoskeleton protein PACSIN 3 (protein kinase C and casein kinase substrate in neurons 3), thereby regulating channel trafficking and preventing or reducing TRPV4 activation by heat, cell swelling and arachidonic acid []. In this context, PACSIN3 can be considered as a TRPV4 channel regulatory protein that regulates both the TRPV4 subcellular localization and its function. Obviously, further studies on cardiomyocytes are needed to better understand the mechanism and the role of this interaction. In addition, previous studies have shown on several heterologous expression systems involving HEK-293, Hela and SJSA-1 cells overexpressing TRPV4 that the TRPV4 N-terminal region is able to interact with human OS-9, an ER-resident lectin, in order to prevent channel trafficking to the plasma membrane []. This point is relevant because it suggests that OS-9 is also an important auxiliary protein for TRPV4 maturation.
The human TRPV4 C-terminal extremity contains different domains: (i) “TRPbox” (carrying the consensus sequence WKFQR) []; (ii) a CaM-binding domain that modulates its tetramerization and its gating in both HEK-293 and T-REX cells [], as well as Ca2+ influx into oocytes []; (iii) a PDZ domain, which interacts with numerous cell auxiliary proteins [,]. Furthermore, the microtubule-associated protein 7 (MAP7) interaction with the C-terminal extremity of TRPV4 increases both expression and functional activity in the plasma membrane in Chinese hamster ovary cells []. Because MAP7 is directly related to the cytoskeletal filaments, this interaction may underlie the TRPV4 mechanotransduction mechanism []; nevertheless, this hypothesis requires further attention in myocytes.
TRPV4 principally assemble as homotetramers [,] to form a functional channel but can also form heterotetramers with other members of the TRP channel family, namely TRPC1 [,] and TRPP2 [,]. It is important to note that these kinds of biophysical studies were conducted only in classical heterologous expression systems (HEK-293 and tsa 201 cells overexpressing TRPV4) and in native endothelial cells (ECs) (human umbilical vein ECs (HUVECs), mouse primary aortic ECs and rat mesenteric artery ECs (MAECs)), undeniably requiring further research in cardiomyocytes to better understand the heteromeric TRPV4 contribution to cardiac cell function. The TRPV4 channel can also interact with the α-subunit of a few ion channels and aquaporins [,,,,,]. This heteromerization was shown to modify the TRPV4 channel’s biophysical properties in HEK-293 and MAECs cells [,,]. The TRPV4 channel shows a lower voltage sensitivity compared to other TRP channels but still harbors an outward rectification current when expressed in heterologous expression systems, constituting the signature of most TRP channels. To date, this weak voltage dependence of TRPV4 is not yet clearly understood. Nevertheless, the low density of positive charges in the S4 voltage sensor domain [] combined with a better understanding of its crystal structure [] provides a plausible mechanism for its gating. Indeed, TRPV4 adopts a domain-swapped arrangement between the S1–S4 domain and the pore domain S5–S6, similarly to TRPV1, TRPV2 and numerous VGICs, whereas the connector between these last two domains adopts an ordered loop structure rather than the α-helix segment present within TRPV1 and TRPV2. This last structural aspect is important, since the VGIC connector acts as a mechanical lever to couple the pore opening and the voltage sensor activation [,], and its absence can change the gating behavior [,]. Taken together, the interface behavior between the S1–S4 and pore domains in TRPV4 is unique among TRP channels and not closely related to the VGICs. In HEK-293 and native mouse aorta ECs, the TRPV4 single channel conductance rates are ~30–60 pS and ~80–100 pS for inward and outward currents, respectively []. Like most of the TRP channels, TRPV4 is mainly permeable to Ca2+ over other ions, and its permeability sequence was established to be as follows: Ca2+ >> Mg2+ > K+ > Na+ (PCa/PNa ~ 10 and PMg/PNa ~ 2–3) [,,]. Surprisingly, the macroscopic TRPV4 current has not yet been unmasked in cardiomyocytes. A recent structural characterization study performed by Yuan’s group reported a larger selectivity filter (10.6–12.6 Å) within the upper gate of TRPV4 compared to other TRPV channels (TRPV1, TRPV2 and TRPV6, ranging from 4.2 to 7.4 Å) (Figure 1 []).
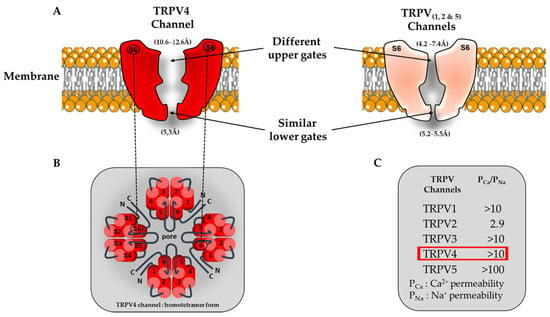
Figure 1.
Schematic view of TRPV channel structures and associated ion permeation pathway. (A) Comparison of TRPV pore structures. For clarity, only two opposing subunits are shown (segment S6). Interestingly the TRPV4 upper gate is larger compared to the other members of the TRPV family (TRPV1, TRPV2 and TRPV5). (B) Homotetrameric structure of TRPV4. (C) Functional characteristics of TRPV channels (permeability ratio PCa/PNa). The red square refers to the Ca2+ permeability of the TRPV4 channel.
Note that the presence of this specificity does not significantly increases the degree of Ca2+ permeability within this ion channel family but should potentially be considered as an important structural feature when TRPV4 has to deal with mechanical forces associated with increased membrane tension. Moreover, this structural specificity opens up the possibility of developing a new generation of drugs that selectively target TRPV4 channels without interfering with other cardiac ion channels from the same family or not. It may, thus, provide a more targeted approach to treat human arrhythmias and minimize off-target effects.
3. Available Tools to Investigate TRPV4’s Roles
The intracellular Ca2+ concentration plays a pivotal role in living organisms because it is involved in several physiological regulation processes but also in responses to various pathological states [,]. In the myocardium, cellular Ca2+ homeostasis maintains normal heart function, which requires various specialized proteins such as ion channels and exchangers []. The central role of Ca2+ in cardiac excitation–contraction is well-established, where Ca2+ is at the interface of the electrical membrane activity (action potential (AP)) and cell contraction []. Any disturbance in intracellular Ca2+ homeostasis may, thus, lead to contractile or electrical defects in the myocardium. In this context, the TRPV4 function related to Ca2+ permeability is evaluated with the greatest attention [,]. Consequently, synthetic specific antagonists of TRPV4 have been designed and evaluated to treat human diseases including cardiac pathologies [,,].
3.1. Pharmacological Modulators
To date, several synthetic molecules that modulate TRPV4 channels have been developed to aim at treating diseases such as osteoarthritis, respiratory diseases, cancers, gastrointestinal disorders and pain []. Recently, some of these compounds entered safety trials to treat heart failure []. Progress in the development of these agents led to the last generation of more potent and specific modulators that have been used to unmask the TRPV4 channel’s pathophysiological roles in the heart.
3.1.1. TRPV4 Agonists
Four generations of TRPV4 agonists (see Table 1) have been used on different tissues and cell types to determine the physiological and pathological functions in the myocardium. The 4α-phorbol 12,13-didecanoate (4α-PDD, a semisynthetic derivative of diterpenoid phorbol), 5,6 epoxyeicosatrienoicacid (5,6-EET, an oxidative metabolite of arachidonic acid), RN-1747 (a benzenesulfonamide) and GSK1016790A agonists (fully synthetic) have been extensively used in cardiovascular investigations [,,,,,]. In addition, Atobe and al. have recently reported on the new TRPV4 agonist quinazolin-4(3 H)-one, a derivative intended to treat osteoarthritis [] (see Table 1).
3.1.2. TRPV4 Antagonists
Over the last decade, several TRPV4 antagonists were used to highlight TRPV4’s roles in the cardiovascular system. The initial studies used several non-selective TRPV4 antagonists such as ruthenium red (RuR) and gadolinium []. Subsequent studies have evaluated the effects of more selective antagonists, including RN-1734, which completely block both ligand- and hypotonicity-activated TRPV4 channels in T-REx and HEK-293 cells without affecting other TRP channels such as TRPV1, TRPV3 and TRPM8 channels [], or the widely-used HC067047 antagonist, which was used in vivo and in vitro in the context of ischemia–reperfusion [,]. Recently, a promising clinical antagonist GSK2193874, which possesses high TRPV4 affinity, has been shown to efficiently prevent and treat lung edema in heart failure models and congestive heart failure [] (Table 2). Several optimized antagonists exhibiting better in vivo availability (RN-9893) [], solubility (GSK3527497) [] or efficacy (novel 2′,4′-dimethyl-[4,5′-bithiazol]-2-yl amino derivatives [,] and GSK2798745 []) or with decreased toxicity (sulfone pyrrolidine sulfonamide) [,] have been developed since then. Knowing that the TRPV4 antagonist GSK2798745 [] was the first inhibitor tested in humans (phase 2), more homologs are likely to appear in the near future (see below Table 2). Indeed, the recently unveiled Cryo-EM structure [] of TRPV4 channels combined with the pharmaceutical industry’s efforts should accelerate the knowledge related to the mechanism of action of current modulators and may help to identify the next generation of TRPV4 drugs.

Table 1.
Overview of the evolution of TRPV4 agonist, the associated physiological processes and their clinical application. Note: 4α-PDD: 4alpha-phorbol 12,13-didecanoate; PKC: protein kinase C; CF: cardiofibroblasts; 5,6-EET: 5,6 epoxyeicosatrienoicacid; mPTP: mitochondrial permeability transition pore; iv: intravenous injection; TNF-α: tumor necrosis factor.
Table 1.
Overview of the evolution of TRPV4 agonist, the associated physiological processes and their clinical application. Note: 4α-PDD: 4alpha-phorbol 12,13-didecanoate; PKC: protein kinase C; CF: cardiofibroblasts; 5,6-EET: 5,6 epoxyeicosatrienoicacid; mPTP: mitochondrial permeability transition pore; iv: intravenous injection; TNF-α: tumor necrosis factor.
| Molecules | Year of Identification | EC50/IC50 | Other Targets | Features | Cardiovascular Effects | Clinical Trials/Uses | References |
|---|---|---|---|---|---|---|---|
| Agonists | |||||||
| 4α-PDD | 2003 | 50 µM |
|
|
|
| [,,,] |
| 5,6-EET | 2003 | 0.13 µM |
|
|
|
| [,,,] |
| RN-1747 | 2009 | 5.9–7.7 µM |
|
|
|
| [] |
| GSK1016790A | 2008 | 1–18 nM |
|
|
|
| [,,,,,,,,,] |
| Quinazolin-4(3H) | 2019 | 280 nM |
|
|
|
| [] |
Table 2.
Overview of the evolution of TRPV4 antagonists, the associated physiological processes and their clinical application. Note: mPTP: mitochondrial permeability transition pore; iv: intravenous injection; RISK: reperfusion injury salvage kinase; DCM-hiPSC-CMs: dilated cardiomyopathy-induced pluripotent stem cell.
Table 2.
Overview of the evolution of TRPV4 antagonists, the associated physiological processes and their clinical application. Note: mPTP: mitochondrial permeability transition pore; iv: intravenous injection; RISK: reperfusion injury salvage kinase; DCM-hiPSC-CMs: dilated cardiomyopathy-induced pluripotent stem cell.
| Molecules | Year of Identification | EC50/IC50 | Other Targets | Features | Cardiovascular Effects | Clinical Trials/Uses | References |
|---|---|---|---|---|---|---|---|
| Antagonists | |||||||
| RN-1734 | 2009 | 2 to 6 µM |
|
|
|
| [,,,,,] |
| HC-067047 | 2010 | 17 to 133 nM |
|
|
|
| [,,,,,,,] |
| RN-9893 | 2015 | 320 to 660 nM |
|
|
|
| [,,,,] |
| GSK2193874 | 2017 | 2 to 50 nM |
|
|
|
| [,,,] |
| GSK3527497 | 2019 | 12 nM |
|
|
|
| [,] |
| GSK2798745 | 2019 | 2 to 16 nM |
|
|
|
| [,,,,,,,] |
| GSK3395879 | 2018 | 1 nM |
|
|
|
| [,,,] |
3.2. TRPV4 Knockout Mice
To our knowledge, two transgenic TRPV4 knockout mouse strains were developed from two different groups. Suzuki et al. used the 129/SvJ strain via a cassette insertion mutagenesis of exon 5 [], whereas Liedtke et al. used the C57bl/6J strain with a Cre-lox-mediated excision of exon 12 []. Both mouse strains were viable and fertile [,] (see Table 3).
