
Dysregulation of Metabolism and Proteostasis in Skeletal Muscle of a Presymptomatic Pompe Mouse Model

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Sample Processing
2.3. LC-MS/MS Analysis
2.4. Analysis of Proteomic Data
2.5. Histological Staining
2.6. Immunofluorescence Studies
2.7. Resin Sections and Electron Microscopy
2.8. Western Blot
2.9. Statistical Analysis
3. Results
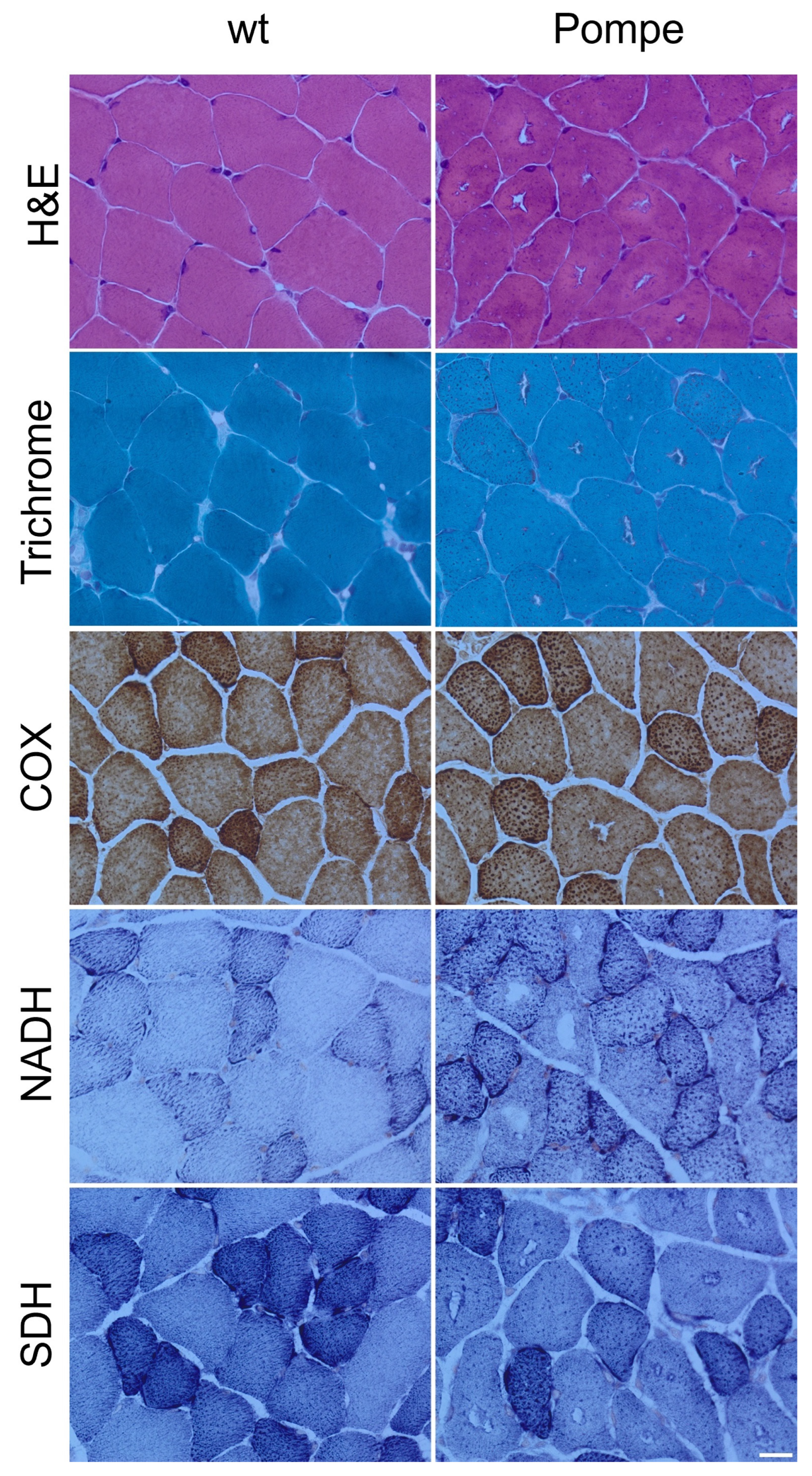
3.1. Histological Staining
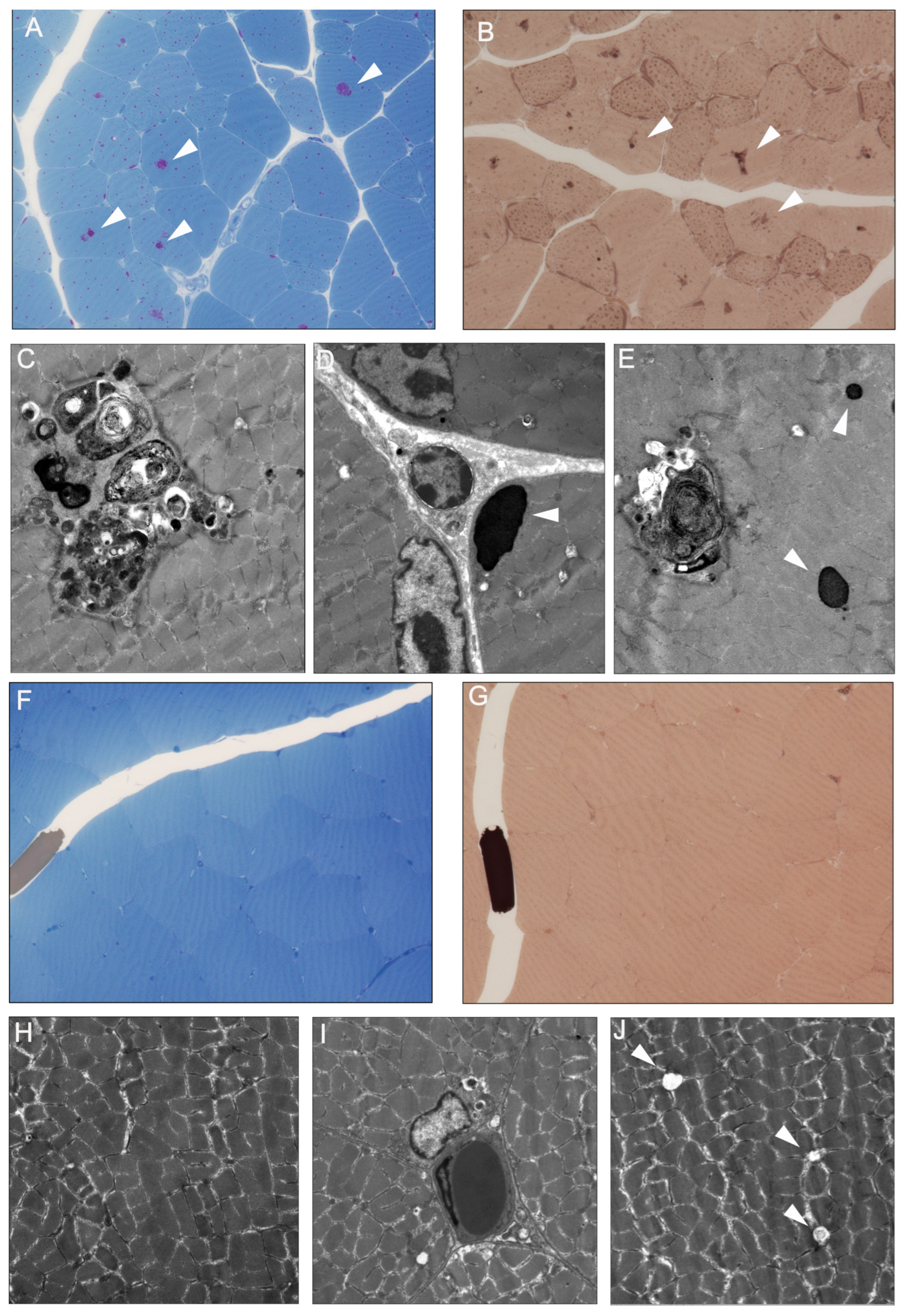
3.2. Resin Sections and Electron Microscopy
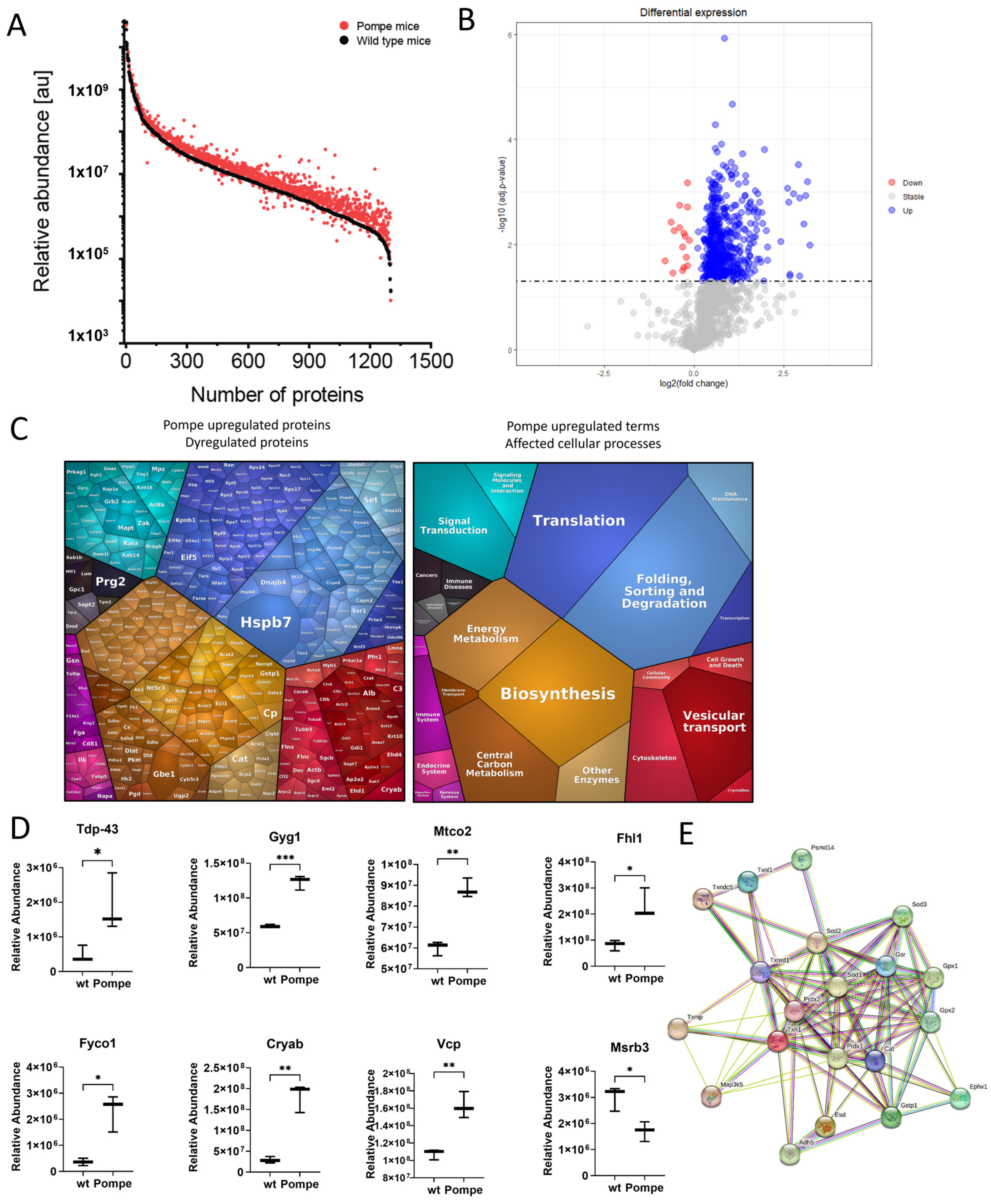
3.3. Overview with GO-Term Analysis
3.4. Similarities to Neurodegenerative Diseases
3.5. Altered Proteostasis
3.6. Metabolism as a Major Upregulated Factor
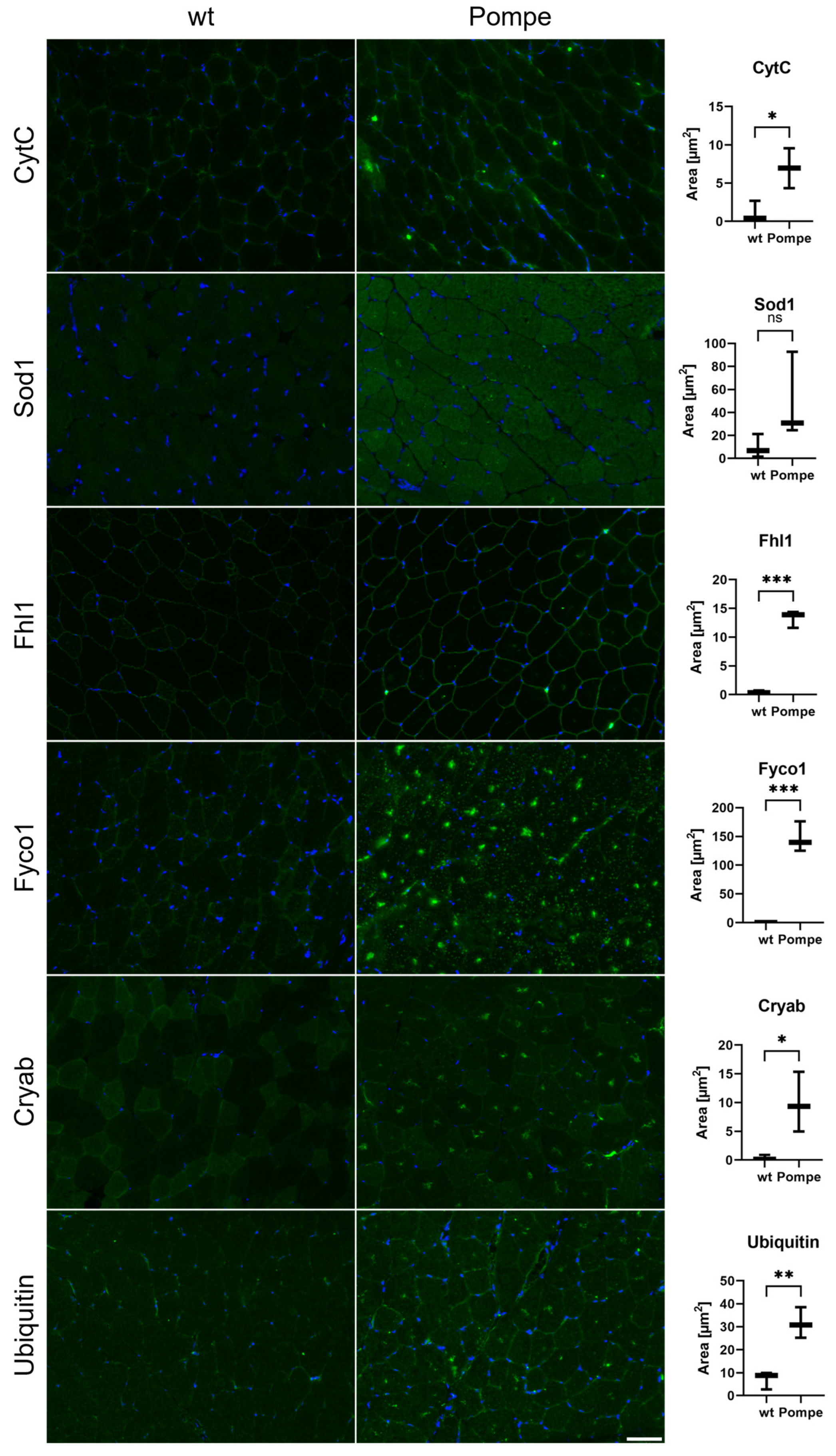
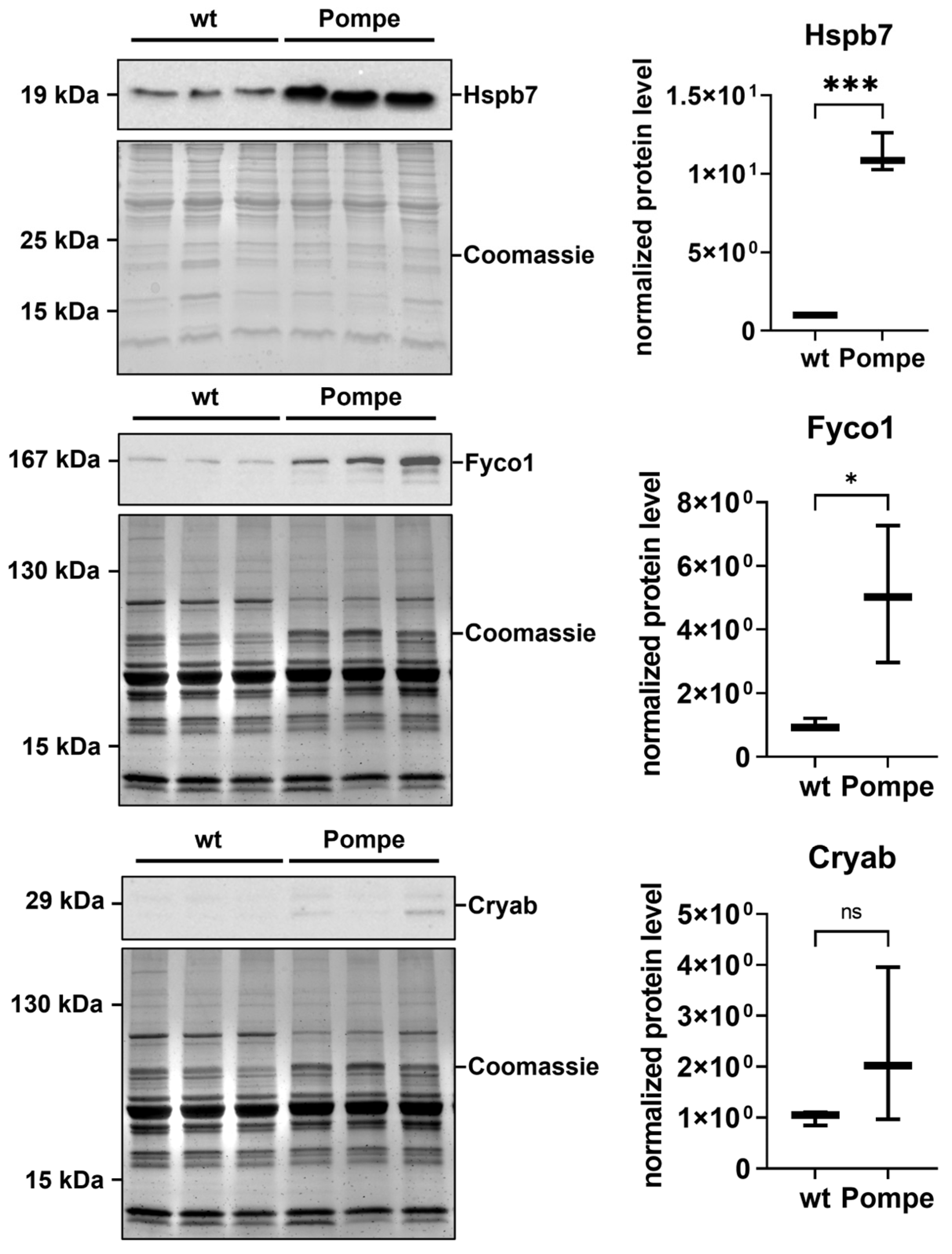
3.7. Immunofluorescence and Western Blot Studies toward Validation of Proteomic Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van der Ploeg, A.T.; Reuser, A.J. Pompe’s Disease. Lancet 2008, 372, 1342–1353. [Google Scholar] [CrossRef]
- Winkel, L.P.F.; Hagemans, M.L.C.; Van Doorn, P.A.; Loonen, M.C.B.; Hop, W.J.C.; Reuser, A.J.J.; Van Der Ploeg, A.T. The Natural Course of Non-Classic Pompe’s Disease; a Review of 225 Published Cases. J. Neurol. 2005, 252, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Bembi, B.; Cerini, E.; Danesino, C.; Donati, M.A.; Gasperini, S.; Morandi, L.; Musumeci, O.; Parenti, G.; Ravaglia, S.; Seidita, F.; et al. Diagnosis of Glycogenosis Type II. Neurology 2008, 71, S4–S11. [Google Scholar] [CrossRef] [PubMed]
