
Recent Advances in CAR-Based Solid Tumor Immunotherapy
 ,
,
Abstract
1. Introduction
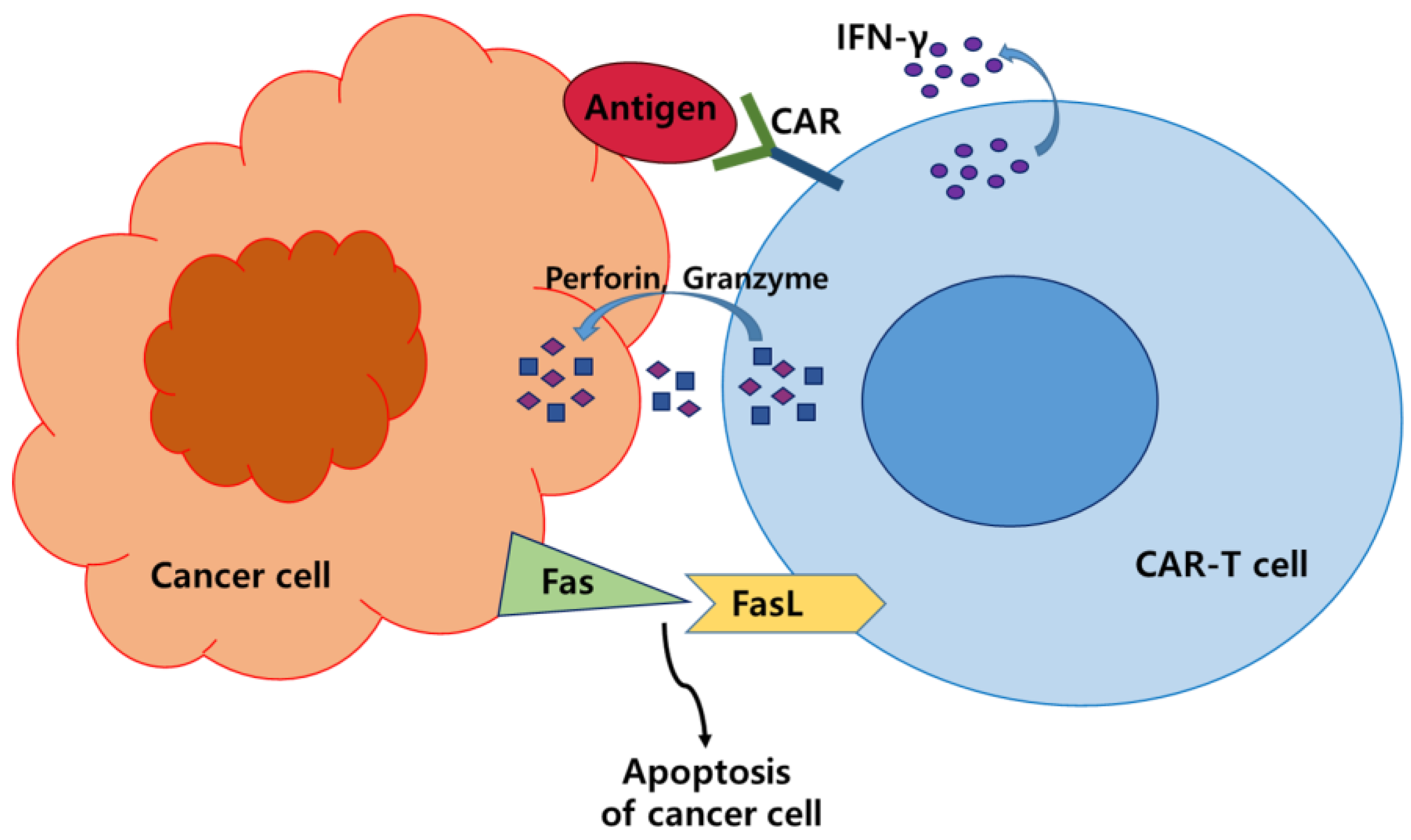
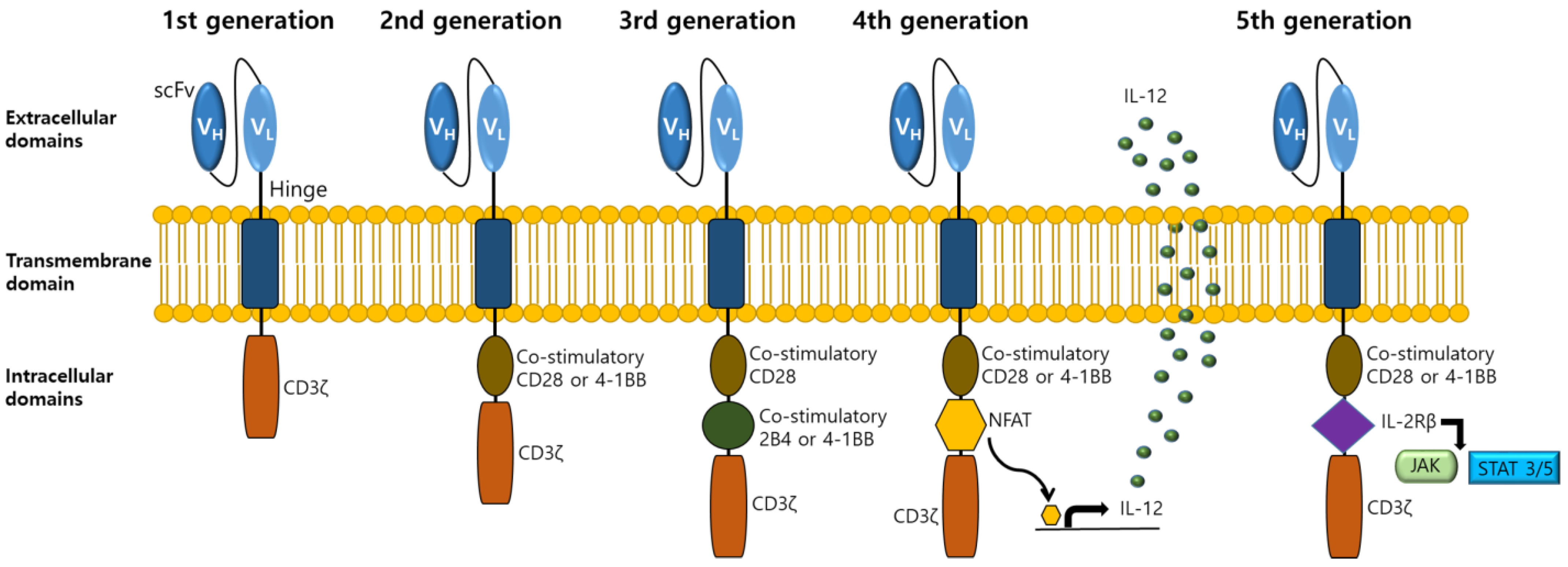
2. CAR-T Cell Therapy
2.1. FDA-Approved CAR-T Cell Therapies
2.2. Clinical Trials of CAR-T Cell Therapies in Solid Tumors
3. CAR-NK Cell Therapy
3.1. NK Cells, a Potential Alternative for CAR-Based Solid Tumor Immunotherapy
3.2. Clinical Trials of CAR-NK Cell Therapies in Solid Tumors
4. CAR-Macrophages
4.1. Macrophage, a Potential Alternative for CAR-Based Solid Tumor Immunotherapy
4.2. Clinical Trials of CAR-M Therapies in Solid Tumors
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, H.; Li, F.; Huang, S.; Chen, F.; Li, Y. Bright future or blind alley? CAR-T cell therapy for solid tumors. Front. Immunol. 2023, 14, 1045024. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, T.J.; Biederstädt, A.; Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 2022, 22, 557–575. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef] [PubMed]
- Casucci, M.; Ciceri, F. A second CD19 CAR T-cell infusion: Yes or no? Blood 2021, 137, 284–286. [Google Scholar] [CrossRef]
- Cappell, K.M.; Kochenderfer, J.N. Long-term outcomes following CAR T cell therapy: What we know so far. Nat. Rev. Clin. Oncol. 2023, 20, 359–371. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Fowler, N.H.; Dickinson, M.; Dreyling, M.; Martinez-Lopez, J.; Kolstad, A.; Butler, J.; Ghosh, M.; Popplewell, L.; Chavez, J.C.; Bachy, E. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: The phase 2 ELARA trial. Nat. Med. 2022, 28, 325–332. [Google Scholar] [CrossRef]
- Hegde, M.; Corder, A.; Chow, K.K.; Mukherjee, M.; Ashoori, A.; Kew, Y.; Zhang, Y.J.; Baskin, D.S.; Merchant, F.A.; Brawley, V.S.; et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 2013, 21, 2087–2101. [Google Scholar] [CrossRef]
- Shah, N.N.; Maatman, T.; Hari, P.; Johnson, B. Multi Targeted CAR-T Cell Therapies for B-Cell Malignancies. Front. Oncol. 2019, 9, 146. [Google Scholar] [CrossRef]
- Furqan, F.; Shah, N.N. Multispecific CAR T Cells Deprive Lymphomas of Escape via Antigen Loss. Annu. Rev. Med. 2023, 74, 279–291. [Google Scholar] [CrossRef]
- Klampatsa, A.; Leibowitz, M.S.; Sun, J.; Liousia, M.; Arguiri, E.; Albelda, S.M. Analysis and Augmentation of the Immunologic Bystander Effects of CAR T Cell Therapy in a Syngeneic Mouse Cancer Model. Mol. Ther. Oncolytics 2020, 18, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, J.; Ruella, M.; Houot, R. Born to survive: How cancer cells resist CAR T cell therapy. J. Hematol. Oncol. 2021, 14, 199. [Google Scholar] [CrossRef]
- Foeng, J.; Comerford, I.; McColl, S.R. Harnessing the chemokine system to home CAR-T cells into solid tumors. Cell Rep. Med. 2022, 3, 100543. [Google Scholar] [CrossRef]
- Trinh, T.; Adams, W.A.; Calescibetta, A.; Tu, N.; Dalton, R.; So, T.; Wei, M.; Ward, G.; Kostenko, E.; Christiansen, S.; et al. CX3CR1 deficiency-induced TIL tumor restriction as a novel addition for CAR-T design in solid malignancies. iScience 2023, 26, 106443. [Google Scholar] [CrossRef]
- Di Stasi, A.; De Angelis, B.; Rooney, C.M.; Zhang, L.; Mahendravada, A.; Foster, A.E.; Heslop, H.E.; Brenner, M.K.; Dotti, G.; Savoldo, B. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 2009, 113, 6392–6402. [Google Scholar] [CrossRef] [PubMed]
- Rapp, M.; Grassmann, S.; Chaloupka, M.; Layritz, P.; Kruger, S.; Ormanns, S.; Rataj, F.; Janssen, K.P.; Endres, S.; Anz, D.; et al. C-C chemokine receptor type-4 transduction of T cells enhances interaction with dendritic cells, tumor infiltration and therapeutic efficacy of adoptive T cell transfer. Oncoimmunology 2016, 5, e1105428. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Rui, W.; Zhao, X.; Lin, X. Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cell. Mol. Immunol. 2021, 18, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, Y.; Wang, P.; Li, S.; Dong, Y.; Zhou, M.; Shi, B.; Jiang, H.; Sun, R.; Li, Z. FAP-targeted CAR-T suppresses MDSCs recruitment to improve the antitumor efficacy of claudin18.2-targeted CAR-T against pancreatic cancer. J. Transl. Med. 2023, 21, 255. [Google Scholar] [CrossRef]