Table 3.
Comparison of 129/SvJ trpv4-/- andC57bl/6J trpv4-/-. Ref.: references.
4. Physiological Roles in Cardiovascular System
4.1. TRPV4 Expression Profile under Physiological Conditions
In mammals, TRPV4 channel expression is distributed in various organs and tissues, including the blood vessels and heart. RT-PCR, immunostaining and functional recordings have demonstrated that the TRPV4 channel is expressed in cardiac cell types such as atrial and ventricular cardiomyocytes [,,,,,,,], human embryonic stem cell-derived cardiomyocytes [], cardiofibroblasts (CFs) [,,,,,], ECs [,,,,,] and smooth muscle cells [71,84,87,120,122–127 ] (see Table 4).
Table 4.
TRPV4 channel expression and demonstrated function in cardiovascular system.
Taken together, the TRPV4 channel is present and functional in the plasma membrane of cardiac cells, where it plays important physiological roles.
4.2. Modulation of Ventricular Electrical Activity
In healthy mammalian hearts, the action potential (AP) waveform initiates and modulates cardiac contractility. It occurs suddenly and transitorily when the resting membrane potential depolarizes and repolarizes according to the successive opening and closing of several ion channels. A lot of attention is paid to the ventricular repolarization phase in preclinical investigations, as AP prolongation is often associated with an increased risk of potentially lethal arrhythmias. Recent evidence has shown that TRP channel opening produces a depolarizing current, since the net flux of cations is inward under physiological conditions []. Therefore, these channels are particularly important in excitable cells, such as cardiomyocytes, where they can both trigger and modulate the AP shape. To date, some TRP channels, such as TRPC3 [,], TRPM4 [,,] and TRPM7 [], have been shown to modulate both the sinus node and cardiac AP. Interestingly, the deletion or mutations of TRPM4 were reported to be associated with several cardiac electrical disorders (Brugada, long QT and progressive cardiac conduction disorders) [,,,,] and hypertrophy []. Since other TRP channels are expressed in cardiac myocytes they may participate in cardiac electrical activity, and their mutations could lead to inherited cardiac electrical disorders. Thus, it appears necessary to specifically describe the effects of each of these channels, including TRPV4, and to evaluate their physiological roles in the cardiovascular system.
TRPV4 protein expression was detected in mouse [,,] and rat [] left ventricular myocytes. It shows membrane-specific expression and locations (plasma membrane and t-tubules), which are age-dependent [,]. In young mice [], the TRPV4 agonist GSK1016790A induced a dose-dependent and transient increase in AP duration in trpv4+/+ left ventricular myocytes. In this study, GSK1016790A was proposed to promote the trafficking of the TRPV4 channel to the membrane. This phenomenon was transient because of the subsequent rapid channel endocytosis [,,]. A similar effect of GSK1016790A was also observed on rat atrial myocytes []. According to its biophysical properties, TRPV4’s potentiation in a physiological or pathological context would prolong AP. Conversely, the TRPV4 inhibitor GSK2193874 (100 nM) significantly shortened the AP duration in trpv4+/+ left ventricular myocytes. The effects of these pharmacological modulators on the ventricular AP were not observed in trpv4-/- mice, showing that the effects observed on WT cardiomyocytes were exclusively due to TRPV4 modulation. These data suggest that TRPV4 channels carry an inward current, which remains to be characterized, during the ventricular AP in basal conditions. Computational modeling was used to predict the shape of the TRPV4 current during AP []. This model considers the channel permeation and open probability depending on the voltage and subsarcolemmal [Ca2+]i that will be sensed by the channel. Since the TRPV4 channel’s open probability increases with depolarization and decreases with [Ca2+]i elevation, the model predicted a transient inward current that developed rapidly after the AP upstroke and declined with increases in [Ca2+]i and membrane repolarization []. The consequence of trpv4 deletion on the cardiac electrical activity was also evaluated. In vivo electrocardiogram recordings revealed a significantly shortened QT interval in trpv4-/- mice compared to their wild-type littermates (C57bl/6J strain). In accordance with these data, a 19% reduction in AP duration was found in ventricular myocytes from trpv4-/- mice compared to trpv4+/+ mice. No change in the properties of the main VGICs participating in AP was observed between trpv4+/+ and trpv4-/- myocytes, suggesting that QT and AP duration shortening in trpv4-/- mice is exclusively attributable to trpv4 deletion []. Thus, TRPV4 channels are constitutively active in cardiomyocytes from young mice under basal conditions and modulate ventricular electrophysiology []. The contribution is likely to increase with age []. Moreover, since TRPV4 is well known to be mechanosensitive [,,], membrane stretching due to contraction–relaxation cycles, especially under conditions of acute increases in ventricular load (e.g., during exercise), may modulate its function. Finally, a recent investigation has shown that the TRPV4 expression level is increased in cardiomyocytes of aged (24–27 months) mice compared with young (3–6 months) mice [] but TRPV4’s contribution to the prolonged repolarization commonly observed with aging remains to be studied.
Together, these studies suggest that TRPV4 modulates ventricular electrical activity under basal conditions and may, thus, be involved in cardiac arrhythmias in aging or under pathological conditions [] (see Section 5 for details).
4.3. Modulation of Cardiac Contractility
Muscular contractility is onset by an increase in cytoplasmic Ca2+. Therefore, Ca2+ handling results from a tight balance between both the Ca2+ influx, intracellular store release and uptake and cell extrusion []. In a ventricular myocyte, Ca2+ influx is mainly mediated by voltage-gated Ca2+ channels (Cav1.2), whereas the Ca2+ extrusion is ensured by the Na/Ca exchanger (NCX1) and plasma membrane Ca2+ pump (PMCA) []. Several TRP channels are present and functional and can constitute alternative sources of Ca2+ entry. Furthermore, it is well established that impaired Ca2+ handling leads to abnormal contractility [,,]. Therefore, a better understanding of the TRP channels involved in Ca2+ handling and contractility will constitute an important step to treat deleterious cardiac diseases involving cytosolic Ca2+ overload in myocytes []. Regarding TRPV4, several studies reported its involvement in these processes, while others did not observe an obvious contribution. Indeed, its contribution to intracellular Ca2+ influx and myocardial contractility was examined by Li et al., using the TRPV4 agonist 4-αPDD on isolated rat papillary muscles. The authors revealed that the 4-αPDD had no effect on contractility []. In the same line, another study revealed in isolated perfused rat hearts that TRPV4 channel potentiation by the agonist GSK1016790A also had no effect on the beating rate and contractility but induced, at the in vivo level, circulatory collapse, which was most likely due to vascular leakage and tissue haemorrhage in the lungs []. On the other hand, our group has recently shown that perfusion of the TRPV4 agonist GSK101679A induced a significant increase in transient Ca2+ current influx in trpv4+/+ but not in trpv4-/- mouse myocytes [], confirming TRPV4’s involvement in modulating the left ventricular intracellular Ca2+ concentration. Recently, Veteto et al. explored the relationship between the TRPV4 function and Ca2+ handling after mechanical stimulation in aged hearts []. To this end, the authors explored the effect of the left ventricular preload elevation in working-heart perfused aged mice hearts and found that following the initial Frank–Starling response, these hearts exhibited a secondary increase in left ventricular maximal pressure, which was absent when perfusing the TRPV4 antagonist HC067047. Interestingly, when the preload elevation was maintained for a longer period of time (20 min), the maximal left ventricular pressure declined less in HC067047-treated than in untreated aged hearts. The authors then studied Ca2+ handling in ventricular myocytes following uniaxial stretching and found a delayed increase in intracellular Ca2+ in myocytes from aged mice, ultimately leading to a contracted state, both of which were prevented by HC067047 treatment. These results suggest that TRPV4 is responsible for Ca2+ entry, leading to a hypercontractile response secondary to myocardial stretching in the aged heart, although when stretched was maintained, the TRPV4-mediated Ca2+ influx was detrimental to cardiac contractility. Further investigations have reported that TRPV4 expression increases in mouse cardiomyocytes with advancing age [] and after in vivo ischemia–reperfusion []. Note that TRPV4 upregulation in aged mice was associated with increased hypoosmotic-stress-mediated contractility, enhanced cell death and increased ischemia–reperfusion injuries []. Finally, the inhibition of TRPV4 by HC067047 exerts a cardioprotective effect during ischemia–reperfusion, attesting the key role of TRPV4 in Ca2+ handling and contractility []. It is important to underline that TRPV4 channel can also dock with other TRP channels and little is known about the contribution of these types of channels in intracellular Ca2+ dynamics and their involvement in the myocyte contractility. In this context, detailed interplay between TRPV4 channels and various α-subunits requires additional research in cardiomyocytes.
Collectively, these data suggest that the TRPV4 channel represents an attractive ion channel target to prevent Ca2+ overload and cardiac contractility dysfunction [,,,]. Indeed, TRPV4 seems to contribute to Ca2+ homeostasis regulation under physiological conditions without impacting the contractility, whereas it may be more deleterious during aging or under pathological conditions and may lead to Ca2+ overload and altered contractility. Further investigations are undoubtedly needed to fully understand TRPV4’s role in cardiac contractility in human cardiovascular diseases.
4.4. Modulation of Vascular Tone
4.4.1. TRPV4 and Vasodilation
TRPV4 is expressed in ECs, where its potentiation results in vasodilatation [,]. TRPV4 is sensitive to shear stress [] and is able to be activated by blood flow and induce Ca2+ entry when expressed in the endothelium. In ECs, Ca2+ entry activates the endothelial nitric oxide synthase (eNOS) pathway and the production of nitric oxide (NO), which can, in turn, diffuse to smooth muscle cells (SMCs). In SMCs, NO activates cyclic GMP/PKG signaling, inducing vasodilatation [], leading to endothelium-derived factors release and causing SMC hyperpolarization and vasodilatation []. Furthermore, Ca2+ entry activates in parallel intermediate (IK)- and small (SK)-conductance Ca2+-sensitive K+ channels in ECs, leading to the hyperpolarization of ECs, and then the hyperpolarization of SMCs via myoendothelial gap junctions []. The genetic deletion or pharmacological blockade of TRPV4 channels inhibits the NO- and endothelium-derived hyperpolarizing factor (EDHF)-dependent relaxation of mouse small mesenteric arteries in response to flow []. Accordingly, the transfection of TRPV4 channels in HEK293 cells confers them sensitivity to flow and induces shear-stress-dependent Ca2+ entry [], confirming the important role of TRPV4 in sensing shear stress. TRPV4-transfected HEK-293T cells are also sensitive to cell confluence, modulating TRPV4’s response to hypoxia at high cell densities, suggesting another important feature of TRPV4 activation, i.e., hypoxia []. Indeed, in vessels, hypoxia induces vasodilatation in the systemic circulation, whereas it induces vasoconstriction in the pulmonary circulation. TRPV4’s potentiation with its agonist GSK1016790A causes the endothelium-dependent relaxation of precontracted rat pulmonary artery rings, and this relaxation is inhibited in de-endothelized vessels. The authors of this study suggested that both NO and EDHF contribute to GSK1016790A-induced relaxation []. Ottolini et al. found that TRPV4 channels in mouse ECs colocalize with IK/SK channels in mesenteric arteries but not in pulmonary arteries, which explains that TRPV4 sparklets activate IK/SK channels in mesenteric arteries but not in pulmonary arteries, where ECs TRPV4 preferentially activate eNOS []. Additionally, flow-induced vasorelaxation in human coronary microvessels requires endothelial TRPV4 activation, which increases mitochondrial ROS production in ECs and induces ROS-dependent vasodilation [,].
