

Interleukin-36 Receptor Signaling Attenuates Epithelial Wound Healing in C57BL/6 Mouse Corneas



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Animals
2.2. Animals and Induction of Diabetes
2.3. Corneal Epithelial Debridement Wounds
2.4. Subconjunctival Injection of Proteins
2.5. PCR Analysis
2.6. Statistical Analysis
3. Results
3.1. IL-36/IL-36R Signaling Plays a Detrimental Role in Corneal Epithelial Wound Closure in Normoglycemia B6 Mouse Corneas
3.2. Wound-Induced Expression of IL-36 Cytokines in B6 Mouse Corneas
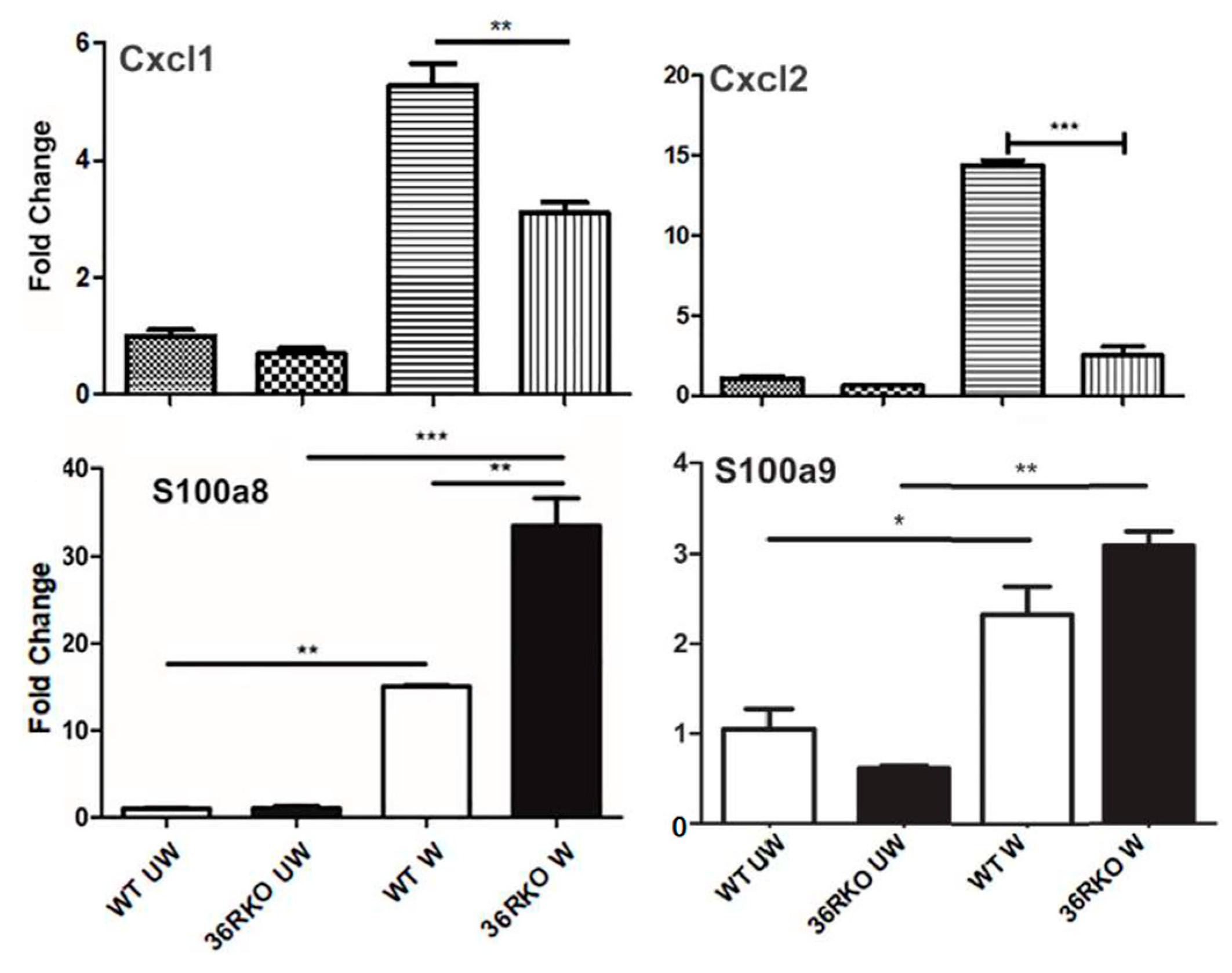
3.3. Effects of IL36R Deficiency on the Expression of Cytokines and Antimicrobial Peptides in Wounded Corneas
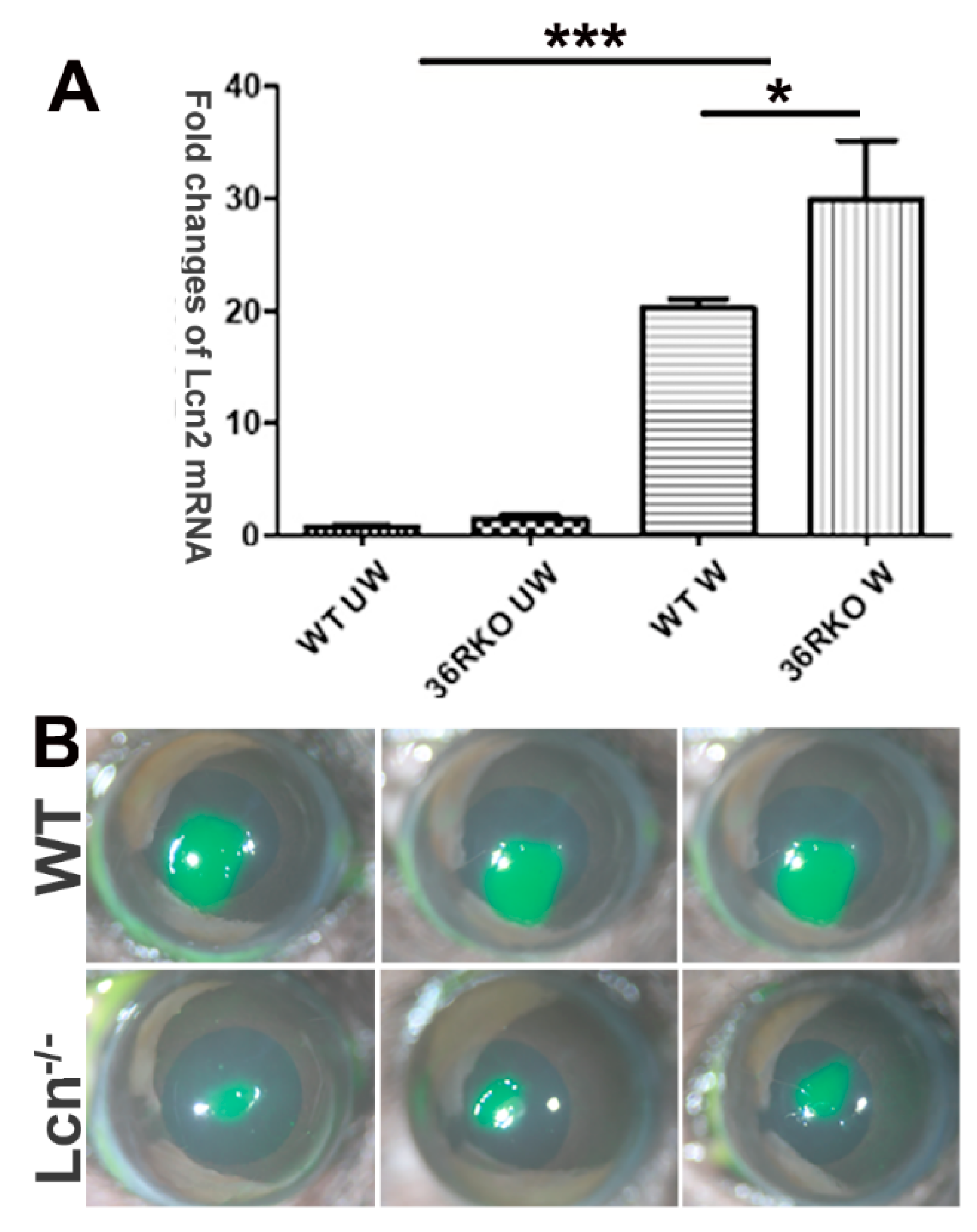
3.4. LCN2 Plays a Detrimental Role in Epithelial Wound Healing in B6 Mouse Corneas
3.5. IL-36R Deficiency Greatly Accelerates Epithelial Wound Closure in DM Corneas of B6 Mice
3.6. IL-36R Deficiency Promotes IL-Ra Expression in DM Corneas
3.7. Recombinant IL-1Ra and IL-36Ra Accelerated Delayed Epithelial Wound Healing in Diabetic B6 Mouse Corneas
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ngo, V.L.; Abo, H.; Kuczma, M.; Szurek, E.; Moore, N.; Medina-Contreras, O.; Nusrat, A.; Merlin, D.; Gewirtz, A.T.; Ignatowicz, L.; et al. IL-36R signaling integrates innate and adaptive immune-mediated protection against enteropathogenic bacteria. Proc. Natl. Acad. Sci. USA 2020, 117, 27540–27548. [Google Scholar] [CrossRef] [PubMed]
- Me, R.; Gao, N.; Zhang, Y.; Lee, P.S.Y.; Wang, J.; Liu, T.; Standiford, T.J.; Mi, Q.-S.; Yu, F.-S.X. IL-36α Enhances Host Defense against Pseudomonas aeruginosa Keratitis in C57BL/6 Mouse Corneas. J. Immunol. 2021, 207, 2868–2877. [Google Scholar] [CrossRef] [PubMed]
- Mahil, S.K.; Catapano, M.; Di Meglio, P.; Dand, N.; Ahlfors, H.; Carr, I.M.; Smith, C.H.; Trembath, R.C.; Peakman, M.; Wright, J.; et al. An analysis of IL-36 signature genes and individuals with IL1RL2 knockout mutations validates IL-36 as a psoriasis therapeutic target. Sci. Transl. Med. 2017, 9, eaan2514. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.D.; Bassoy, E.Y.; Caruso, A.; Palomo, J.; Rodriguez, E.; Lemeille, S.; Gabay, C. IL-36 signaling in keratinocytes controls early IL-23 production in psoriasis-like dermatitis. Life Sci. Alliance 2020, 3, e202000688. [Google Scholar] [CrossRef] [PubMed]