- Yoon, D.H.; Osborn, M.J.; Tolar, J.; Kim, C.J. Incorporation of Immune Checkpoint Blockade into Chimeric Antigen Receptor T Cells (CAR-Ts): Combination or Built-In CAR-T. Int. J. Mol. Sci. 2018, 19, 340. [Google Scholar] [CrossRef]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert. Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Zhang, Z.; Miao, L.; Ren, Z.; Tang, F.; Li, Y. Gene-Edited Interleukin CAR-T Cells Therapy in the Treatment of Malignancies: Present and Future. Front. Immunol. 2021, 12, 718686. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, Z.; Dang, Q.; Xu, H.; Lv, J.; Li, H.; Han, X. Immunosuppression in tumor immune microenvironment and its optimization from CAR-T cell therapy. Theranostics 2022, 12, 6273–6290. [Google Scholar] [CrossRef]
- Grosser, R.; Cherkassky, L.; Chintala, N.; Adusumilli, P.S. Combination Immunotherapy with CAR T Cells and Checkpoint Blockade for the Treatment of Solid Tumors. Cancer Cell 2019, 36, 471–482. [Google Scholar] [CrossRef]
- Xue, L.; Yi, Y.; Xu, Q.; Wang, L.; Yang, X.; Zhang, Y.; Hua, X.; Chai, X.; Yang, J.; Chen, Y.; et al. Chimeric antigen receptor T cells self-neutralizing IL6 storm in patients with hematologic malignancy. Cell Discov. 2021, 7, 84. [Google Scholar] [CrossRef]
- Amatya, C.; Pegues, M.A.; Lam, N.; Vanasse, D.; Geldres, C.; Choi, S.; Hewitt, S.M.; Feldman, S.A.; Kochenderfer, J.N. Development of CAR T Cells Expressing a Suicide Gene Plus a Chimeric Antigen Receptor Targeting Signaling Lymphocytic-Activation Molecule F7. Mol. Ther. 2021, 29, 702–717. [Google Scholar] [CrossRef]
- Sidaway, P. Allogeneic CAR T cells show promise. Nat. Rev. Clin. Oncol. 2022, 19, 748. [Google Scholar] [CrossRef]
- Yan, T.; Zhu, L.; Chen, J. Current advances and challenges in CAR T-Cell therapy for solid tumors: Tumor-associated antigens and the tumor microenvironment. Exp. Hematol. Oncol. 2023, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell 2017, 9, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E.; et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Katz, S.C.; Hardaway, J.; Prince, E.; Guha, P.; Cunetta, M.; Moody, A.; Wang, L.J.; Armenio, V.; Espat, N.J.; Junghans, R.P. HITM-SIR: Phase Ib trial of intraarterial chimeric antigen receptor T-cell therapy and selective internal radiation therapy for CEA+ liver metastases. Cancer Gene Ther. 2020, 27, 341–355. [Google Scholar] [CrossRef]
- Pietrobon, V.; Todd, L.A.; Goswami, A.; Stefanson, O.; Yang, Z.; Marincola, F. Improving CAR T-Cell Persistence. Int. J. Mol. Sci. 2021, 22, 10828. [Google Scholar] [CrossRef]
- Andrea, A.E.; Chiron, A.; Mallah, S.; Bessoles, S.; Sarrabayrouse, G.; Hacein-Bey-Abina, S. Advances in CAR-T Cell Genetic Engineering Strategies to Overcome Hurdles in Solid Tumors Treatment. Front. Immunol. 2022, 13, 309. [Google Scholar] [CrossRef]
- Xiao, B.-F.; Zhang, J.-T.; Zhu, Y.-G.; Cui, X.-R.; Lu, Z.-M.; Yu, B.-T.; Wu, N. Chimeric Antigen Receptor T-Cell Therapy in Lung Cancer: Potential and Challenges. Front. Immunol. 2021, 12, 4556. [Google Scholar] [CrossRef] [PubMed]
- Yeo, D.; Giardina, C.; Saxena, P.; Rasko, J.E.J. The next wave of cellular immunotherapies in pancreatic cancer. Mol. Ther.-Oncolytics 2022, 24, 561–576. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 1 June 2023).
- Włodarczyk, M.; Pyrzynska, B. CAR-NK as a Rapidly Developed and Efficient Immunotherapeutic Strategy against Cancer. Cancers 2022, 15, 117. [Google Scholar] [CrossRef]
- Pan, K.; Farrukh, H.; Chittepu, V.C.S.R.; Xu, H.; Pan, C.-X.; Zhu, Z. CAR race to cancer immunotherapy: From CAR T, CAR NK to CAR macrophage therapy. J. Exp. Clin. Cancer Res. 2022, 41, 119. [Google Scholar] [CrossRef]
- Shin, M.H.; Kim, J.; Lim, S.A.; Kim, J.; Kim, S.J.; Lee, K.M. NK Cell-Based Immunotherapies in Cancer. Immune Netw. 2020, 20, e14. [Google Scholar] [CrossRef]
- Li, H.; Song, W.; Li, Z.; Zhang, M. Preclinical and clinical studies of CAR-NK-cell therapies for malignancies. Front. Immunol. 2022, 13, 992232. [Google Scholar] [CrossRef] [PubMed]
- Abdin, S.M.; Paasch, D.; Morgan, M.; Lachmann, N. CARs and beyond: Tailoring macrophage-based cell therapeutics to combat solid malignancies. J. Immunother. Cancer 2021, 9, e002741. [Google Scholar] [CrossRef] [PubMed]
- Maalej, K.M.; Merhi, M.; Inchakalody, V.P.; Mestiri, S.; Alam, M.; Maccalli, C.; Cherif, H.; Uddin, S.; Steinhoff, M.; Marincola, F.M.; et al. CAR-cell therapy in the era of solid tumor treatment: Current challenges and emerging therapeutic advances. Mol. Cancer 2023, 22, 20. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, M.T. Macrophages get a CAR. Nat. Rev. Cancer 2020, 20, 300. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Tan, Y.; Wang, M.; Zhang, Y.; Ge, S.; Zhong, F.; Xia, G.; Sun, C. Tumor-Associated Macrophages: A Potential Target for Cancer Therapy. Front. Oncol. 2021, 11, 693517. [Google Scholar] [CrossRef] [PubMed]