4.4.2. TRPV4 and Vasoconstriction
TRPV4 is also expressed in SMCs, where it participates in vascular contraction, cell migration and proliferation [,]. TRPV4 activation in pulmonary artery SMCs results in Ca2+ entry and Ca2+ release from the SR through the activation of ryanodine receptors []. This TRPV4-mediated Ca2+ elevation leading to SMC contraction [,] can be triggered by serotonin [,] or mechanical stimuli such as flow shear stress []. The dysregulation of TRPV4 expression between ECs and SMCs could lead to an impaired balance between vasorelaxation and vasoconstriction and result in an altered myogenic tone [,,].
4.4.3. TRPV4 and Mechanosensitivity
In vascular physiology, cells undergo mechanical stimulation induced by blood pressure, shear stress, stretching or parietal tension, which can promote TRPV4 opening. In accordance, TRPV4 was shown to be involved in the regulation of myogenic tone. In PAECs, TRPV4 can be activated by shear stress, leading to vasodilation []. It was also shown that TRPV4 could be activated downstream of Piezo1 in ECs, where shear stress resulted in an elevation of the intracellular Ca2+ concentration []. The elevation of intracellular Ca2+ due to Piezo1 was transient, whereas the TRPV4-induced Ca2+ response was sustained, resulting in the modification of adherent junctions or actin cytoskeleton remodeling []. In SMCs, intraluminal blood pressure can activate TRPV4, whose subsequent Ca2+ entry triggers contraction, migration and proliferation phenomena [,,,,]. TRPV4 activity can also be regulated by membrane stiffness, as it was shown that cyclic stretching could lead to a lower cholesterol/phosphatidylcholine ratio in membranes and that cholesterol modulates TRPV4 activation to GSK1016790A or stretching []. An increase in mechanical stress in the vessels could lead to a dysregulation of TRPV4 signaling and to impaired physiological responses such as constriction, proliferation or migration, which are features of pulmonary hypertension.
5. Pathological Implications of TRPV4 Channels
5.1. Expression Remodeling under Pathological Condition
TRPV4 expression increases under certain pathophysiological conditions, such as aging [], pressure overload [,], ischemia–reperfusion [,], increased membrane tension [,] and pericarditis in rats and patients with atrial fibrillation []. In this context, it seems to be important to investigate the TRPV4 expression profile during life but also during the development of cardiac diseases.
5.2. Arrhythmias
Cardiac arrhythmias refer to abnormal heart rhythms or significant irregularities in the electrical signals, which may alter the cardiac function or lead to sudden cardiac death. Atrial fibrillation is a common supraventricular arrhythmia characterized by rapid and irregular electrical activity in the upper chambers of the heart (atria). This arrhythmia can have various etiologies, including structural heart abnormalities, hypertension, heart valve disorders, coronary artery disease and thyroid dysfunction [], and is associated, in part, with Ca2+ handling defects (connell et al.). entricular fibrillation is a life-threatening arrhythmia that occurs in the lower chambers of the heart (ventricles), causing electrical storms that can lead to torsades de pointes and sudden cardiac death (Solomon et al., Ludhwani et al.). Ventricular fibrillation is typically a consequence of an underlying heart disease, such as coronary artery disease, MI, cardiomyopathy; a primary electrical disorder; or electrolyte imbalances (Antzelevitch et al.) Unfortunately, current antiarrhythmic drugs for the treatment of atrial and ventricular fibrillation are not sufficiently specific and effective and are most of the time associated with both intra and extra-cardiac effects, which may, in turn, offset their therapeutic benefits. In this context, a deeper understanding of the maintenance and evolution of arrhythmia phenotypes may help to find adaptive therapies for cardiac patients. To date, the current therapeutic strategies use pharmacological drugs to target ion channels or limit Ca2+ overload and catheter-based ablation approaches. Among these channels, the TRP channel family is under the spotlight because of their biophysical properties and more specifically their permeability to Ca2+ [].
Since the TRPV4 channels are expressed in native cardiac cells, including atrial cardiomyocytes, their implication in atrial fibrillation was evaluated []. In a model of a rat sterile pericarditis-related atrial fibrillation phenotype, the TRPV4 expression level markedly increased within the atrial tissue over 2 weeks after surgery compared to the sham condition. This result is interesting because it suggests that TRPV4 channels can be directly linked to the atrial fibrillation phenotype in the early phase of its development. The TRPV4 agonist GK1016790A perfusion on atrial myocytes increased both the action potential duration and intracellular Ca2+ levels, whereas the TRPV4 channel inhibition by GSK2193874 had an opposite effect. Interestingly, the authors have shown in vivo that the blockade of TRPV4 limited abnormal electrophysiological changes, protected the heart against cardiac fibrosis and inflammation and decreased the vulnerability to atrial fibrillation without explaining the accurate mechanism of this positive effect [,]. In this context, TRPV4 may constitute an interesting therapeutic target to treat atrial and ventricular fibrillation. Additional research is needed to identify TRPV4’s involvement in human arrhythmias
At the left ventricular myocyte level, another recent investigation pointed out the importance of considering aging and its potential negative effects on Ca2+ handling, the resting membrane potential and the risk of developing ventricular arrhythmia after ischemia–reperfusion. Because theTRPV4 channel is upregulated in cardiomyocytes of aged mice [], the perfusion of the TRPV4 antagonist HC067047 after ischemia–reperfusion reduces the rate of pro-arrhythmic diastolic Ca2+ signaling, maintains the resting membrane potential and decreases the ventricular arrhythmia score []. Therefore, TRPV4 blockade may also constitute a promising therapeutic option to limit the occurrence of arrhythmogenic events for aged populations following ischemia–reperfusion and MI.
It is important to underline that the precise role of TRPV4 channels in different types of cardiac arrhythmia and their potential as therapeutic targets is still an area of active investigation. Further research is needed to fully understand the mechanisms by which TRPV4 channels contribute to arrhythmogenesis and to develop safe and effective strategies to modulate them.
5.3. Cardiac Remodeling and Fibrosis
There is some evidence that TRPV4 may be involved in cardiac pathological remodeling and could, thus, be an interesting therapeutic target in these contexts. In a recent study, TRPV4 channel expression was found to be significantly increased in the ventricles of mice with left ventricular pressure overload-induced hypertrophy and in failing human ventricles []. Interestingly, the pressure overload resulted in reduced cardiac hypertrophy, cardiac dysfunction, fibrosis and inflammation in TRPV4 knockout mice compared to wild-type animals. Moreover, treatment with GSK2193874 (TRPV4 antagonist) prevented the pressure-overload-induced remodeling and dysfunction, further confirming TRPV4’s involvement. In the same study, the authors showed that the increase in CaMKII phosphorylation found in the pressure-overloaded left ventricle in wild-type mice was absent in TRPV4 knockout mice. They suggested that this TRPV4-related increase in CaMKII phosphorylation leads to NF-κB phosphorylation and NLRP3 activation, which both contribute to the pro-inflammatory remodeling found in these pressure-overloaded hearts. Therefore, inhibiting TRPV4 appears promising to limit CaMKII phosphorylation and its multiple consequences and inflammation in cardiac hypertrophy. Despite the convincing evidence in animal models, it must be noted that TRPV4 expression regulation in human heart failure remains uncertain, with some studies showing increased expression levels [], while others found no significant regulation []. Such discrepancies may be related to the heart failure etiology, as the first study [] selected patients with dilated cardiomyopathy only, while the second [] used a broader range of heart failure etiologies. TRPV4’s expression and function in human heart failure will require further investigations. The role of TRPV4 in cardiac remodeling has also been studied in the context of MI and subsequent fibrosis through channels expressed in CFs.
CFs originate from mesenchymal stem cells and represent two-thirds of the cardiac cell population []. They contribute to maintaining the structural, biochemical, mechanical and electrical properties of the healthy myocardium and constitute the main source of extracellular matrix (ECM) protein production (collagen types I and III and metalloproteases) []. Fibrosis is known to be related to an expansion of the cardiac interstitial space due to an accumulation of ECM proteins. During this phenomenon, CFs proliferate, migrate and differentiate into myofibroblasts [] under the effect of a variety of factors, including transforming growth factor β1 (TGF-β1), tumor necrosis factor-α (TNFα), angiotensin II (Ang II), platelet-derived growth factor (PDGF), endothelin 1 (ET-1) and mechanical (stretching and stiffness) factors [,]. Interestingly, these changes in the heart eventually lead to an increased matrix stiffness, structural damaging effect and electrical disorders [] and predispose it to diastolic dysfunction [,]. Moreover, intracellular Ca2+ signaling, which is involved in fibroblast proliferation and differentiation, is mainly activated in CFs through TRP channels, including TRPV4 channels [,,,,]. Indeed, the accumulating evidence hints that the TRPV4 mechanosensor is an important factor in the progression of fibrosis or prevention of fibroproliferative disorders in several organs such as the skin, lungs [], liver, kidneys, brain, blood vessels and heart []. The role of TRPV4 mechanotransduction in cardiac fibrosis was demonstrated using a trpv4-/- mouse model []. The results suggested that the TRPV4 deletion preserves cardiac function and reduces fibrosis in trpv4-/- mice but not in WT mice after transverse aortic constriction and MI []. Interestingly, the trpv4-/- mice displayed a marked decrease in fibrosis, as well as a decrease in the pro-fibrotic gene expression levels, including for col1A2, α-SMA, NFAT, TGF-β1 and MRTF, a mechanosensitive transcription factor. Conversely, the WT mice subjected to MI or congestive heart failure showed an upregulation of TRPV4 channels and profibrotic genes compared to the untreated WT mice, which led to increased intracellular Ca2+ levels and amplified the pathological fibrosis process. These findings demonstrated that TRPV4 is functional in mouse CFs and directly related to the fibrosis process insofar as its genetic deletion preserves cardiac function and protects the heart against adverse fibrosis effects []. In line with these results, several in vitro investigations of rat or human CFs have shown a rapid but sustained increase in Ca2+ influx in response to TRPV4 agonist perfusion (4αPDD [] or GSK1016790A []), which promotes the fibroblasts’ differentiation into myofibroblasts. Consistently, the blockade of TRPV4 by different antagonists (AB159908 [], RuR [] or GSK2193874 []) or TRPV4 short-interference RNA (TRPV4-siRNA) [] inhibits TGF-β-induced fibroblast differentiation [], abolishes Ca2+ conductance [] or decreases the Ca2+ influx level [,]. Recently, and in agreement with the previous investigations, treatment with the pro-fibrotic cytokine TGF-β increased the TRPV4 channel expression in human ventricular CFs []. Simultaneously, the authors noted both an increase in a fibrosis biomarker (the plasminogen activator inhibitor-1) and an increase in a fibroblast differentiation biomarker (the α-smooth muscle actin, α-SMA) []. In addition, and via the MAPK/ERK pathway, TRPV4 channel stimulation by its specific agonist (GSK1016790A) triggers the CFs’ transformation into myofibroblasts, while its antagonist (RN-9893) combined with a Ca2+ chelating agent (BAPTA-AM) reduces it []. These different points are important because they show that the Ca2+ permeability of TRPV4 channels is an essential component of human ventricular CF differentiation. A recent investigation demonstrated that theTRPV4 channel expression level was higher in CFs isolated from Sprague–Dawley neonatal diabetic rats and from Sprague–Dawley adult rats cultured in glucose-rich medium. Interestingly, the perfusion of the TRPV4 channel agonist HC067047 significantly abolished these CF changes. It has also been reported that HC067047 decreases collagen I synthesis and suppresses the presence of the growth factor TGF-β necessary for the differentiation of CFs into myofibroblasts. In this context, different approaches could be used directly or indirectly to manage the TRPV4 channel function and imagine new therapeutic strategies to limit fibrosis within cardiac diabetic patients [] or in various cardiac pathologies involving many different patient populations. Finally, the link between fibrosis and the increased risk for atrial [,,,] or ventricular [] arrhythmias has been reported in several cardiac diseases [,]. In this context, the anti-fibrosis approach via TRPV4 inhibition constitutes a promising therapeutic way to limit adverse outcomes and prevent arrhythmias in the heart.