- van der Ploeg, A.T.; Kruijshaar, M.E.; Toscano, A.; Laforêt, P.; Angelini, C.; Lachmann, R.H.; Pascual Pascual, S.I.; Roberts, M.; Rösler, K.; Stulnig, T.; et al. European Consensus for Starting and Stopping Enzyme Replacement Therapy in Adult Patients with Pompe Disease: A 10-Year Experience. Eur. J. Neurol. 2017, 24, 768-e31. [Google Scholar] [CrossRef]
- Raben, N.; Nagaraju, K.; Lee, E.; Plotz, P. Modulation of Disease Severity in Mice with Targeted Disruption of the Acid α-Glucosidase Gene. Neuromuscul. Disord. 2000, 10, 283–291. [Google Scholar] [CrossRef]
- Sidman, R.L.; Taksir, T.; Fidler, J.; Zhao, M.; Dodge, J.C.; Passini, M.A.; Raben, N.; Thurberg, B.L.; Cheng, S.H.; Shihabuddin, L.S. Temporal Neuropathologic and Behavioral Phenotype of 6neo/6neo Pompe Disease Mice. J. Neuropathol. Exp. Neurol. 2008, 67, 803–818. [Google Scholar] [CrossRef] [PubMed]
- Raben, N.; Nagaraju, K.; Lee, E.; Kessler, P.; Byrne, B.; Lee, L.; LaMarca, M.; King, C.; Ward, J.; Sauer, B.; et al. Targeted Disruption of the Acid α-Glucosidase Gene in Mice Causes an Illness with Critical Features of Both Infantile and Adult Human Glycogen Storage Disease Type II. J. Biol. Chem. 1998, 273, 19086–19092. [Google Scholar] [CrossRef]
- Roos, A.; Thompson, R.; Horvath, R.; Lochmüller, H.; Sickmann, A. Intersection of Proteomics and Genomics to “Solve the Unsolved” in Rare Disorders such as Neurodegenerative and Neuromuscular Diseases. Proteom. Clin. Appl. 2018, 12, 1700073. [Google Scholar] [CrossRef]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and Chemical Approaches to Diseases of Proteostasis Deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef]