- FDA Approval Brings First Gene Therapy to the United States. Available online: https://www.fda.gov/news-events/press-announcements/fda-approval-brings-first-gene-therapy-united-states (accessed on 1 June 2023).
- Boccalatte, F.; Mina, R.; Aroldi, A.; Leone, S.; Suryadevara, C.M.; Placantonakis, D.G.; Bruno, B. Advances and Hurdles in CAR T Cell Immune Therapy for Solid Tumors. Cancers 2022, 14, 5108. [Google Scholar] [CrossRef]
- Roex, G.; Timmers, M.; Wouters, K.; Campillo-Davo, D.; Flumens, D.; Schroyens, W.; Chu, Y.; Berneman, Z.N.; Lion, E.; Luo, F.; et al. Safety and clinical efficacy of BCMA CAR-T-cell therapy in multiple myeloma. J. Hematol. Oncol. 2020, 13, 164. [Google Scholar] [CrossRef]
- Xie, Y.J.; Dougan, M.; Jailkhani, N.; Ingram, J.; Fang, T.; Kummer, L.; Momin, N.; Pishesha, N.; Rickelt, S.; Hynes, R.O.; et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc. Natl. Acad. Sci. USA 2019, 116, 7624–7631. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Ma, J.; Zheng, B.; Goswami, S.; Meng, L.; Zhang, D.; Cao, C.; Li, T.; Zhu, F.; Ma, L.; Zhang, Z.; et al. PD1Hi CD8+ T cells correlate with exhausted signature and poor clinical outcome in hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 331. [Google Scholar] [CrossRef]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Li, A.M.; Hucks, G.E.; Dinofia, A.M.; Seif, A.E.; Teachey, D.T.; Baniewicz, D.; Callahan, C.; Fasano, C.; McBride, B.; Gonzalez, V.; et al. Checkpoint Inhibitors Augment CD19-Directed Chimeric Antigen Receptor (CAR) T Cell Therapy in Relapsed B-Cell Acute Lymphoblastic Leukemia. Blood 2018, 132, 556. [Google Scholar] [CrossRef]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.-H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK–STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Abila, B.; Mostafa Kamel, Y. CAR-T: What Is Next? Cancers 2023, 15, 663. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Brudno, J.N. CAR T-Cell Therapy in Hematologic Malignancies: Clinical Role, Toxicity, and Unanswered Questions. Am. Soc. Clin. Oncol. Educ. Book. 2021, 41, e246–e265. [Google Scholar] [CrossRef]
- Young, R.M.; Engel, N.W.; Uslu, U.; Wellhausen, N.; June, C.H. Next-Generation CAR T-cell Therapies. Cancer Discov. 2022, 12, 1625–1633. [Google Scholar] [CrossRef]
- Kwon, M.; Iacoboni, G.; Reguera, J.L.; Corral, L.L.; Morales, R.H.; Ortiz-Maldonado, V.; Guerreiro, M.; Caballero, A.C.; Domínguez, M.L.G.; Pina, J.M.S.; et al. Axicabtagene ciloleucel compared to tisagenlecleucel for the treatment of aggressive B-cell lymphoma. Haematologica 2023, 108, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.W.; Maude, S.L.; Rives, S.; Hiramatsu, H.; Bittencourt, H.; Bader, P.; Baruchel, A.; Boyer, M.; De Moerloose, B.; Qayed, M.; et al. Three-Year Update of Tisagenlecleucel in Pediatric and Young Adult Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia in the ELIANA Trial. J. Clin. Oncol. 2023, 41, 1664–1669. [Google Scholar] [CrossRef]
- Schultz, L.M.; Eaton, A.; Baggott, C.; Rossoff, J.; Prabhu, S.; Keating, A.K.; Krupski, C.; Pacenta, H.; Philips, C.L.; Talano, J.A.; et al. Outcomes After Nonresponse and Relapse Post-Tisagenlecleucel in Children, Adolescents, and Young Adults With B-Cell Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2023, 41, 354–363. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Jacobson, C.A.; Ghobadi, A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Five-year follow-up of ZUMA-1 supports the curative potential of axicabtagene ciloleucel in refractory large B-cell lymphoma. Blood 2023, 141, 2307–2315. [Google Scholar] [CrossRef]
- Locke, F.L.; Miklos, D.B.; Jacobson, C.A.; Perales, M.-A.; Kersten, M.-J.; Oluwole, O.O.; Ghobadi, A.; Rapoport, A.P.; McGuirk, J.; Pagel, J.M.; et al. Axicabtagene Ciloleucel as Second-Line Therapy for Large B-Cell Lymphoma. N. Engl. J. Med. 2021, 386, 640–654. [Google Scholar] [CrossRef]
- Seyfrid, M.; Maich, W.T.; Shaikh, M.V.; Tatari, N.; Upreti, D.; Piyasena, D.; Subapanditha, M.; Savage, N.; McKenna, D.; Mikolajewicz, N.; et al. CD70 as an actionable immunotherapeutic target in recurrent glioblastoma and its microenvironment. J. Immunother. Cancer 2022, 10, e003289. [Google Scholar] [CrossRef]
- Hickman, T.L.; Choi, E.; Whiteman, K.R.; Muralidharan, S.; Pai, T.; Johnson, T.; Parikh, A.; Friedman, T.; Gilbert, M.; Shen, B.; et al. BOXR1030, an anti-GPC3 CAR with exogenous GOT2 expression, shows enhanced T cell metabolism and improved anti-cell line derived tumor xenograft activity. PLoS ONE 2022, 17, e0266980. [Google Scholar] [CrossRef]