- Medina-Contreras, O.; Harusato, A.; Nishio, H.; Flannigan, K.L.; Ngo, V.; Leoni, G.; Neumann, P.-A.; Geem, D.; Lili, L.N.; Ramadas, R.A.; et al. Cutting Edge: IL-36 Receptor Promotes Resolution of Intestinal Damage. J. Immunol. 2016, 196, 34–38. [Google Scholar] [CrossRef] [Green Version]
- Scheibe, K.; Backert, I.; Wirtz, S.; Hueber, A.; Schett, G.; Vieth, M.; Probst, H.C.; Bopp, T.; Neurath, M.F.; Neufert, C. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut 2016, 66, 823–838. [Google Scholar] [CrossRef]
- Jiang, Z.; Liu, Y.; Li, C.; Chang, L.; Wang, W.; Wang, Z.; Gao, X.; Ryffel, B.; Wu, Y.; Lai, Y. IL-36gamma Induced by the TLR3-SLUG-VDR Axis Promotes Wound Healing via REG3A. J. Investig. Dermatol. 2017, 137, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Bal, E.; Lim, A.C.; Shen, M.; Douangpanya, J.; Madrange, M.; Gazah, R.; Tauber, M.; Beghdadi, W.; Casanova, J.L.; Bourrat, E.; et al. Mutation in IL36RN impairs the processing and regulatory function of the interleukin-36-receptor antagonist and is associated with DITRA syndrome. Exp. Dermatol. 2019, 28, 1114–1117. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Iwata, Y.; Fukushima, H.; Watanabe, S.; Tanaka, Y.; Hasegawa, Y.; Akiyama, M.; Sugiura, K. IL-36 receptor antagonist deficiency resulted in delayed wound healing due to excessive recruitment of immune cells. Sci. Rep. 2020, 10, 14772. [Google Scholar] [CrossRef]
- Tanaka, Y.; Iwata, Y.; Saito, K.; Fukushima, H.; Watanabe, S.; Hasegawa, Y.; Akiyama, M.; Sugiura, K. Cutaneous ischemia-reperfusion injury is exacerbated by IL-36 receptor antagonist deficiency. J. Eur. Acad. Dermatol. Venereol. 2021, 36, 295–304. [Google Scholar] [CrossRef]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [Green Version]