- Al-Amrani, S.; Al-Jabri, Z.; Al-Zaabi, A.; Alshekaili, J.; Al-Khabori, M. Proteomics: Concepts and Applications in Human Medicine. World J. Biol. Chem. 2021, 12, 57–69. [Google Scholar] [CrossRef]
- Lim, J.A.; Sun, B.; Puertollano, R.; Raben, N. Therapeutic Benefit of Autophagy Modulation in Pompe Disease. Mol. Ther. 2018, 26, 1783–1796. [Google Scholar] [CrossRef]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef]
- Liebermeister, W.; Noor, E.; Flamholz, A.; Davidi, D.; Bernhardt, J.; Milo, R. Visual Account of Protein Investment in Cellular Functions. Proc. Natl. Acad. Sci. USA 2014, 111, 8488–8493. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Schänzer, A.; Kaiser, A.-K.; Mühlfeld, C.; Kulessa, M.; Paulus, W.; von Pein, H.; Rohrbach, M.; Viergutz, L.; Mengel, E.; Marquardt, T.; et al. Quantification of Muscle Pathology in Infantile Pompe Disease. Neuromuscul. Disord. 2017, 27, 141–152. [Google Scholar] [CrossRef]
- Kulessa, M.; Weyer-Menkhoff, I.; Viergutz, L.; Kornblum, C.; Claeys, K.G.; Schneider, I.; Plöckinger, U.; Young, P.; Boentert, M.; Vielhaber, S.; et al. An Integrative Correlation of Myopathology, Phenotype and Genotype in Late Onset Pompe Disease. Neuropathol. Appl. Neurobiol. 2020, 46, 359–374. [Google Scholar] [CrossRef]