- Nikiforow, S.; Werner, L.; Murad, J.; Jacobs, M.; Johnston, L.; Patches, S.; White, R.; Daley, H.; Negre, H.; Reder, J.; et al. Safety Data from a First-in-Human Phase 1 Trial of NKG2D Chimeric Antigen Receptor-T Cells in AML/MDS and Multiple Myeloma. Blood 2016, 128, 4052. [Google Scholar] [CrossRef]
- Kozlowska, A.; Zhang, Y.; Fritz, J.; Wang, S.; Codde, R.; Argus, E.; Ibitokou, S.; Richardson, V.; Jain, S.; Richter, M.; et al. 120 P-MUC1C-ALLO1: An allogeneic car-t for multiple solid tumor indications. J. Immunother. Cancer 2020, 8, A73. [Google Scholar] [CrossRef]
- Innovent Announces First Patient Dosing of Universal “Modular” CAR-T Cell Product IBI345. Available online: https://www.prnewswire.com/news-releases/innovent-announces-first-patient-dosing-of-universal-modular-car-t-cell-product-ibi345-301486172.html. (accessed on 1 June 2023).
- Batra, S.A.; Rathi, P.; Guo, L.; Courtney, A.N.; Fleurence, J.; Balzeau, J.; Shaik, R.S.; Nguyen, T.P.; Wu, M.F.; Bulsara, S.; et al. Glypican-3-Specific CAR T Cells Coexpressing IL15 and IL21 Have Superior Expansion and Antitumor Activity against Hepatocellular Carcinoma. Cancer Immunol. Res. 2020, 8, 309–320. [Google Scholar] [CrossRef]
- Qi, C.; Gong, J.; Li, J.; Liu, D.; Qin, Y.; Ge, S.; Zhang, M.; Peng, Z.; Zhou, J.; Cao, Y.; et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: Phase 1 trial interim results. Nat. Med. 2022, 28, 1189–1198. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, J.; Yang, X.; Liu, Y.; Zou, C.; Lv, W.; Chen, C.; Cheng, K.K.-y.; Chen, T.; Chang, L.-J.; et al. Safety and antitumor activity of GD2-Specific 4SCAR-T cells in patients with glioblastoma. Mol. Cancer 2023, 22, 3. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Z.; Yang, Z.; Wang, M.; Li, S.; Li, Y.; Zhang, R.; Xiong, Z.; Wei, Z.; Shen, J.; et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA(+) Metastatic Colorectal Cancers. Mol. Ther. 2017, 25, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Del Bufalo, F.; De Angelis, B.; Caruana, I.; Del Baldo, G.; De Ioris, M.A.; Serra, A.; Mastronuzzi, A.; Cefalo, M.G.; Pagliara, D.; Amicucci, M.; et al. GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N. Engl. J. Med. 2023, 388, 1284–1295. [Google Scholar] [CrossRef] [PubMed]
- Prapa, M.; Chiavelli, C.; Golinelli, G.; Grisendi, G.; Bestagno, M.; Di Tinco, R.; Dall’Ora, M.; Neri, G.; Candini, O.; Spano, C.; et al. GD2 CAR T cells against human glioblastoma. NPJ Precis. Oncol. 2021, 5, 93. [Google Scholar] [CrossRef] [PubMed]
- Tumino, N.; Weber, G.; Besi, F.; Del Bufalo, F.; Bertaina, V.; Paci, P.; Quatrini, L.; Antonucci, L.; Sinibaldi, M.; Quintarelli, C.; et al. Polymorphonuclear myeloid-derived suppressor cells impair the anti-tumor efficacy of GD2.CAR T-cells in patients with neuroblastoma. J. Hematol. Oncol. 2021, 14, 191. [Google Scholar] [CrossRef] [PubMed]
- Pang, N.; Shi, J.; Qin, L.; Chen, A.; Tang, Y.; Yang, H.; Huang, Y.; Wu, Q.; Li, X.; He, B.; et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J. Hematol. Oncol. 2021, 14, 118. [Google Scholar] [CrossRef]
- Sakamoto, J.; Furukawa, K.; Cordon-Cardo, C.; Yin, B.W.; Rettig, W.J.; Oettgen, H.F.; Old, L.J.; Lloyd, K.O. Expression of Lewisa, Lewisb, X, and Y blood group antigens in human colonic tumors and normal tissue and in human tumor-derived cell lines. Cancer Res. 1986, 46, 1553–1561. [Google Scholar] [PubMed]
- Meyran, D.; Zhu, J.; Buttler, J.; Tantalo, D.; Neeson, M.; Ekert, P.; Kershaw, M.; Trapani, J.; Darcy, P.; Neeson, P. Development of next generation car’s targeting the lewis y antigen for the treatment of cancer. Cytotherapy 2019, 21, e15. [Google Scholar] [CrossRef]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.; Gileadi, T.; et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Sci. Transl. Med. 2020, 12, eabd6169. [Google Scholar] [CrossRef]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug. Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Glasner, A.; Ghadially, H.; Gur, C.; Stanietsky, N.; Tsukerman, P.; Enk, J.; Mandelboim, O. Recognition and prevention of tumor metastasis by the NK receptor NKp46/NCR1. J. Immunol. 2012, 188, 2509–2515. [Google Scholar] [CrossRef]
- Cantoni, C.; Bottino, C.; Vitale, M.; Pessino, A.; Augugliaro, R.; Malaspina, A.; Parolini, S.; Moretta, L.; Moretta, A.; Biassoni, R. NKp44, a triggering receptor involved in tumor cell lysis by activated human natural killer cells, is a novel member of the immunoglobulin superfamily. J. Exp. Med. 1999, 189, 787–796. [Google Scholar] [CrossRef]