Taken together, these findings are promising and identify TRPV4 as a potential therapeutic target to attenuate cardiac fibrosis, cardiac dysfunction and arrhythmias in heart failure and myocardial infarction. However, further work in human samples and patients is required to confirm these results and ensure translation to the clinic.
5.4. TRPV4 Channelopathies
Numerous members of the transient receptor potential channel family (TRPA1, TRPC6, TRPM1, 2, 3, 4, 6, 7, TRPML1 and TRPV3, 4) have been described as being implicated in hereditary channelopathies. In several cases, mutations disrupt ion channel function and are causal for the disease pathogenesis [,,]. To date, no trpv4 mutation has been uncovered in primary cardiac diseases and electrical disorders. However, mutations carried by the trpv4 gene are directly associated to neurodegenerative disorders (Charcot–Marie–Tooth disease type 2C, scapuloperoneal spinal muscular atrophy, distal spinal motor neuropathy, distal spinal muscular atrophy) [,,,,] and various skeletal displasisas ranging from mild autosomal dominant brachyolmia to severe metatropic dysplasia [,,]. Moreover, it is important to underline that there is a crosstalk between both neuropathy and skeletal dysplasia, as some TRPV4 mutations, such as A217S, E278K, V620I and P799R, are responsible for linking the two phenotypes [,]. Interestingly, these pathologies are often associated with cardiac dysfunction and arrhythmias However, a direct link between mutated TRPV4 channels and the cardiac phenotype remains to be demonstrated.
6. Conclusions and Perspectives
The evidence for a crucial role of the TRPV4 channel has emerged from a large range of experiments in which its inhibition preserves the physiological intracellular Ca2+ dynamics and protects the heart form several dysfunctions, including pathological cardiac disorders (Figure 2), HF and arrhythmia. Unfortunately, the molecular mechanism involving the transition from a healthy heart towards a cardiac pathology is poorly understood. The use of specific TRPV4 modulators combined with transgenic animal models has given valuable information about TRPV4’s involvement in physiological or pathological cardiovascular remodeling. Note that the recent availability of the crystal structure of the TRPV4 channel will facilitate drug development to counteract diseases, including potentially pathological cardiac phenotypes. Furthermore, various genetic diseases involving abnormal TRPV4 channel function have highlighted the key role of this ion channel in a broad spectrum of cellular processes. Surprisingly, no trpv4 mutation was found in patients with cardiac diseases so far. It would be relevant to further investigate the role of TRPV4 in the elderly population, given the increase in its expression and the risk of cardiac diseases (ischemia–reperfusion lesion, MI, pressure overload, arrhythmia) found in this population. Future work in this promising field may help to better understand the function and regulation of this channel and identify its interactions with other channels and the implications for cardiac physiological and pathological processes.
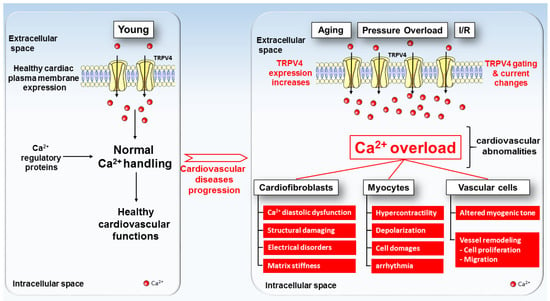
Figure 2.
TRPV4 expression and function model in healthy (left) and pathologic (right) cardiac cells. Under physiological conditions, TRPV4 channels (yellow channels) present a low expression level in the plasma membranes of cardiac cells. With aging, pressure overload, ischemia–reperfusion and others deleterious factors, TRPV4 exhibits increases in both expression and activity in the plasma membranes of cardiac cells, which tend towards the progression of cardiovascular diseases.
Author Contributions
Conceptualization, S.C.; methodology, S.C., R.G. and D.B.; Writing—review and editing, S.C., S.B., T.D., R.G. and D.B.; supervision, S.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work received financial support from the French Government as part of the “Investments of the Future” program managed by the National Research Agency (ANR-10-IAHU-04). SB and TD are supported by the French ANR CANALBRET (19-CE44-0010-02) and the New Aquitaine regional council PHYSTRIG (AAPR2020I-2019-8151810). Finally, DB is supported by the ANR MEGAVOLT (18CE14002701).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moran, M.M.; Xu, H.; Clapham, D.E. TRP ion channels in the nervous system. Curr. Opin. Neurobiol. 2004, 14, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.M. TRP Channels as Potential Drug Targets. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 309–330. [Google Scholar] [CrossRef] [PubMed]
- Hof, T.; Chaigne, S.; Récalde, A.; Sallé, L.; Brette, F.; Guinamard, R. Transient receptor potential channels in cardiac health and disease. Nat. Rev. Cardiol. 2019, 16, 344–360. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.M.; Cibelli, M.; Urban, L.; Nilius, B.; McGeown, J.G.; Nagy, I. TRPV4: Molecular Conductor of a Diverse Orchestra. Physiol. Rev. 2016, 96, 911–973. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Koch, S.E.; Veteto, A.; Domeier, T.; Rubinstein, J. Role of Known Transient Receptor Potential Vanilloid Channels in Modulating Cardiac Mechanobiology. Front. Physiol. 2021, 12, 734113. [Google Scholar] [CrossRef]
- Randhawa, P.K.; Jaggi, A.S. TRPV4 channels: Physiological and pathological role in cardiovascular system. Basic Res. Cardiol. 2015, 110, 54. [Google Scholar] [CrossRef] [PubMed]
- Inoue, R.; Kurahara, L.-H.; Hiraishi, K. TRP channels in cardiac and intestinal fibrosis. Semin. Cell Dev. Biol. 2019, 94, 40–49. [Google Scholar] [CrossRef]
- Chaigne, S.; Cardouat, G.; Louradour, J.; Vaillant, F.; Charron, S.; Sacher, F.; Ducret, T.; Guinamard, R.; Vigmond, E.; Hof, T. Transient receptor potential vanilloid 4 channel participates in mouse ventricular electrical activity. Am. J. Physiol. Circ. Physiol. 2021, 320, H1156–H1169. [Google Scholar] [CrossRef]
- Gorbunov, A.S.; Maslov, L.N.; Jaggi, A.S.; Singh, N.; De Petrocellis, L.; Boshchenko, A.A.; Roohbakhsh, A.; Bezuglov, V.V.; Oeltgen, P.R. Physiological and Pathological Role of TRPV1, TRPV2 and TRPV4 Channels in Heart. Curr. Cardiol. Rev. 2019, 15, 244–251. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, H.; Jiang, Y.; Wei, H.; Liu, P.; Wang, W.; Niu, W. Unusual localization and translocation of TRPV4 protein in cultured ventricular myocytes of the neonatal rat. Eur. J. Histochem. 2012, 56, e32. [Google Scholar] [CrossRef]
- Jones, J.L.; Peana, D.; Veteto, A.B.; Lambert, M.D.; Nourian, Z.; Karasseva, N.G.; Hill, M.A.; Lindman, B.R.; Baines, C.P.; Krenz, M.; et al. TRPV4 increases cardiomyocyte calcium cycling and contractility yet contributes to damage in the aged heart following hypoosmotic stress. Cardiovasc. Res. 2019, 115, 46–56. [Google Scholar] [CrossRef]
- Arniges, M.; Fernández-Fernández, J.M.; Albrecht, N.; Schaefer, M.; Valverde, M.A. Human TRPV4 channel splice variants revealed a key role of ankyrin domains in multimerization and trafficking. J. Biol. Chem. 2006, 281, 1580–1586. [Google Scholar] [CrossRef]
- Shigematsu, H.; Sokabe, T.; Danev, R.; Tominaga, M.; Nagayama, K. A 3.5-nm structure of rat TRPV4 cation channel revealed by Zernike phase-contrast cryoelectron microscopy. J. Biol. Chem. 2010, 285, 11210–11218. [Google Scholar] [CrossRef] [PubMed]
- Inada, H.; Procko, E.; Sotomayor, M.; Gaudet, R. Structural and biochemical consequences of disease-causing mutations in the ankyrin repeat domain of the human TRPV4 channel. Biochemistry 2012, 51, 6195–6206. [Google Scholar] [CrossRef]
- Takahashi, N.; Hamada-Nakahara, S.; Itoh, Y.; Takemura, K.; Shimada, A.; Ueda, Y.; Kitamata, M.; Matsuoka, R.; Hanawa-Suetsugu, K.; Senju, Y.; et al. TRPV4 channel activity is modulated by direct interaction of the ankyrin domain to PI(4,5)P(2). Nat. Commun. 2014, 5, 4994. [Google Scholar] [CrossRef]
- Deng, Z.; Paknejad, N.; Maksaev, G.; Sala-Rabanal, M.; Nichols, C.G.; Hite, R.K.; Yuan, P. Cryo-EM and X-ray structures of TRPV4 reveal insight into ion permeation and gating mechanisms. Nat. Struct. Mol. Biol. 2018, 25, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Voets, T. The puzzle of TRPV4 channelopathies. EMBO Rep. 2013, 14, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, N.; Albrecht, N.; Harteneck, C.; Schultz, G.; Schaefer, M. Homo- and heteromeric assembly of TRPV channel subunits. J. Cell Sci. 2005, 118 Pt 5, 917–928. [Google Scholar] [CrossRef]
- Jin, X.; Touhey, J.; Gaudet, R. Structure of the N-terminal ankyrin repeat domain of the TRPV2 ion channel. J. Biol. Chem. 2006, 281, 25006–25010. [Google Scholar] [CrossRef]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef]
- Lishko, P.V.; Procko, E.; Jin, X.; Phelps, C.B.; Gaudet, R. The ankyrin repeats of TRPV1 bind multiple ligands and modulate channel sensitivity. Neuron 2007, 54, 905–918. [Google Scholar] [CrossRef]
- McCleverty, C.J.; Koesema, E.; Patapoutian, A.; Lesley, S.A.; Kreusch, A. Crystal structure of the human TRPV2 channel ankyrin repeat domain. Protein Sci. 2006, 15, 2201–2206. [Google Scholar] [CrossRef]
- D’Hoedt, D.; Owsianik, G.; Prenen, J.; Cuajungco, M.P.; Grimm, C.; Heller, S.; Voets, T.; Nilius, B. Stimulus-specific modulation of the cation channel TRPV4 by PACSIN 3. J. Biol. Chem. 2008, 283, 6272–6280. [Google Scholar] [CrossRef]
- Wang, Y.; Fu, X.; Gaiser, S.; Köttgen, M.; Kramer-Zucker, A.; Walz, G.; Wegierski, T. OS-9 regulates the transit and polyubiquitination of TRPV4 in the endoplasmic reticulum. J. Biol. Chem. 2007, 282, 36561–36570. [Google Scholar] [CrossRef]