- Mercer, E.J.; Lin, Y.F.; Cohen-Gould, L.; Evans, T. Hspb7 Is a Cardioprotective Chaperone Facilitating Sarcomeric Proteostasis. Dev. Biol. 2018, 435, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Güttsches, A.K.; Brady, S.; Krause, K.; Maerkens, A.; Uszkoreit, J.; Eisenacher, M.; Schreiner, A.; Galozzi, S.; Mertens-Rill, J.; Tegenthoff, M.; et al. Proteomics of Rimmed Vacuoles Define New Risk Allele in Inclusion Body Myositis. Ann. Neurol. 2017, 81, 227. [Google Scholar] [CrossRef] [PubMed]
- Guettsches, A.K.; Meyer, N.; Zahedi, R.P.; Evangelista, T.; Muentefering, T.; Ruck, T.; Lacene, E.; Heute, C.; Gonczarowska-Jorge, H.; Schoser, B.; et al. FYCO1 Increase and Effect of Arimoclomol–Treatment in Human VCP–Pathology. Biomedicines 2022, 10, 2443. [Google Scholar] [CrossRef]
- Yamamoto, S.; Yamashita, A.; Arakaki, N.; Nemoto, H.; Yamazaki, T. Prevention of Aberrant Protein Aggregation by Anchoring the Molecular Chaperone AB-Crystallin to the Endoplasmic Reticulum. Biochem. Biophys. Res. Commun. 2014, 455, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.M.; Meyers, E.; Phipps, M.; Kishnani, P.S.; Cheng, S.H.; Scheule, R.K.; Moreland, R.J. Dysregulation of Multiple Facets of Glycogen Metabolism in a Murine Model of Pompe Disease. PLoS ONE 2013, 8, e56181. [Google Scholar] [CrossRef] [PubMed]
- Canibano-Fraile, R.; Harlaar, L.; Dos Santos, C.A.; Hoogeveen-Westerveld, M.; Demmers, J.A.A.; Snijders, T.; Lijnzaad, P.; Verdijk, R.M.; van der Beek, N.A.M.E.; van Doorn, P.A.; et al. Lysosomal Glycogen Accumulation in Pompe Disease Results in Disturbed Cytoplasmic Glycogen Metabolism. J. Inherit. Metab. Dis. 