- Wu, X.; Matosevic, S. Gene-edited and CAR-NK cells: Opportunities and challenges with engineering of NK cells for immunotherapy. Mol. Ther. Oncolytics 2022, 27, 224–238. [Google Scholar] [CrossRef]
- Ramírez-Labrada, A.; Pesini, C.; Santiago, L.; Hidalgo, S.; Calvo-Pérez, A.; Oñate, C.; Andrés-Tovar, A.; Garzón-Tituaña, M.; Uranga-Murillo, I.; Arias, M.A.; et al. All About (NK Cell-Mediated) Death in Two Acts and an Unexpected Encore: Initiation, Execution and Activation of Adaptive Immunity. Front. Immunol. 2022, 13, 896228. [Google Scholar] [CrossRef] [PubMed]
- Zamai, L.; Ahmad, M.; Bennett, I.M.; Azzoni, L.; Alnemri, E.S.; Perussia, B. Natural killer (NK) cell-mediated cytotoxicity: Differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J. Exp. Med. 1998, 188, 2375–2380. [Google Scholar] [CrossRef]
- Smyth, M.J.; Cretney, E.; Takeda, K.; Wiltrout, R.H.; Sedger, L.M.; Kayagaki, N.; Yagita, H.; Okumura, K. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J. Exp. Med. 2001, 193, 661–670. [Google Scholar] [CrossRef]
- Montel, A.H.; Bochan, M.R.; Hobbs, J.A.; Lynch, D.H.; Brahmi, Z. Fas involvement in cytotoxicity mediated by human NK cells. Cell. Immunol. 1995, 166, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.; Mishra, D.P. Trailing TRAIL Resistance: Novel Targets for TRAIL Sensitization in Cancer Cells. Front. Oncol. 2015, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Zhao, X.; Li, Z.; Hu, Y.; Wang, H. From CAR-T Cells to CAR-NK Cells: A Developing Immunotherapy Method for Hematological Malignancies. Front. Oncol. 2021, 11, 720501. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Kaufman, D.S. An Improved Method to Produce Clinical-Scale Natural Killer Cells from Human Pluripotent Stem Cells; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural killer cells for immunotherapy–advantages of the NK-92 cell line over blood NK cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef]
- Oberoi, P.; Kamenjarin, K.; Villena Ossa, J.F.; Uherek, B.; Bönig, H.; Wels, W.S. Directed differentiation of mobilized hematopoietic stem and progenitor cells into functional NK cells with enhanced antitumor activity. Cells 2020, 9, 811. [Google Scholar] [CrossRef]
- Condiotti, R.; Zakai, Y.B.; Barak, V.; Nagler, A. Ex vivo expansion of CD56+ cytotoxic cells from human umbilical cord blood. Exp. Hematol. 2001, 29, 104–113. [Google Scholar] [CrossRef]
- Valeri, A.; García-Ortiz, A.; Castellano, E.; Córdoba, L.; Maroto-Martín, E.; Encinas, J.; Leivas, A.; Río, P.; Martínez-López, J. Overcoming tumor resistance mechanisms in CAR-NK cell therapy. Front. Immunol. 2022, 13, 4208. [Google Scholar] [CrossRef]
- Ingegnere, T.; Mariotti, F.R.; Pelosi, A.; Quintarelli, C.; De Angelis, B.; Tumino, N.; Besi, F.; Cantoni, C.; Locatelli, F.; Vacca, P. Human CAR NK cells: A new non-viral method allowing high efficient transfection and strong tumor cell killing. Front. Immunol. 2019, 10, 957. [Google Scholar] [CrossRef]
- Gong, Y.; Klein Wolterink, R.G.J.; Wang, J.; Bos, G.M.J.; Germeraad, W.T.V. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J. Hematol. Oncol. 2021, 14, 73. [Google Scholar] [CrossRef]
- Gurney, M.; O’Reilly, E.; Corcoran, S.; Brophy, S.; Hardwicke, D.; Krawczyk, J.; Hermanson, D.; Childs, R.W.; Szegezdi, E.; O’Dwyer, M.E. Tc Buster Transposon Engineered CLL-1 CAR-NK Cells Efficiently Target Acute Myeloid Leukemia. Blood 2021, 138, 1725. [Google Scholar] [CrossRef]
- Müller, S.; Bexte, T.; Gebel, V.; Kalensee, F.; Stolzenberg, E.; Hartmann, J.; Koehl, U.; Schambach, A.; Wels, W.S.; Modlich, U.; et al. High Cytotoxic Efficiency of Lentivirally and Alpharetrovirally Engineered CD19-Specific Chimeric Antigen Receptor Natural Killer Cells Against Acute Lymphoblastic Leukemia. Front. Immunol. 2019, 10, 3123. [Google Scholar] [CrossRef] [PubMed]
- Ojo, E.O.; Sharma, A.A.; Liu, R.; Moreton, S.; Checkley-Luttge, M.-A.; Gupta, K.; Lee, G.; Lee, D.A.; Otegbeye, F.; Sekaly, R.-P. Membrane bound IL-21 based NK cell feeder cells drive robust expansion and metabolic activation of NK cells. Sci. Rep. 2019, 9, 14916. [Google Scholar] [CrossRef] [PubMed]
- Fujisaki, H.; Kakuda, H.; Shimasaki, N.; Imai, C.; Ma, J.; Lockey, T.; Eldridge, P.; Leung, W.H.; Campana, D. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009, 69, 4010–4017. [Google Scholar] [CrossRef]