- Toft-Bertelsen, T.L.; Larsen, B.R.; MacAulay, N. Sensing and regulation of cell volume—We know so much and yet understand so little: TRPV4 as a sensor of volume changes but possibly without a volume-regulatory role? Channels 2018, 12, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Strotmann, R.; Schultz, G.; Plant, T.D. Plant, Ca2+-dependent potentiation of the nonselective cation channel TRPV4 is mediated by a C-terminal calmodulin binding site. J. Biol. Chem. 2003, 278, 26541–26549. [Google Scholar] [CrossRef]
- Loukin, S.H.; Teng, J.; Kung, C. A channelopathy mechanism revealed by direct calmodulin activation of TrpV4. Proc. Natl. Acad. Sci. USA 2015, 112, 9400–9405. [Google Scholar] [CrossRef]
- Suzuki, M.; Hirao, A.; Mizuno, A. Microtubule-associated [corrected] protein 7 increases the membrane expression of transient receptor potential vanilloid 4 (TRPV4). J. Biol. Chem. 2003, 278, 51448–51453. [Google Scholar] [CrossRef]
- Stewart, A.P.; Smith, G.D.; Sandford, R.N.; Edwardson, J.M. Atomic force microscopy reveals the alternating subunit arrangement of the TRPP2-TRPV4 heterotetramer. Biophys. J. 2010, 99, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Cheng, K.-T.; Wong, C.-O.; O’neil, R.G.; Birnbaumer, L.; Ambudkar, I.S.; Yao, X. Heteromeric TRPV4-C1 channels contribute to store-operated Ca(2+) entry in vascular endothelial cells. Cell Calcium 2011, 50, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Ma, X.; Shen, B.; Huang, Y.; Birnbaumer, L.; Yao, X. TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J. 2014, 28, 4677–4685. [Google Scholar] [CrossRef] [PubMed]
- Benfenati, V.; Caprini, M.; Dovizio, M.; Mylonakou, M.N.; Ferroni, S.; Ottersen, O.P.; Amiry-Moghaddam, M. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 2563–2568. [Google Scholar] [CrossRef]
- Liu, X.; Bandyopadhyay, B.C.; Nakamoto, T.; Singh, B.B.; Liedtke, W.; Melvin, J.E.; Ambudkar, I.S. A role for AQP5 in activation of TRPV4 by hypotonicity: Concerted involvement of AQP5 and TRPV4 in regulation of cell volume recovery. J. Biol. Chem. 2006, 281, 15485–15495. [Google Scholar] [CrossRef]
- Galizia, L.; Pizzoni, A.; Fernandez, J.; Rivarola, V.; Capurro, C.; Ford, P. Functional interaction between AQP2 and TRPV4 in renal cells. J. Cell. Biochem. 2012, 113, 580–589. [Google Scholar] [CrossRef]
- Verkerk, A.O.; Lodder, E.M.; Wilders, R. Aquaporin Channels in the Heart-Physiology and Pathophysiology. Int. J. Mol. Sci. 2019, 20, 2039. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Lee, E.J.; Chun, J.; Hyun, S.; Kang, S.S. Phosphorylation on TRPV4 Serine 824 Regulates Interaction with STIM1. Open Biochem. J. 2015, 9, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.K.; Kim, J.; Ahn, S.; Xiao, K.; Shenoy, S.K.; Liedtke, W.; Lefkowitz, R.J. Arresting a transient receptor potential (TRP) channel: Beta-arrestin 1 mediates ubiquitination and functional down-regulation of TRPV4. J. Biol. Chem. 2010, 285, 30115–30125. [Google Scholar] [CrossRef]
- Ma, X.; Nilius, B.; Wong, J.W.-Y.; Huang, Y.; Yao, X. Electrophysiological properties of heteromeric TRPV4-C1 channels. Biochim. Biophys. Acta 2011, 1808, 2789–2797. [Google Scholar] [CrossRef] [PubMed]
- Köttgen, M.; Buchholz, B.; Garcia-Gonzalez, M.A.; Kotsis, F.; Fu, X.; Doerken, M.; Boehlke, C.; Steffl, D.; Tauber, R.; Wegierski, T.; et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J. Cell Biol. 2008, 182, 437–447. [Google Scholar] [CrossRef]
- Brauchi, S.; Orio, P. Voltage sensing in thermo-TRP channels. In Transient Receptor Potential Channels; Springer: Dordrecht, The Netherlands, 2011; Volume 704, pp. 517–530. [Google Scholar]
- Long, S.B.; Campbell, E.B.; MacKinnon, R. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science 2005, 309, 903–908. [Google Scholar] [CrossRef]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 2005, 309, 897–903. [Google Scholar] [CrossRef]
- Whicher, J.R.; MacKinnon, R. Structure of the voltage-gated K(+) channel Eag1 reveals an alternative voltage sensing mechanism. Science 2016, 353, 664–669. [Google Scholar] [CrossRef]
- Lee, C.-H.; MacKinnon, R. Structures of the Human HCN1 Hyperpolarization-Activated Channel. Cell 2017, 168, 111–120.e11. [Google Scholar] [CrossRef]
- Watanabe, H.; Vriens, J.; Suh, S.H.; Benham, C.D.; Droogmans, G.; Nilius, B. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J. Biol. Chem. 2002, 277, 47044–47051. [Google Scholar] [CrossRef] [PubMed]
- Voets, T.; Prenen, J.; Vriens, J.; Watanabe, H.; Janssens, A.; Wissenbach, U.; Bödding, M.; Droogmans, G.; Nilius, B. Molecular determinants of permeation through the cation channel TRPV4. J. Biol. Chem. 2002, 277, 33704–33710. [Google Scholar] [CrossRef]
- Nilius, B.; Vriens, J.; Prenen, J.; Droogmans, G.; Voets, T. TRPV4 calcium entry channel: A paradigm for gating diversity. Am. J. Physiol. Physiol. 2004, 286, C195–C205. [Google Scholar] [CrossRef]
- Lawhorn, B.G.; Brnardic, E.J.; Behm, D.J. TRPV4 antagonists: A patent review (2015–2020). Expert Opin. Ther. Pat. 2021, 31, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, W.A. Channel-mediated calcium current in the heart. Cardiovasc. Drugs Ther. 1988, 1, 447–459. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Wu, Q.; Qian, C.; Zhao, N.; Zhao, Z.; Lu, K.; Zhang, S.; Dong, Q.; Chen, L.; Li, Q.; et al. TRPV4 blockade suppresses atrial fibrillation in sterile pericarditis rats. J. Clin. Investig. 2020, 5, e137528. [Google Scholar] [CrossRef] [PubMed]
- Goyal, N.; Skrdla, P.; Schroyer, R.; Kumar, S.; Fernando, D.; Oughton, A.; Norton, N.; Sprecher, D.L.; Cheriyan, J. Clinical Pharmacokinetics, Safety, and Tolerability of a Novel, First-in-Class TRPV4 Ion Channel Inhibitor, GSK2798745, in Healthy and Heart Failure Subjects. Am. J. Cardiovasc. Drugs 2019, 19, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Lawhorn, B.G.; Brnardic, E.J.; Behm, D.J. Recent advances in TRPV4 agonists and antagonists. Bioorganic Med. Chem. Lett. 2020, 30, 127022. [Google Scholar] [CrossRef] [PubMed]
- Thorneloe, K.S.; Cheung, M.; Holt, D.A.; Willette, R.N. Properties of the TRPV4 Activator GSK1016790A and the TRPV4 Antagonist GSK2193874. Physiol. Rev. 2017, 97, 1231–1232. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.-F.; Qian, C.; Zhao, N.; Dong, Q.; Li, J.; Wang, B.-B.; Chen, L.; Yu, L.; Han, B.; Du, Y.-M.; et al. Activation of transient receptor potential vanilloid 4 involves in hypoxia/reoxygenation injury in cardiomyocytes. Cell Death Dis. 2017, 8, e2828. [Google Scholar] [CrossRef] [PubMed]
- Baratchi, S.; Keov, P.; Darby, W.G.; Lai, A.; Khoshmanesh, K.; Thurgood, P.; Vahidi, P.; Ejendal, K.; McIntyre, P. The TRPV4 Agonist GSK1016790A Regulates the Membrane Expression of TRPV4 Channels. Front. Pharmacol. 2019, 10, 6. [Google Scholar] [CrossRef]
- Liu, L.; Guo, M.; Lv, X.; Wang, Z.; Yang, J.; Li, Y.; Yu, F.; Wen, X.; Feng, L.; Zhou, T. Role of Transient Receptor Potential Vanilloid 4 in Vascular Function. Front. Mol. Biosci. 2021, 8, 677661. [Google Scholar] [CrossRef]
- Atobe, M.; Nagami, T.; Muramatsu, S.; Ohno, T.; Kitagawa, M.; Suzuki, H.; Ishiguro, M.; Watanabe, A.; Kawanishi, M. Discovery of Novel Transient Receptor Potential Vanilloid 4 (TRPV4) Agonists as Regulators of Chondrogenic Differentiation: Identification of Quinazolin-4(3 H)-ones and in Vivo Studies on a Surgically Induced Rat Model of Osteoarthritis. J. Med. Chem. 2019, 62, 1468–1483. [Google Scholar] [CrossRef]
- Grace, M.S.; Bonvini, S.J.; Belvisi, M.G.; McIntyre, P. Modulation of the TRPV4 ion channel as a therapeutic target for disease. Pharmacol. Ther. 2017, 177, 9–22. [Google Scholar] [CrossRef]
- Vincent, F.; Acevedo, A.; Nguyen, M.T.; Dourado, M.; DeFalco, J.; Gustafson, A.; Spiro, P.; Emerling, D.E.; Kelly, M.G.; Duncton, M.A. Identification and characterization of novel TRPV4 modulators. Biochem. Biophys. Res. Commun. 2009, 389, 490–494. [Google Scholar] [CrossRef]
- Dong, Q.; Li, J.; Wu, Q.-F.; Zhao, N.; Qian, C.; Ding, D.; Wang, B.-B.; Chen, L.; Guo, K.-F.; Fu, D.; et al. Blockage of transient receptor potential vanilloid 4 alleviates myocardial ischemia/reperfusion injury in mice. Sci. Rep. 2017, 7, 42678. [Google Scholar] [CrossRef]
- Hilfiker, M.A.; Hoang, T.H.; Cornil, J.; Eidam, H.S.; Matasic, D.S.; Roethke, T.J.; Klein, M.; Thorneloe, K.S.; Cheung, M. Optimization of a Novel Series of TRPV4 Antagonists with In Vivo Activity in a Model of Pulmonary Edema. ACS Med. Chem. Lett. 2013, 4, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.-L.; Nguyen, M.T.; O’mahony, D.J.; Acevedo, A.; Zipfel, S.; Zhang, Q.; Liu, L.; Dourado, M.; Chi, C.; Yip, V.; et al. Identification of orally-bioavailable antagonists of the TRPV4 ion-channel. Bioorganic Med. Chem. Lett. 2015, 25, 4011–4015. [Google Scholar] [CrossRef]
- Brooks, C.A.; Barton, L.S.; Behm, D.J.; Brnardic, E.J.; Costell, M.H.; Holt, D.A.; Jolivette, L.J.; Matthews, J.M.; McAtee, J.J.; McCleland, B.W.; et al. Discovery of GSK3527497: A Candidate for the Inhibition of Transient Receptor Potential Vanilloid-4 (TRPV4). J. Med. Chem. 2019, 62, 9270–9280. [Google Scholar] [CrossRef] [PubMed]
- Tsuno, N.; Yukimasa, A.; Yoshida, O.; Ichihashi, Y.; Inoue, T.; Ueno, T.; Yamaguchi, H.; Matsuda, H.; Funaki, S.; Yamanada, N.; et al. Discovery of novel 2’,4’-dimethyl-[4,5’-bithiazol]-2-yl amino derivatives as orally bioavailable TRPV4 antagonists for the treatment of pain: Part 1. Bioorganic Med. Chem. Lett. 2016, 26, 4930–4935. [Google Scholar] [CrossRef]
- Tsuno, N.; Yukimasa, A.; Yoshida, O.; Suzuki, S.; Nakai, H.; Ogawa, T.; Fujiu, M.; Takaya, K.; Nozu, A.; Yamaguchi, H.; et al. Discovery of novel 2’,4’-dimethyl-[4,5’-bithiazol]-2-yl amino derivatives as orally bioavailable TRPV4 antagonists for the treatment of pain: Part 2. Bioorganic Med. Chem. Lett. 2016, 26, 4936–4941. [Google Scholar] [CrossRef] [PubMed]