2023, 46, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Moriggi, M.; Capitanio, D.; Torretta, E.; Barbacini, P.; Bragato, C.; Sartori, P.; Moggio, M.; Maggi, L.; Mora, M.; Gelfi, C. Muscle Proteomic Profile before and after Enzyme Replacement Therapy in Late-Onset Pompe Disease. Int. J. Mol. Sci. 2021, 22, 2850. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Kido, J.; Nakamura, K. Newborn Screening for Pompe Disease. Int. J. Neonatal Screen. 2020, 6, 31. [Google Scholar] [CrossRef]
- Shimada, Y.; Kobayashi, H.; Kawagoe, S.; Aoki, K.; Kaneshiro, E.; Shimizu, H.; Eto, Y.; Ida, H.; Ohashi, T. Endoplasmic Reticulum Stress Induces Autophagy through Activation of P38 MAPK in Fibroblasts from Pompe Disease Patients Carrying c.546G>T Mutation. Mol. Genet. Metab. 2011, 104, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.B.; Sommerville, R.B.; Allred, P.; Bell, S.; Ma, D.; Cooper, P.; Lopate, G.; Pestronk, A.; Weihl, C.C.; Baloh, R.H. Exome Sequencing Reveals DNAJB6 Mutations in Dominantly-Inherited Myopathy. Ann. Neurol. 2012, 71, 407. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Kimura, E.; Tawara, N.; Sakaguchi, H.; Nakama, T.; Maeda, Y.; Hirano, T.; Uchino, M.; Ando, Y. Optineurin Is Potentially Associated with TDP-43 and Involved in the Pathogenesis of Inclusion Body Myositis. Neuropathol. Appl. Neurobiol. 2013, 39, 406–416. [Google Scholar] [CrossRef]