- Wang, X.; Yang, X.; Yuan, X.; Wang, W.; Wang, Y. Chimeric antigen receptor-engineered NK cells: New weapons of cancer immunotherapy with great potential. Exp. Hematol. Oncol. 2022, 11, 85. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cheng, L.; Liu, L.; Li, X. NK cells are never alone: Crosstalk and communication in tumour microenvironments. Mol. Cancer 2023, 22, 34. [Google Scholar] [CrossRef]
- He, B.; Mai, Q.; Pang, Y.; Deng, S.; He, Y.; Xue, R.; Xu, N.; Zhou, H.; Liu, X.; Xuan, L.; et al. Cytokines induced memory-like NK cells engineered to express CD19 CAR exhibit enhanced responses against B cell malignancies. Front. Immunol. 2023, 14, 1130442. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Quintarelli, C.; Sivori, S.; Caruso, S.; Carlomagno, S.; Falco, M.; Boffa, I.; Orlando, D.; Guercio, M.; Abbaszadeh, Z.; Sinibaldi, M.; et al. Efficacy of third-party chimeric antigen receptor modified peripheral blood natural killer cells for adoptive cell therapy of B-cell precursor acute lymphoblastic leukemia. Leukemia 2020, 34, 1102–1115. [Google Scholar] [CrossRef]
- Owens, G.L.; Sheard, V.E.; Kalaitsidou, M.; Blount, D.; Lad, Y.; Cheadle, E.J.; Edmondson, R.J.; Kooner, G.; Gilham, D.E.; Harrop, R. Preclinical Assessment of CAR T-Cell Therapy Targeting the Tumor Antigen 5T4 in Ovarian Cancer. J. Immunother. 2018, 41, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zheng, H.; Luo, W.; Zhang, Q.; Liu, J.; Yao, K. 5T4-specific chimeric antigen receptor modification promotes the immune efficacy of cytokine-induced killer cells against nasopharyngeal carcinoma stem cell-like cells. Sci. Rep. 2017, 7, 4859. [Google Scholar] [CrossRef]
- Xia, N.; Haopeng, P.; Gong, J.U.; Lu, J.; Chen, Z.; Zheng, Y.; Wang, Z.; Sun, Y.U.; Yang, Z.; Hoffman, R.M.; et al. Robo1-specific CAR-NK Immunotherapy Enhances Efficacy of (125)I Seed Brachytherapy in an Orthotopic Mouse Model of Human Pancreatic Carcinoma. Anticancer Res. 2019, 39, 5919–5925. [Google Scholar] [CrossRef]
- Xiao, L.; Cen, D.; Gan, H.; Sun, Y.; Huang, N.; Xiong, H.; Jin, Q.; Su, L.; Liu, X.; Wang, K.; et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol. Ther. 2019, 27, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef]
- Zou, Z.; Lin, H.; Li, M.; Lin, B. Tumor−associated macrophage polarization in the inflammatory tumor microenvironment. Front. Oncol. 2023, 13, 228. [Google Scholar] [CrossRef]
- Kadomoto, S.; Izumi, K.; Mizokami, A. Macrophage Polarity and Disease Control. Int. J. Mol. Sci. 2021, 23, 144. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.-H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef] [PubMed]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Hasan, M.N.; Capuk, O.; Patel, S.M.; Sun, D. The Role of Metabolic Plasticity of Tumor-Associated Macrophages in Shaping the Tumor Microenvironment Immunity. Cancers 2022, 14, 3331. [Google Scholar] [CrossRef] [PubMed]
- Carisma Drives CAR-M Engineered Macrophage Cancer Therapy Forward. Available online: https://www.nature.com/articles/d43747-020-01096-y (accessed on 1 June 2023).
- Sloas, C.; Gill, S.; Klichinsky, M. Engineered CAR-Macrophages as Adoptive Immunotherapies for Solid Tumors. Front. Immunol. 2021, 12, 783305. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Lei, A.; Wang, X.; Lu, H.; Wang, S.; Yang, Y.; Li, N.; Zhang, Y.; Zhang, J. Induced CAR-Macrophages as a Novel Therapeutic Cell Type for Cancer Immune Cell Therapies. Cells 2022, 11, 1652. [Google Scholar] [CrossRef]
- Bobadilla, S.; Sunseri, N.; Landau, N.R. Efficient transduction of myeloid cells by an HIV-1-derived lentiviral vector that packages the Vpx accessory protein. Gene Ther. 2013, 20, 514–520. [Google Scholar] [CrossRef]
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhu, M.; Xu, J.; Zhao, W.; Zhu, Y. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J. Hematol. Oncol. 2020, 13, 153. [Google Scholar] [CrossRef]
- Daigneault, M.; Preston, J.A.; Marriott, H.M.; Whyte, M.K.; Dockrell, D.H. The identification of markers of macrophage differentiation in PMA-stimulated THP-1 cells and monocyte-derived macrophages. PLoS ONE 2010, 5, e8668. [Google Scholar] [CrossRef]
- Fuss, I.J.; Kanof, M.E.; Smith, P.D.; Zola, H. Isolation of whole mononuclear cells from peripheral blood and cord blood. Curr. Protoc. Immunol. 2009, 85, 7.1.1–7.1.8. [Google Scholar] [CrossRef]
- Carisma Therapeutics Announces U.S. Food and Drug Administration Grants Fast Track Designation to CT-0508 for the Treatment of Patients with Solid Tumors. Available online: https://carismatx.com/carisma-therapeutics-announces-u-s-food-and-drug-administration-grants-fast-track-designation-to-ct-0508-for-the-treatment-of-patients-with-solid-tumors/ (accessed on 1 June 2023).