- Pero, J.E.; Matthews, J.M.; Behm, D.J.; Brnardic, E.J.; Brooks, C.; Budzik, B.W.; Costell, M.H.; Donatelli, C.A.; Eisennagel, S.H.; Erhard, K.; et al. Design and Optimization of Sulfone Pyrrolidine Sulfonamide Antagonists of Transient Receptor Potential Vanilloid-4 with in Vivo Activity in a Pulmonary Edema Model. J. Med. Chem. 2018, 61, 11209–11220. [Google Scholar] [CrossRef]
- Xu, F.; Satoh, E.; Iijima, T. Protein kinase C-mediated Ca2+ entry in HEK 293 cells transiently expressing human TRPV4. Br. J. Pharmacol. 2003, 140, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Duncton, M. Chapter 12—Small Molecule Agonists and Antagonists of TRPV4. In TRP Channels as Therapeutic Targets; Academic Press: Cambridge, MA, USA, 2015; pp. 205–219. [Google Scholar]
- Alexander, R.; Kerby, A.; Aubdool, A.; Power, A.; Grover, S.; Gentry, C.; Grant, A. 4alpha-phorbol 12,13-didecanoate activates cultured mouse dorsal root ganglia neurons independently of TRPV4. Br. J. Pharmacol. 2013, 168, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Dahan, D.; Ducret, T.; Quignard, J.-F.; Marthan, R.; Savineau, J.-P.; Estève, E. Implication of the ryanodine receptor in TRPV4-induced calcium response in pulmonary arterial smooth muscle cells from normoxic and chronically hypoxic rats. Am. J. Physiol. Cell. Mol. Physiol. 2012, 303, L824–L833. [Google Scholar] [CrossRef]
- Watanabe, H.; Vriens, J.; Prenen, J.; Droogmans, G.; Voets, T.; Nilius, B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature 2003, 424, 434–438. [Google Scholar] [CrossRef]
- Katragadda, D.; Batchu, S.N.; Cho, W.J.; Chaudhary, K.R.; Falck, J.R.; Seubert, J.M. Epoxyeicosatrienoic acids limit damage to mitochondrial function following stress in cardiac cells. J. Mol. Cell. Cardiol. 2009, 46, 867–875. [Google Scholar] [CrossRef]
- Lai, J.; Chen, C. The Role of Epoxyeicosatrienoic Acids in Cardiac Remodeling. Front. Physiol. 2021, 12, 642470. [Google Scholar] [CrossRef]
- Yang, L.; Mäki-Petäjä, K.; Cheriyan, J.; McEniery, C.; Wilkinson, I.B. The role of epoxyeicosatrienoic acids in the cardiovascular system. Br. J. Clin. Pharmacol. 2015, 80, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Thorneloe, K.S.; Cheung, M.; Bao, W.; Alsaid, H.; Lenhard, S.; Jian, M.-Y.; Costell, M.; Maniscalco-Hauk, K.; Krawiec, J.A.; Olzinski, A.; et al. An orally active TRPV4 channel blocker prevents and resolves pulmonary edema induced by heart failure. Sci. Transl. Med. 2012, 4, 159ra148. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Liu, B.; Yin, M.; Koroleva, M.; Mastrangelo, M.; Ture, S.; Morrell, C.N.; Zhang, D.X.; Fisher, E.A.; Jin, Z.G. A novel TRPV4-specific agonist inhibits monocyte adhesion and atherosclerosis. Oncotarget 2016, 7, 37622–37635. [Google Scholar] [CrossRef] [PubMed]
- Pankey, E.A.; Zsombok, A.; Lasker, G.F.; Kadowitz, P.J. Analysis of responses to the TRPV4 agonist GSK1016790A in the pulmonary vascular bed of the intact-chest rat. Am. J. Physiol. Circ. Physiol. 2014, 306, H33–H40. [Google Scholar] [CrossRef]
- Zhang, S.; Lu, K.; Yang, S.; Wu, Y.; Liao, J.; Lu, Y.; Wu, Q.; Zhao, N.; Dong, Q.; Chen, L.; et al. Activation of transient receptor potential vanilloid 4 exacerbates myocardial ischemia-reperfusion injury via JNK-CaMKII phosphorylation pathway in isolated mice hearts. Cell Calcium 2021, 100, 102483. [Google Scholar] [CrossRef]
- Willette, R.N.; Bao, W.; Nerurkar, S.; Yue, T.-L.; Doe, C.P.; Stankus, G.; Turner, G.H.; Ju, H.; Thomas, H.; Fishman, C.E.; et al. Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: Part 2. J. Pharmacol. Exp. Ther. 2008, 326, 443–452. [Google Scholar] [CrossRef]
- Adapala, R.K.; Thoppil, R.J.; Luther, D.J.; Paruchuri, S.; Meszaros, J.G.; Chilian, W.M.; Thodeti, C.K. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J. Mol. Cell. Cardiol. 2013, 54, 45–52. [Google Scholar] [CrossRef]
- Ahn, M.S.; Eom, Y.W.; Oh, J.E.; Cha, S.K.; Park, K.S.; Son, J.W.; Lee, J.W.; Youn, Y.J.; Ahn, S.G.; Kim, J.Y.; et al. Transient receptor potential channel TRPV4 mediates TGF-beta1-induced differentiation of human ventricular fibroblasts. Cardiol. J. 2020, 27, 162–170. [Google Scholar] [CrossRef]
- Doñate-Macian, P.; Duarte, Y.; Rubio-Moscardo, F.; Pérez-Vilaró, G.; Canan, J.; Díez, J.; González-Nilo, F.; Valverde, M.A. Structural determinants of TRPV4 inhibition and identification of new antagonists with antiviral activity. Br. J. Pharmacol. 2022, 179, 3576–3591. [Google Scholar] [CrossRef]
- Filosa, J.A.; Yao, X.; Rath, G. TRPV4 and the regulation of vascular tone. J. Cardiovasc. Pharmacol. 2013, 61, 113–119. [Google Scholar] [CrossRef]
- Greenberg, H.Z.; Carlton-Carew, S.R.; Khan, D.M.; Zargaran, A.K.; Jahan, K.S.; Ho, W.S.V.; Albert, A.P. Heteromeric TRPV4/TRPC1 channels mediate calcium-sensing receptor-induced nitric oxide production and vasorelaxation in rabbit mesenteric arteries. Vasc. Pharmacol. 2017, 96–98, 53–62. [Google Scholar] [CrossRef]
- Lu, J.; Lee, Y.-K.; Ran, X.; Lai, W.-H.; Li, R.A.; Keung, W.; Tse, K.; Tse, H.-F.; Yao, X. An abnormal TRPV4-related cytosolic Ca(2+) rise in response to uniaxial stretch in induced pluripotent stem cells-derived cardiomyocytes from dilated cardiomyopathy patients. Biochim. Biophys. Acta (BBA) -Mol. Basis Dis. 2017, 1863, 2964–2972. [Google Scholar] [CrossRef]
- Xia, Y.; Fu, Z.; Hu, J.; Huang, C.; Paudel, O.; Cai, S.; Liedtke, W.; Sham, J.S. TRPV4 channel contributes to serotonin-induced pulmonary vasoconstriction and the enhanced vascular reactivity in chronic hypoxic pulmonary hypertension. Am. J. Physiol. Cell Physiol. 2013, 305, C704–C715. [Google Scholar] [CrossRef]
- Gevaert, T.; Vriens, J.; Segal, A.; Everaerts, W.; Roskams, T.; Talavera, K.; Owsianik, G.; Liedtke, W.; Daelemans, D.; Dewachter, I.; et al. Deletion of the transient receptor potential cation channel TRPV4 impairs murine bladder voiding. J. Clin. Investig. 2007, 117, 3453–3462. [Google Scholar] [CrossRef]
- Vizin, R.C.L.; Scarpellini, C.D.S.; Ishikawa, D.T.; Correa, G.M.; de Souza, C.O.; Gargaglioni, L.H.; Carrettiero, D.C.; Bícego, K.C.; Almeida, M.C. TRPV4 activates autonomic and behavioural warmth-defence responses in Wistar rats. Acta Physiol. 2015, 214, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Xiao, C.; Sheng, D.; Yang, M.; Cheng, Q.; Wu, J.; Zhang, S. TRPV4 Mediates Cardiac Fibrosis via the TGF-beta1/Smad3 Signaling Pathway in Diabetic Rats. Cardiovasc. Toxicol. 2020, 20, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Veteto, A.B.; Lambert, M.D.; McDonald, K.S.; Domeier, T.L. TRPV4 Contributes to Stretch-Induced Hypercontractility and Time-Dependent Dysfunction in the Aged Heart. Biophys. J. 2019, 116, 382a–383a. [Google Scholar] [CrossRef]
- Cheung, M.; Bao, W.; Behm, D.J.; Brooks, C.A.; Bury, M.J.; Dowdell, S.E.; Eidam, H.S.; Fox, R.M.; Goodman, K.B.; Holt, D.A.; et al. Discovery of GSK2193874: An Orally Active, Potent, and Selective Blocker of Transient Receptor Potential Vanilloid 4. ACS Med. Chem. Lett. 2017, 8, 549–554. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, F.; Staunton, C.A.; Barrett-Jolley, R. Systemic application of the transient receptor potential vanilloid-type 4 antagonist GSK2193874 induces tail vasodilation in a mouse model of thermoregulation. Biol. Lett. 2022, 18, 20220129. [Google Scholar] [CrossRef]
- Pero, J.E.; McAtee, J.J.; Behm, D.J.; Briand, J.; Graczyk-Millbrandt, G.; Erhard, K.; Roberts, A.D.; Rivero, R.A.; Holt, D.A.; Lawhorn, B.G. Identification, Synthesis, and Characterization of a Major Circulating Human Metabolite of TRPV4 Antagonist GSK2798745. ACS Med. Chem. Lett. 2021, 12, 1498–1502. [Google Scholar] [CrossRef] [PubMed]
- Brnardic, E.J.; Ye, G.; Brooks, C.; Donatelli, C.; Barton, L.; McAtee, J.; Sanchez, R.M.; Shu, A.; Erhard, K.; Terrell, L.; et al. Discovery of Pyrrolidine Sulfonamides as Selective and Orally Bioavailable Antagonists of Transient Receptor Potential Vanilloid-4 (TRPV4). J. Med. Chem. 2018, 61, 9738–9755. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Mizuno, A.; Kodaira, K.; Imai, M. Impaired pressure sensation in mice lacking TRPV4. J. Biol. Chem. 2003, 278, 22664–22668. [Google Scholar] [CrossRef]
- Liedtke, W.; Friedman, J.M. Abnormal osmotic regulation in trpv4-/- mice. Proc. Natl. Acad. Sci. USA 2003, 100, 13698–13703. [Google Scholar] [CrossRef]
- Zhang, D.X.; Mendoza, S.A.; Bubolz, A.H.; Mizuno, A.; Ge, Z.-D.; Li, R.; Warltier, D.C.; Suzuki, M.; Gutterman, D.D. Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension 2009, 53, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Vriens, J.; Owsianik, G.; Fisslthaler, B.; Suzuki, M.; Janssens, A.; Voets, T.; Morisseau, C.; Hammock, B.D.; Fleming, I.; Busse, R.; et al. Modulation of the Ca2 permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ. Res. 2005, 97, 908–915. [Google Scholar] [CrossRef]
- Mendoza, S.A.; Fang, J.; Gutterman, D.D.; Wilcox, D.A.; Bubolz, A.H.; Li, R.; Suzuki, M.; Zhang, D.X.; Naik, J.S.; Osmond, J.M.; et al. TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am. J. Physiol. Circ. Physiol. 2010, 298, H466–H476. [Google Scholar] [CrossRef]
- Sonkusare, S.K.; Bonev, A.D.; Ledoux, J.; Liedtke, W.; Kotlikoff, M.I.; Heppner, T.J.; Hill-Eubanks, D.C.; Nelson, M.T. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 2012, 336, 597–601. [Google Scholar] [CrossRef]
- Earley, S.; Pauyo, T.; Drapp, R.; Tavares, M.J.; Liedtke, W.; Brayden, J.E.; Morrison, H.W.; Filosa, J.A.; Onishi, M.; Yamanaka, K.; et al. TRPV4-dependent dilation of peripheral resistance arteries influences arterial pressure. Am. J. Physiol. Circ. Physiol. 2009, 297, H1096–H1102. [Google Scholar] [CrossRef]
- Loot, A.E.; Popp, R.; Fisslthaler, B.; Vriens, J.; Nilius, B.; Fleming, I. Role of cytochrome P450-dependent transient receptor potential V4 activation in flow-induced vasodilatation. Cardiovasc. Res. 2008, 80, 445–452. [Google Scholar] [CrossRef]