- Walsh, P.T.; Fallon, P.G. The emergence of the IL-36 cytokine family as novel targets for inflammatory diseases. Ann. N. Y. Acad. Sci. 2016, 1417, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.; Arend, W.; Sims, J.; Smith, D.; Blumberg, H.; O’Neill, L.; Goldbach-Mansky, R.; Pizarro, T.; Hoffman, H.; Bufler, P.; et al. IL-1 family nomenclature. Nat. Immunol. 2010, 11, 973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Towne, J.E.; Garka, K.E.; Renshaw, B.R.; Virca, G.D.; Sims, J.E. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J. Biol. Chem. 2004, 279, 13677–13688. [Google Scholar] [CrossRef] [Green Version]
- Aoyagi, T.; Newstead, M.; Zeng, X.; Kunkel, S.; Kaku, M.; Standiford, T. IL-36 receptor deletion attenuates lung injury and decreases mortality in murine influenza pneumonia. Mucosal Immunol. 2017, 10, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segueni, N.; Vigne, S.; Palmer, G.; Bourigault, M.-L.; Olleros, M.L.; Vesin, D.; Garcia, I.; Ryffel, B.; Quesniaux, V.F.J.; Gabay, C. Limited Contribution of IL-36 versus IL-1 and TNF Pathways in Host Response to Mycobacterial Infection. PLoS ONE 2015, 10, e0126058. [Google Scholar] [CrossRef] [Green Version]
- Kovach, M.A.; Singer, B.; Martinez-Colon, G.; Newstead, M.W.; Zeng, X.; Mancuso, P.; Moore, T.A.; Kunkel, S.L.; Peters-Golden, M.; Moore, B.B.; et al. IL-36gamma is a crucial proximal component of protective type-1-mediated lung mucosal immunity in Gram-positive and -negative bacterial pneumonia. Mucosal Immunol. 2017, 10, 1320–1334. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Lin, S.; Yan, X.; Wang, C.; Tu, H.; Yin, Y.; Cao, J. Interleukin 38 Protects Against Lethal Sepsis. J. Infect. Dis. 2018, 218, 1175–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, V.L.; Abo, H.; Maxim, E.; Harusato, A.; Geem, D.; Medina-Contreras, O.; Merlin, D.; Gewirtz, A.T.; Nusrat, A.; Denning, T.L. A cytokine network involving IL-36gamma, IL-23, and IL-22 promotes antimicrobial defense and recovery from intestinal barrier damage. Proc. Natl. Acad. Sci. USA 2018, 115, E5076–E5085. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Fang, H.; Dang, E.; Xue, K.; Zhang, J.; Li, B.; Qiao, H.; Cao, T.; Zhuang, Y.; Shen, S.; et al. Neutrophil Extracellular Traps Promote Inflammatory Responses in Psoriasis via Activating Epidermal TLR4/IL-36R Crosstalk. Front. Immunol. 2019, 10, 746. [Google Scholar] [CrossRef] [Green Version]