{kind=link}
{kind=link}
| Advantages |
|
| Limitations -Strategies |
|
| Brand Name | Generic Name | Target Antigen | Indications | Manufacturer | Date of FDA Approval |
|---|---|---|---|---|---|
| Kymriah | Tisagenlecleucel | CD19 |
| Novartis Pharmaceuticals Corporation (Basel, Switzerland) | 30 August 2017 |
| Yescarta | Axicabtagene ciloleucel | CD19 |
| Kite Pharma, Inc. (Los Angeles, CA, USA) | 18 October 2017 |
| Tecartus | Brexucabtagene autoleucel | CD19 |
| Kite Pharma, Inc. | 24 July 2020 |
| Breyanzi | Lisocabtagene maraleucel | CD19 |
| Juno Therapeutics, Inc., (Seattle, WA, USA) Bristol-Myers Squibb Company (New York, NY, USA) | 5 February 2021 |
| Abecma | Idecabtagene vicleucel | BCMA |
| Celgene Corporation, (Summit, NJ, USA) Bristol-Myers Squibb Company | 26 March 2021 |
| Carvykti | Ciltacabtagene autoleucel | BCMA |
| Janssen Biotech, Inc. (Horsham, PA, USA) | 28 February 2022 |
| National Clinical Trial (NCT) Number | Title | Status | Conditions | CAR-T Product | Targeted Antigen | Modification to Overcome the Limitations | Phase | Start Date |
|---|---|---|---|---|---|---|---|---|
| NCT 05693844 | CD40 ligand-expressing MSLN CAR-T cell therapy in MSLN-positive advanced/metastatic solid tumors | Recruiting | Advanced or metastatic solid tumors | CD40 ligand expressing MSLN-CAR-T cells | MSLN | Targeting both CD40 and MSLN | Phase 1 Phase 2 | 20 January 2023 |
| NCT 05621486 | A clinical study to evaluate B4T2-001 CAR-T cells in the treatment of advanced solid tumors | Recruiting | Advanced solid Tumor | B4T2-001 CAR-T cells | BT-001 | Targeting novel-, self-antigen | Phase 1 | 14 September 2022 |
| NCT 05518253 | A clinical study of CD70-targeted CAR-T in the treatment of CD70-positive advanced/metastatic solid tumors | Recruiting | Metastatic tumor, Advanced solid tumor, Renal cell carcinoma, Ovarian cancer, Cervix cancer | CD70 CAR-T cells | CD70 | Administratio method: Intravenous infusion versus Intraperitoneal injection | Phase 1 | 30 May 2022 |
| NCT 05120271 | BOXR1030 T cells in subjects with advanced GPC3-positive solid tumors | Recruiting | Hepatocellular carcinoma, Squamous cell carcinoma of the lung, Merkel cell carcinoma, Myxoid/round cell liposarcoma | GPC3-CAR-T cells | GPC3 | Expressing exogenous GOT2 (Glutamic-oxaloacetic transaminase 2) for optimal T cell activity | Phase 1 Phase 2 | 18 May 2022 |
| NCT 05382377 | NKG2D CAR-T(KD-025) in the treatment of advanced NKG2DL+ solid tumors | Recruiting | CRC, Solid tumor | NKG2D CAR-T cells (KD-025) | NKG2DL | Potent anti-tumor activity with upregula- ting TNFa, IFN-γ, IL-10 and IL-2 cytokines | Early Phase 1 | 17 May 2022 |
| NCT 05239143 | P-MUC1C-ALLO1 allogeneic CAR-T cells in the treatment of subjects with advanced or metastatic solid tumors | Recruiting | Breast cancer, Ovarian cancer, Non-small cell lung cancer, Colorectal cancer, Pancreatic cancer, Renal cell carcinoma, Nasopharyngeal cancer, Head and neck squamous cell carcinoma, Gastric cancer | P-MUC1C-ALLO1 CAR-T cells | MUC1-C | Allogeneic CAR-T cells | Phase 1 | 15 February 2022 |
| NCT 05199519 | Study to Evaluate the Safety, Tolerance, Pharmacokinetics and Preliminary Efficacy of IBI345 | Completed | CLDN18.2 Positive Solid Tumors | IBI345 (IBI345 CAR-T cell) | CLDN18.2 | Allogeneic CAR-T cells | Phase 1 | 13 December 2021 |
| NCT 05103631 | Interleukin-15 armored Glypican 3-specific chimeric antigen receptor expressed in autologous T cells for hepatocellular carcinoma | Recruiting | Liver cell carcinoma, Solid tumor, Wilms tumor, Malignant rhabdoid tumor, Yolk sac tumor, Rhabdomyosarcoma, Liposarcoma, Embryonal sarcoma of the liver | Interleukin-15 armored Glypican 3-specific CAR-T cells | GPC3 | Engineered to express IL-15 to enhance anti-tumor activity | Phase 1 | 17 June 2021 |
| NCT 04581473 | A study to evaluate the efficacy, safety, and pharmacokinetics of CT041 autologous CAR-T cell injection | Recruiting | Gastric adenocarcinoma Pancreatic cancer, Gastroesophageal junction adenocarcinoma | CT041 CAR-T cells | CLDN18.2 | CLDN18.2, a digestive system cancer-specific biomarker | Phase 1 Phase 2 | 23 October 2020 |
| NCT 04511871 | A phase I trial of CCT303-406 in patients with relapsed or refractory HER2-positive solid tumors | Recruiting | Solid tumor, Gastric cancer, Breast cancer, Ovarian cancer, Sarcoma | HER2 CAR-T cells | HER2 | Requiring both target antigen and TME to activate CAR-T cells | Phase 1 | 9 July 2020 |
| NCT 03170141 | Immunogene-modified T (IgT) Cells Against Glioblastoma Multiforme | Enrolling by invitation | Glioblastoma Multiforme of Brain, Glioblastoma Multiforme | AutologousGD2-specific fourth-generation safety-designed chimeric antigen receptor (4SCAR)-T cells | GD2 | Contains a suicide gene safety switch (namely inducible Caspase 9) | Phase 1 | 31 May 2020 |
| NCT 04348643 | Safety and efficacy of CEA-targeted CAR-T therapy for relapsed/refractory CEA+ cancer | Recruiting | Solid tumor, Lung cancer, Colorectal cancer, Liver cancer, Pancreatic cancer, Gastric cancer, Breast cancer | CEA CAR-T cells | CEA | CEA, a digestive tract cancer-specific biomarker | Phase 1 Phase 2 | 20 February 2020 |
| NCT 05812326 | PD-1 Knockout Anti-MUC1 CAR-T Cells in the Treatment of Advanced Breast Cancer | Completed | Advanced Breast Cancer, Breast Neoplasm Malignant Female | AJMUC1- PD-1 gene knockout anti- MUC1 CAR-T cells | MUC1 | PD-1 knockout | Phase 1 Phase 2 | 17 May 2019 |