- Salajegheh, M.; Pinkus, J.L.; Taylor, J.P.; Amato, A.; Nazareno, R.; Baloh, R.H.; Greenberg, S.A. Sarcoplasmic Redistribution of Nuclear TDP-43 in Inclusion Body Myositis. Muscle Nerve 2009, 40, 19–31. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 Is a Component of Ubiquitin-Positive Tau-Negative Inclusions in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Selcen, D.; Bromberg, M.B.; Chin, S.S.; Engel, A.G. Reducing Bodies and Myofibrillar Myopathy Features in FHL1 Muscular Dystrophy. Neurology 2011, 77, 1951–1959. [Google Scholar] [CrossRef]
- Friedrich, F.W.; Carrier, L. Genetics of Hypertrophic and Dilated Cardiomyopathy. Curr. Pharm. Biotechnol. 2012, 13, 2467–2476. [Google Scholar] [CrossRef]
- Keßler, M.; Kieltsch, A.; Kayvanpour, E.; Katus, H.A.; Schoser, B.; Schessl, J.; Just, S.; Rottbauer, W. A Zebrafish Model for FHL1-Opathy Reveals Loss-of-Function Effects of Human FHL1 Mutations. Neuromuscul. Disord. 2018, 28, 521–531. [Google Scholar] [CrossRef]
- Han, S.; Cui, C.; He, H.; Shen, X.; Chen, Y.; Wang, Y.; Li, D.; Zhu, Q.; Yin, H. FHL1 Regulates Myoblast Differentiation and Autophagy through Its Interaction with LC3. J. Cell. Physiol. 2020, 235, 4667–4678. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.A.; Lamark, T.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. FYCO1 Is a Rab7 Effector That Binds to LC3 and PI3P to Mediate Microtubule plus End–Directed Vesicle Transport. J. Cell. Biol. 2010, 188, 253. [Google Scholar] [CrossRef] [PubMed]