- Hartmannsgruber, V.; Heyken, W.-T.; Kacik, M.; Kaistha, A.; Grgic, I.; Harteneck, C.; Liedtke, W.; Hoyer, J.; Kohler, R. Arterial response to shear stress critically depends on endothelial TRPV4 expression. PLoS ONE 2007, 2, e827. [Google Scholar] [CrossRef]
- Mizuno, A.; Matsumoto, N.; Imai, M.; Suzuki, M. Impaired osmotic sensation in mice lacking TRPV4. Am. J. Physiol. Physiol. 2003, 285, C96–C101. [Google Scholar] [CrossRef]
- Tabuchi, K.; Suzuki, M.; Mizuno, A.; Hara, A. Hearing impairment in TRPV4 knockout mice. Neurosci. Lett. 2005, 382, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, D.F.; King, J.A.; Weber, D.; Addison, E.; Liedtke, W.; Townsley, M.I. Transient receptor potential vanilloid 4-mediated disruption of the alveolar septal barrier: A novel mechanism of acute lung injury. Circ. Res. 2006, 99, 988–995. [Google Scholar] [CrossRef]
- Taniguchi, J.; Tsuruoka, S.; Mizuno, A.; Sato, J.-I.; Fujimura, A.; Suzuki, M. TRPV4 as a flow sensor in flow-dependent K+ secretion from the cortical collecting duct. Am. J. Physiol. Physiol. 2007, 292, F667–F673. [Google Scholar] [CrossRef] [PubMed]
- Gualdani, R.; Seghers, F.; Yerna, X.; Schakman, O.; Tajeddine, N.; Achouri, Y.; Tissir, F.; Devuyst, O.; Gailly, P. Mechanical activation of TRPV4 channels controls albumin reabsorption by proximal tubule cells. Sci. Signal. 2020, 13, eabc6967. [Google Scholar] [CrossRef]
- Masuyama, R.; Vriens, J.; Voets, T.; Karashima, Y.; Owsianik, G.; Vennekens, R.; Lieben, L.; Torrekens, S.; Moermans, K.; Bosch, A.V.; et al. TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab. 2008, 8, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Seghers, F.; Yerna, X.; Zanou, N.; Devuyst, O.; Vennekens, R.; Nilius, B.; Gailly, P. TRPV4 participates in pressure-induced inhibition of renin secretion by juxtaglomerular cells. J. Physiol. 2016, 594, 7327–7340. [Google Scholar] [CrossRef] [PubMed]
- Kunert-Keil, C.; Bisping, F.; Krüger, J.; Brinkmeier, H. Tissue-specific expression of TRP channel genes in the mouse and its variation in three different mouse strains. BMC Genom. 2006, 7, 159. [Google Scholar] [CrossRef] [PubMed]
- Veteto, A.B.; Peana, D.; Lambert, M.D.; McDonald, K.S.; Domeier, T.L. Transient receptor potential vanilloid-4 contributes to stretch-induced hypercontractility and time-dependent dysfunction in the aged heart. Cardiovasc. Res. 2020, 116, 1887–1896. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Li, Z.; Kong, C.-W.; Tang, N.L.; Huang, Y.; Li, R.A.; Yao, X. Uniaxial cyclic stretch stimulates TRPV4 to induce realignment of human embryonic stem cell-derived cardiomyocytes. J. Mol. Cell. Cardiol. 2015, 87, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Thodeti, C.K.; Paruchuri, S.; Meszaros, J.G. TRP to cardiac fibroblast differentiation. Channels 2013, 7, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Hatano, N.; Itoh, Y.; Muraki, K. Cardiac fibroblasts have functional TRPV4 activated by 4alpha-phorbol 12,13-didecanoate. Life Sci. 2009, 85, 808–814. [Google Scholar] [CrossRef]
- Du, J.; Xie, J.; Zhang, Z.; Tsujikawa, H.; Fusco, D.; Silverman, D.; Liang, B.; Yue, L. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ. Res. 2010, 106, 992–1003. [Google Scholar] [CrossRef]
- Köhler, R.; Heyken, W.-T.; Heinau, P.; Schubert, R.; Si, H.; Kacik, M.; Busch, C.; Grgic, I.; Maier, T.; Hoyer, J. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arter. Thromb. Vasc. Biol. 2006, 26, 1495–1502. [Google Scholar] [CrossRef]
- Sullivan, M.N.; Francis, M.; Pitts, N.L.; Taylor, M.S.; Earley, S. Optical recording reveals novel properties of GSK1016790A-induced vanilloid transient receptor potential channel TRPV4 activity in primary human endothelial cells. Mol. Pharmacol. 2012, 82, 464–472. [Google Scholar] [CrossRef]
- Sukumaran, S.V.; Singh, T.U.; Parida, S.; Narasimha Reddy, C.E.; Thangamalai, R.; Kandasamy, K.; Singh, V.; Mishra, S.K. TRPV4 channel activation leads to endothelium-dependent relaxation mediated by nitric oxide and endothelium-derived hyperpolarizing factor in rat pulmonary artery. Pharmacol. Res. 2013, 78, 18–27. [Google Scholar] [CrossRef]
- Bubolz, A.H.; Mendoza, S.A.; Zheng, X.; Zinkevich, N.S.; Li, R.; Gutterman, D.D.; Zhang, D.X.; Allaqaband, H.; Kadlec, A.O.; Soni, H.; et al. Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: Role of Ca2+ entry and mitochondrial ROS signaling. Am. J. Physiol. Circ. Physiol. 2012, 302, H634–H642. [Google Scholar] [CrossRef]
- Yang, X.-R.; Lin, A.H.Y.; Hughes, J.M.; Flavahan, N.A.; Cao, Y.-N.; Liedtke, W.; Sham, J.S.K.; Marshall, J.D.; Bazan, I.; Zhang, Y.; et al. Upregulation of osmo-mechanosensitive TRPV4 channel facilitates chronic hypoxia-induced myogenic tone and pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2012, 302, L555–L568. [Google Scholar] [CrossRef] [PubMed]
- Parpaite, T.; Cardouat, G.; Mauroux, M.; Gillibert-Duplantier, J.; Robillard, P.; Quignard, J.-F.; Marthan, R.; Savineau, J.-P.; Ducret, T. Effect of hypoxia on TRPV1 and TRPV4 channels in rat pulmonary arterial smooth muscle cells. Pflug. Arch. 2016, 468, 111–130. [Google Scholar] [CrossRef]
- Baylie, R.L.; Brayden, J.E. TRPV channels and vascular function. Acta Physiol. 2011, 203, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, M.; Daneva, Z.; Chen, Y.L.; Cope, E.L.; Kasetti, R.B.; Zode, G.S.; Sonkusare, S.K. Mechanisms underlying selective coupling of endothelial Ca(2+) signals with eNOS vs. IK/SK channels in systemic and pulmonary arteries. J. Physiol. 2020, 598, 3577–3596. [Google Scholar] [CrossRef] [PubMed]
- Ducret, T.; Guibert, C.; Marthan, R.; Savineau, J.-P. Serotonin-induced activation of TRPV4-like current in rat intrapulmonary arterial smooth muscle cells. Cell Calcium 2008, 43, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Yamamura, A.; Yamamura, H.; Ayon, R.J.; Smith, K.A.; Tang, H.; Makino, A.; Yuan, J.X.-J. Flow shear stress enhances intracellular Ca2+ signaling in pulmonary artery smooth muscle cells from patients with pulmonary arterial hypertension. Am. J. Physiol. Physiol. 2014, 307, C373–C383. [Google Scholar] [CrossRef] [PubMed]
- Barbeau, S.; Gilbert, G.; Cardouat, G.; Baudrimont, I.; Freund-Michel, V.; Guibert, C.; Marthan, R.; Vacher, P.; Quignard, J.-F.; Ducret, T. Mechanosensitivity in Pulmonary Circulation: Pathophysiological Relevance of Stretch-Activated Channels in Pulmonary Hypertension. Biomolecules 2021, 11, 1389. [Google Scholar] [CrossRef]
- Doleschal, B.; Primessnig, U.; Wolkart, G.; Wolf, S.; Schernthaner, M.; Lichtenegger, M.; Glasnov, T.N.; Kappe, C.O.; Mayer, B.; Antoons, G.; et al. TRPC3 contributes to regulation of cardiac contractility and arrhythmogenesis by dynamic interaction with NCX1. Cardiovasc. Res. 2015, 106, 163–173. [Google Scholar] [CrossRef]
- Qi, Z.; Wong, C.K.; Suen, C.H.; Wang, J.; Long, C.; Sauer, H.; Yao, X.; Tsang, S.Y. TRPC3 regulates the automaticity of embryonic stem cell-derived cardiomyocytes. Int. J. Cardiol. 2016, 203, 169–181. [Google Scholar] [CrossRef]
- Guinamard, R.; Bouvagnet, P.; Hof, T.; Liu, H.; Simard, C.; Sallé, L. TRPM4 in cardiac electrical activity. Cardiovasc. Res. 2015, 108, 21–30. [Google Scholar] [CrossRef]
- Hof, T.; Sallé, L.; Coulbault, L.; Richer, R.; Alexandre, J.; Rouet, R.; Manrique, A.; Guinamard, R. TRPM4 non-selective cation channels influence action potentials in rabbit Purkinje fibres. J. Physiol. 2016, 594, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Hof, T.; Simard, C.; Rouet, R.; Sallé, L.; Guinamard, R. Implication of the TRPM4 nonselective cation channel in mammalian sinus rhythm. Hear. Rhythm. 2013, 10, 1683–1689. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.; Mesirca, P.; Van den Boogert, M.; Rosen, J.; Mably, J.; Mangoni, M.E.; Clapham, D.E. Ion channel-kinase TRPM7 is required for maintaining cardiac automaticity. Proc. Natl. Acad. Sci. USA 2013, 110, E3037–E3046. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nakamura, K.; Nishi, N.; Igawa, O.; Yoshida, M.; Miyoshi, T.; Watanabe, A.; Morita, H.; Ito, H. TRPM4 Mutation in Patients With Ventricular Noncompaction and Cardiac Conduction Disease. Circ. Genom. Precis. Med. 2018, 11, e002103. [Google Scholar] [CrossRef]
- Bianchi, B.; Ozhathil, L.C.; Medeiros-Domingo, A.; Gollob, M.H.; Abriel, H. Four TRPM4 Cation Channel Mutations Found in Cardiac Conduction Diseases Lead to Altered Protein Stability. Front. Physiol. 2018, 9, 177. [Google Scholar] [CrossRef]
- Liu, H.; El Zein, L.; Kruse, M.; Guinamard, R.; Beckmann, A.; Bozio, A.; Kurtbay, G.; Mégarbané, A.; Ohmert, I.; Blaysat, G.; et al. Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ. Cardiovasc. Genet. 2010, 3, 374–385. [Google Scholar] [CrossRef]
- Stallmeyer, B.; Zumhagen, S.; Denjoy, I.; Duthoit, G.; Hébert, J.-L.; Ferrer, X.; Maugenre, S.; Schmitz, W.; Kirchhefer, U.; Schulze-Bahr, E.; et al. Mutational spectrum in the Ca(2+)—Activated cation channel gene TRPM4 in patients with cardiac conductance disturbances. Hum. Mutat. 2012, 33, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Palladino, A.; Papa, A.A.; Petillo, R.; Scutifero, M.; Morra, S.; Passamano, L.; Nigro, V.; Politano, L. The Role of TRPM4 Gene Mutations in Causing Familial Progressive Cardiac Conduction Disease: A Further Contribution. Genes 2022, 13, 258. [Google Scholar] [CrossRef] [PubMed]
- Demion, M.; Thireau, J.; Gueffier, M.; Finan, A.; Khoueiry, Z.; Cassan, C.; Serafini, N.; Aimond, F.; Granier, M.; Pasquié, J.-L.; et al. Trpm4 gene invalidation leads to cardiac hypertrophy and electrophysiological alterations. PLoS ONE 2014, 9, e115256. [Google Scholar] [CrossRef]