| NCT 03373097 | Anti-GD2 CAR-T cells in pediatric patients affected by high-risk and/or relapsed/refractory neuroblastoma or other GD2-positive solid tumors | Recruiting | Neuroblastoma, Recurrent neuroblastoma, GD2-positive solid tumors, Osteosarcoma, Ewing sarcoma, Sarcoma | GD2 CAR-T cells | GD2 | Contains a suicide gene safety switch (namely inducible Caspase 9) | Phase 1 Phase 2 | 5 January 2018 |
| NCT 03198546 | GPC3-CAR-T Cells for Immunotherapy of Cancer With GPC3 Expression | Recruiting | Hepatocellular Carcinoma Immunotherapy, CAR GPC3 Gene Inactivation, T Cell, Squamous Cell, Lung Cancer | GPC3 targeting CAR-T cells | GPC3 | Engineered to secrete human IL-7 and CCL19 for enhanced anti-tumor activity | Phase 1 | 1 July 2017 |
| NCT 03851146 | A Study of Anti-Lewis Y Chimeric Antigen Receptor-T Cells (LeY-CAR-T) in Patients With Solid Tumours (LeY-CAR-T) | Completed | Advanced cancer | LeY CAR T cells | LeY (Lewis Y) | High levels of IFN-γ, TNFα and IL-2 secretion following direct stimulation through LeY+ tumor targets. | Phase 1 | 24 November 2016 |
| NCT 02761915 | A Phase I Trial of Anti-GD2 T-cells (1RG-CART) | Completed | Relapsed or Refractory Neuroblastoma | 1RG-CART | GD2 | RQR8 suicide gene is incorporated | Phase 1 | 29 February 2016 |
| Advantages | |
| Limitations -Strategies |
|
| National Clinical Trial (NCT) Number | Title | Status | Conditions | CAR-NK Product | Targeted Antigen | Modification to Overcome the Limitations | Phase | Start Date |
|---|---|---|---|---|---|---|---|---|
| NCT 05528341 | NKG2D-CAR-NK92 cell immunotherapy for solid tumors | Recruiting | Relapsed/refractory solid tumors | NKG2D CAR-NK92 cells | NKG2DL | Off-the-shelf NK92 cell line-based CAR-NK | Phase 1 | 26 January 2023 |
| NCT 05410717 | CLDN6-CAR-NK cell therapy for advanced solid tumors | Recruiting | Stage IV ovarian cancer, Testicular cancer, refractory Endometrial cancer, recurrent | Claudin6 targeting CAR-NK cells | Claudin6 | Engineered to express IL7/CCL19 and/or scfv against PD1/CTLA4/Lag3 | Phase 1 Phase 2 | 1 June 2022 |
| NCT 05194709 | Study of anti-5T4 CAR-NK cell therapy in advanced solid tumors | Recruiting | Advanced solid tumors | Anti-5T4 CAR-NK Cells | Oncofetal trophoblast glycoprotein (5T4) | Targeting 5T4 (oncofetal antigen) which allow survival of tumor in its host | Early Phase 1 | 30 December 2021 |
| NCT 05137275 | Study of anti-5T4 CARraNK cell therapy in locally advanced or metastatic solid tumors | Recruiting | Locally advanced or metastatic solid tumors | Anti-5T4 CAR-raNK (allogeneic NK) Cells | Oncofetal trophoblast glycoprotein (5T4) | Allogeneic CAR-NK cells | Early Phase 1 | 24 November 2021 |
| NCT 03940820 | Clinical Research of ROBO1 Specific CAR-NK Cells on Patients With Solid Tumors | Unknown | Solid tumor-Pancreatic cancer | ROBO1 CAR-NK cells | ROBO1 (Roundabout homolog 1) | Off-the-shelf NK92 cell line-based CAR-NK | Phase 1 Phase 2 | May 2019 |
| NCT 03415100 | Pilot Study of NKG2D-Ligand Targeted CAR-NK Cells in Patients With Metastatic Solid Tumours | Unknown | Metastatic solid tumors | CAR-NK cells targeting NKG2D ligands | NKG2DL | Allogeneic CAR-NK cells | Phase 1 | 2 January 2018 |
| NCT 02839954 | CAR-pNK Cell Immunotherapy in MUC1 Positive Relapsed or Refractory Solid Tumor | Unknown | Hepatocellular Carcinoma, Non-small Cell Lung Cancer, Pancreatic Carcinoma, Triple-Negative Invasive Breast Carcinoma, Malignant Glioma of Brain, Colorectal Carcinoma, Gastric Carcinoma | anti-MUC1 CAR-pNK cells | MUC1 (Mucin 1) | Targeting MUC1 (Mucin 1) on epithelial surfaces for enhanced tumor infiltration | Phase 1 Phase 2 | July 2016 |
| Advantages | |
| Limitations -Strategies |
|
| Advantages | |
| Disadvantages |
| Advantages | |
| Disadvantage |
|
| National Clinical Trial (NCT) Number | Title | Status | Conditions | CAR-T Product | Targeted Antigen | Modification to Overcome the Limitations | Phase | Start Date |
|---|---|---|---|---|---|---|---|---|
| NCT 04660929 | CAR-macrophages for the treatment of HER2-overexpressing solid tumors | Recruiting | HER2-positive Adenocarcinoma, Bile duct cancer, Biliary tract cancer, Bladder cancer, Breast cancer, Breast neoplasm, Carcinoma, ductal Carcinoma, hepatocellular and 21 more | Anti-HER2 CAR macrophages | HER2 | Adenoviral vector CAR, Combination treatment wirh pembrolizumab | Phase 1 | 2 February 2021 |
| NCT 05007379 | Cohort study to determine the anti-tumor activity of new CAR macrophages in the derived organoids of breast cancer patients | Not yet recruiting | Breast cancer | Anti-HER2 CAR macrophages | HER2 | CAR-M efficacy testing against patient-derived organoids | 1 September 2021 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, M.H.; Oh, E.; Kim, Y.; Nam, D.-H.; Jeon, S.Y.; Yu, J.H.; Minn, D. Recent Advances in CAR-Based Solid Tumor Immunotherapy. Cells 2023, 12, 1606. https://doi.org/10.3390/cells12121606
Shin MH, Oh E, Kim Y, Nam D-H, Jeon SY, Yu JH, Minn D. Recent Advances in CAR-Based Solid Tumor Immunotherapy. Cells. 2023; 12(12):1606. https://doi.org/10.3390/cells12121606
Chicago/Turabian StyleShin, Min Hwa, Eunha Oh, Yunjeong Kim, Dae-Hwan Nam, So Young Jeon, Jin Hyuk Yu, and Dohsik Minn. 2023. "Recent Advances in CAR-Based Solid Tumor Immunotherapy" Cells 12, no. 12: 1606. https://doi.org/10.3390/cells12121606
APA StyleShin, M. H., Oh, E., Kim, Y., Nam, D.-H., Jeon, S. Y., Yu, J. H., & Minn, D. (2023). Recent Advances in CAR-Based Solid Tumor Immunotherapy. Cells, 12(12), 1606. https://doi.org/10.3390/cells12121606