- Vicart, P.; Caron, A.; Guicheney, P.; Li, Z.; Prévost, M.C.; Faure, A.; Chateau, D.; Chapon, F.; Tomé, F.; Dupret, J.M.; et al. A Missense Mutation in the AB-Crystallin Chaperone Gene Causes a Desmin-Related Myopathy. Nat. Genet. 1998, 20, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Yamasaki, T.; Hasebe, R.; Suzuki, A.; Horiuchi, M. Enhanced Phosphorylation of PERK in Primary Cultured Neurons as an Autonomous Neuronal Response to Prion Infection. PLoS ONE 2020, 15, e0234147. [Google Scholar] [CrossRef]
- Juo, L.Y.; Liao, W.C.; Shih, Y.L.; Yang, B.Y.; Liu, A.B.; Yan, Y.T. HSPB7 Interacts with Dimerized FLNC and Its Absence Results in Progressive Myopathy in Skeletal Muscles. J. Cell Sci. 2016, 129, 1661–1670. [Google Scholar] [CrossRef]
- Fukuda, T.; Ewan, L.; Bauer, M.; Mattaliano, R.J.; Zaal, K.; Ralston, E.; Plotz, P.H.; Raben, N. Dysfunction of Endocytic and Autophagic Pathways in a Lysosomal Storage Disease. Ann. Neurol. 2006, 59, 700–708. [Google Scholar] [CrossRef]
- Taverna, S.; Cammarata, G.; Colomba, P.; Sciarrino, S.; Zizzo, C.; Francofonte, D.; Zora, M.; Scalia, S.; Brando, C.; Curto, A.L.; et al. Pompe Disease: Pathogenesis, Molecular Genetics and Diagnosis. Aging 2020, 12, 15856–15874. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody | Manufactory | Dilution |
| anti-SOD1 | GTX100554, Biozol, Eching, Germany | 1:200 |
| anti-CytC | sc-13156, Santa Cruz Biotechnology, Dallas, TX, USA | 1:200 |
| anti-FHL1 | ab255828, Abcam, Cambridge, UK | 1:300 |
| anti-FYCO1 | HPA035526, Sigma–Aldrich, St. Louis, MO, USA | 1:500 |
| anti-CRYAB | CPTC-CRYAB-2, deposited to the DSHB by Clinical Proteomics Technologies for Cancer | 0.5 ng/µL |
| anti-Ubiquitin | ab19247, Abcam, Cambridge, UK | 1:200 |