- Jin, M.; Wu, Z.; Chen, L.; Jaimes, J.; Collins, D.; Walters, E.T.; O’Neil, R.G. Determinants of TRPV4 activity following selective activation by small molecule agonist GSK1016790A. PLoS ONE 2011, 6, e16713. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Abi-Samra, F.; Gutterman, D. Cardiac contractility modulation: A novel approach for the treatment of heart failure. Hear. Fail. Rev. 2016, 21, 645–660. [Google Scholar] [CrossRef]
- Ezeani, M. TRP Channels Mediated Pathological Ca2+-Handling and Spontaneous Ectopy. Front. Cardiovasc. Med. 2019, 6, 83. [Google Scholar] [CrossRef] [PubMed]
- Freichel, M.; Berlin, M.; Schürger, A.; Mathar, I.; Bacmeister, L.; Medert, R.; Frede, W.; Marx, A.; Segin, S.; Londoño, J.E.C. TRP Channels in the Heart. In Neurobiology of TRP Channels; Emir, T.L.R., Ed.; CRC Press: Boca Raton, FL, USA, 2017; pp. 149–185. [Google Scholar]
- Li, J.; Wang, M.-H.; Wang, L.; Tian, Y.; Duan, Y.-Q.; Luo, H.-Y.; Hu, X.-W.; Hescheler, J.; Tang, M. Role of transient receptor potential vanilloid 4 in the effect of osmotic pressure on myocardial contractility in rat. Sheng Li Xue Bao Acta Physiol. Sin. 2008, 60, 181–188. [Google Scholar]
- Wu, Q.; Lu, K.; Zhao, Z.; Wang, B.; Liu, H.; Zhang, S.; Liao, J.; Zeng, Y.; Dong, Q.; Zhao, N.; et al. Blockade of Transient Receptor Potential Vanilloid 4 Enhances Antioxidation after Myocardial Ischemia/Reperfusion. Oxidative Med. Cell. Longev. 2019, 2019, 7283683. [Google Scholar] [CrossRef]
- Turner, G.H.; Bao, W.; Jucker, B.M.; Lepore, J.J.; Willette, R.N.; Thorneloe, K. Preservation of Cardiac Function and Attenuation of Remodelling in Transient Receptor Potential Vanilloid 4 Knockout Mice Following Myocardial Infarction. Clin. Exp. Cardiol. 2015, 6, 3. [Google Scholar]
- Rosenbaum, T.; Benítez-Angeles, M.; Sánchez-Hernández, R.; Morales-Lázaro, S.L.; Hiriart, M.; Morales-Buenrostro, L.E.; Torres-Quiroz, F. TRPV4: A Physio and Pathophysiologically Significant Ion Channel. Int. J. Mol. Sci. 2020, 21, 3837. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Sonkusare, S.K. Endothelial TRPV4 channels and vasodilator reactivity. Curr. Top Membr. 2020, 85, 89–117. [Google Scholar] [PubMed]
- Barbeau, S.; Joushomme, A.; Chappe, Y.; Cardouat, G.; Baudrimont, I.; Freund-Michel, V.; Guibert, C.; Marthan, R.; Berger, P.; Vacher, P.; et al. Cell Confluence Modulates TRPV4 Channel Activity in Response to Hypoxia. Biomolecules 2022, 12, 954. [Google Scholar] [CrossRef]
- Earley, S.; Heppner, T.J.; Nelson, M.T.; Brayden, J.E. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ. Res. 2005, 97, 1270–1279. [Google Scholar] [CrossRef]
- Suresh, K.; Servinsky, L.; Jiang, H.; Bigham, Z.; Yun, X.; Kliment, C.; Huetsch, J.; Damarla, M.; Shimoda, L.A. Reactive oxygen species induced Ca(2+) influx via TRPV4 and microvascular endothelial dysfunction in the SU5416/hypoxia model of pulmonary arterial hypertension. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2018, 314, L893–L907. [Google Scholar] [CrossRef]
- Swain, S.M.; Liddle, R.A. Piezo1 acts upstream of TRPV4 to induce pathological changes in endothelial cells due to shear stress. J. Biol. Chem. 2021, 296, 100171. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Wang, C. A Novel Mechanism of Sildenafil Improving the Excessive Proliferation and H2S Production in Pulmonary Arterial Smooth Muscle Cells. J. Cardiovasc. Pharmacol. 2019, 74, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Lakk, M.; Hoffmann, G.F.; Gorusupudi, A.; Enyong, E.; Lin, A.; Bernstein, P.S.; Toft-Bertelsen, T.; MacAulay, N.; Elliott, M.H.; Križaj, D. Membrane cholesterol regulates TRPV4 function, cytoskeletal expression, and the cellular response to tension. J. Lipid Res. 2021, 62, 100145. [Google Scholar] [CrossRef] [PubMed]
- Morine, K.J.; Paruchuri, V.; Qiao, X.; Aronovitz, M.; Huggins, G.S.; DeNofrio, D.; Kiernan, M.S.; Karas, R.H.; Kapur, N.K. Endoglin selectively modulates transient receptor potential channel expression in left and right heart failure. Cardiovasc. Pathol. 2016, 25, 478–482. [Google Scholar] [CrossRef]
- Zou, Y.; Zhang, M.; Wu, Q.; Zhao, N.; Chen, M.; Yang, C.; Du, Y.; Han, B. Activation of transient receptor potential vanilloid 4 is involved in pressure overload-induced cardiac hypertrophy. Elife 2022, 11, e74519. [Google Scholar] [CrossRef]
- Hobbs, F.D.R.; Bankhead, C.; Mukhtar, T.; Stevens, S.; Perera-Salazar, R.; Holt, T.; Salisbury, C. Clinical workload in UK primary care: A retrospective analysis of 100 million consultations in England, 2007-14. Lancet 2016, 387, 2323–2330. [Google Scholar] [CrossRef]
- Liao, J.; Yang, S.T.; Lu, K.; Lu, Y.; Wu, Y.W.; YM, D. Oral administration of TRPV4 inhibitor improves atrial calcium handling abnormalities in sterile pericarditis rats. Sheng Li Xue Bao Acta Physiol. Sin. 2022, 74, 188–200. [Google Scholar]
- Peana, D.; Polo-Parada, L.; Domeier, T.L. Arrhythmogenesis in the aged heart following ischaemia-reperfusion: Role of transient receptor potential vanilloid 4. Cardiovasc. Res. 2022, 118, 1126–1137. [Google Scholar] [CrossRef]
- van Nieuwenhoven, F.A.; Turner, N.A. The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vascul. Pharmacol. 2013, 58, 182–188. [Google Scholar] [CrossRef]
- Banerjee, I.; Yekkala, K.; Borg, T.K.; A Baudino, T. Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Ann. N. Y. Acad. Sci. 2006, 1080, 76–84. [Google Scholar] [CrossRef]
- Disertori, M.; Masè, M.; Ravelli, F. Myocardial fibrosis predicts ventricular tachyarrhythmias. Trends Cardiovasc. Med. 2017, 27, 363–372. [Google Scholar] [CrossRef]
- Adapala, R.K.; Katari, V.; Teegala, L.R.; Thodeti, S.; Paruchuri, S.; Thodeti, C.K. TRPV4 Mechanotransduction in Fibrosis. Cells 2021, 10, 3053. [Google Scholar] [CrossRef]
- Biernacka, A.; Frangogiannis, N.G. Aging and Cardiac Fibrosis. Aging Dis. 2011, 2, 158–173. [Google Scholar]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Adapala, R.K.; Kanugula, A.K.; Paruchuri, S.; Chilian, W.M.; Thodeti, C.K. TRPV4 deletion protects heart from myocardial infarction-induced adverse remodeling via modulation of cardiac fibroblast differentiation. Basic Res. Cardiol. 2020, 115, 14. [Google Scholar] [CrossRef]
- Cussac, L.-A.; Cardouat, G.; Pillai, N.T.; Campagnac, M.; Robillard, P.; Montillaud, A.; Guibert, C.; Gailly, P.; Marthan, R.; Quignard, J.-F.; et al. TRPV4 channel mediates adventitial fibroblast activation and adventitial remodeling in pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2020, 318, L135–L146. [Google Scholar] [CrossRef] [PubMed]
- Verheule, S.; Sato, T.; Everett IV, T.; Engle, S.K.; Otten, D.; Rubart-von der Lohe, M.; Nakajima, H.O.; Nakajima, H.; Field, L.J.; Olgin, J.E. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF-beta1. Circ. Res. 2004, 94, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Verheule, S.; Tuyls, E.; van Hunnik, A.; Kuiper, M.; Schotten, U.; Allessie, M. Fibrillatory conduction in the atrial free walls of goats in persistent and permanent atrial fibrillation. Circ. Arrhythmia Electrophysiol. 2010, 3, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.K.; Chang, P.C.; Lee, Y.S.; Lin, S.F.; Zhu, W.; Maruyama, M.; Fishbein, M.C.; Chen, Z.; Rubart-von der Lohe, M.; Field, L.J. Triggered firing and atrial fibrillation in transgenic mice with selective atrial fibrosis induced by overexpression of TGF-beta1. Circ. J. 2012, 76, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Verheule, S.; Schotten, U. Electrophysiological Consequences of Cardiac Fibrosis. Cells 2021, 10, 3220. [Google Scholar] [CrossRef]
- Nilius, B.; Owsianik, G. Channelopathies converge on TRPV4. Nat. Genet. 2010, 42, 98–100. [Google Scholar] [CrossRef]
- Nilius, B.; Owsianik, G. Transient receptor potential channelopathies. Pflug. Arch. 2010, 460, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Xu, H. TRP channels in health and disease at a glance. J. Cell Sci. 2021, 134, jcs258372. [Google Scholar] [CrossRef] [PubMed]
- Auer-Grumbach, M.; Olschewski, A.; Papić, L.; Kremer, H.; E McEntagart, M.; Uhrig, S.; Fischer, C.; Fröhlich, E.; Bálint, Z.; Tang, B.; et al. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat. Genet. 2010, 42, 160–164. [Google Scholar] [CrossRef]
- Deng, H.-X.; Klein, C.J.; Yan, J.; Shi, Y.; Wu, Y.; Fecto, F.; Yau, H.-J.; Yang, Y.; Zhai, H.; Siddique, N.; et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat. Genet. 2010, 42, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, C.; Moro, F.; Brisca, G.; Astrea, G.; Nesti, C.; Bálint, Z.; Olschewski, A.; Meschini, M.C.; Guelly, C.; Auer-Grumbach, M.; et al. TRPV4 mutations in children with congenital distal spinal muscular atrophy. Neurogenetics 2012, 13, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Lamandé, S.R.; Yuan, Y.; Gresshoff, I.L.; Rowley, L.; Belluoccio, D.; Kaluarachchi, K.; Little, C.B.; Botzenhart, E.; Zerres, K.; Amor, D.J.; et al. Mutations in TRPV4 cause an inherited arthropathy of hands and feet. Nat. Genet. 2011, 43, 1142–1146. [Google Scholar] [CrossRef]
- Landouré, G.; A Zdebik, A.; Martinez, T.L.; Burnett, B.G.; Stanescu, H.C.; Inada, H.; Shi, Y.; A Taye, A.; Kong, L.; Munns, C.H.; et al. Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C. Nat. Genet. 2010, 42, 170–174. [Google Scholar] [CrossRef]
- Toft-Bertelsen, T.L.; MacAulay, N. TRPing to the Point of Clarity: Understanding the Function of the Complex TRPV4 Ion Channel. Cells 2021, 10, 165. [Google Scholar] [CrossRef]
- McCray, B.A.; Schindler, A.; Hoover-Fong, J.E.; Sumner, C.J. Autosomal Dominant TRPV4 Disorders; in, GeneReviews((R)), Adam, M.P., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Taga, A.; Peyton, M.A.; Goretzki, B.; Gallagher, T.Q.; Ritter, A.; Harper, A.; Crawford, T.O.; Hellmich, U.A.; Sumner, C.J.; McCray, B.A. TRPV4 mutations causing mixed neuropathy and skeletal phenotypes result in severe gain of function. Ann. Clin. Transl. Neurol. 2022, 9, 375–391. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).