| Secondary Antibody | Manufactory | Dilution |
| Alexa green 488 anti-rabbit | RRID: AB_2313584, Jackson ImmunoResearch Laboratories, Pennsylvania, PA, USA | 1:1500 |
| Alexa green 488 anti-mouse | RRID: AB_2307324, Jackson ImmunoResearch Laboratories, Pennsylvania, PA, USA | 1:1000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rohm, M.; Volke, L.; Schlaffke, L.; Rehmann, R.; Südkamp, N.; Roos, A.; Schänzer, A.; Hentschel, A.; Vorgerd, M. Dysregulation of Metabolism and Proteostasis in Skeletal Muscle of a Presymptomatic Pompe Mouse Model. Cells 2023, 12, 1602. https://doi.org/10.3390/cells12121602
Rohm M, Volke L, Schlaffke L, Rehmann R, Südkamp N, Roos A, Schänzer A, Hentschel A, Vorgerd M. Dysregulation of Metabolism and Proteostasis in Skeletal Muscle of a Presymptomatic Pompe Mouse Model. Cells. 2023; 12(12):1602. https://doi.org/10.3390/cells12121602
Chicago/Turabian StyleRohm, Marlena, Leon Volke, Lara Schlaffke, Robert Rehmann, Nicolina Südkamp, Andreas Roos, Anne Schänzer, Andreas Hentschel, and Matthias Vorgerd. 2023. "Dysregulation of Metabolism and Proteostasis in Skeletal Muscle of a Presymptomatic Pompe Mouse Model" Cells 12, no. 12: 1602. https://doi.org/10.3390/cells12121602
APA StyleRohm, M., Volke, L., Schlaffke, L., Rehmann, R., Südkamp, N., Roos, A., Schänzer, A., Hentschel, A., & Vorgerd, M. (2023). Dysregulation of Metabolism and Proteostasis in Skeletal Muscle of a Presymptomatic Pompe Mouse Model. Cells, 12(12), 1602. https://doi.org/10.3390/cells12121602