- Gao, N.; Me, R.; Dai, C.; Seyoum, B.; Yu, F.-S.X. Opposing Effects of IL-1Ra and IL-36Ra on Innate Immune Response to Pseudomonas aeruginosa Infection in C57BL/6 Mouse Corneas. J. Immunol. 2018, 201, 688–699. [Google Scholar] [CrossRef] [Green Version]
- Ljubimov, A.V. Diabetic complications in the cornea. Vis. Res. 2017, 139, 138–152. [Google Scholar] [CrossRef]
- Ljubimov, A.V.; Saghizadeh, M. Progress in corneal wound healing. Prog. Retin. Eye Res. 2015, 49, 17–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikbova, G.; Oshitari, T.; Tawada, A.; Yamamoto, S. Corneal Changes in Diabetes Mellitus. Curr. Diabetes Rev. 2012, 8, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, A.; Hopeross, M.W.; Chell, P.B. Neurotrophic keratopathy and diabetes mellitus. Eye 2005, 20, 837–839. [Google Scholar] [CrossRef]
- Frank, R.N. Diabetic retinopathy. N. Engl. J. Med. 2004, 350, 48–58. [Google Scholar] [CrossRef]
- Yu, F.-S.; Yin, J.; Lee, P.S.; Hwang, F.S.; McDermott, M. Sensory nerve regeneration after epithelium wounding in normal and diabetic corneas. Expert Rev. Ophthalmol. 2015, 10, 383–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovach, M.A.; Singer, B.H.; Newstead, M.W.; Zeng, X.; Moore, T.A.; White, E.S.; Kunkel, S.L.; Peters-Golden, M.; Standiford, T.J. IL-36gamma is secreted in microparticles and exosomes by lung macrophages in response to bacteria and bacterial components. J. Leukoc. Biol. 2016, 100, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Yu, F.-S.X. Impaired Epithelial Wound Healing and EGFR Signaling Pathways in the Corneas of Diabetic Rats. Investig. Opthalmol. Vis. Sci. 2011, 52, 3301–3308. [Google Scholar] [CrossRef]
- Yin, J.; Huang, J.; Chen, C.; Gao, N.; Wang, F.; Yu, F.-S.X. Corneal Complications in Streptozocin-Induced Type I Diabetic Rats. Investig. Opthalmol. Vis. Sci. 2011, 52, 6589–6596. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Inoue, K.; Kato, J.; Minamishima, S.; Morisaki, H. Functions and regulation of lipocalin-2 in gut-origin sepsis: A narrative review. Crit. Care 2019, 23, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, Q.; Ku, A.T.; Nishino, Y.; Howard, J.M.; Rao, A.S.; Shaver, T.M.; Garcia, G.E.; Le, D.N.; Karlin, K.L.; Westbrook, T.F.; et al. Tcf3 promotes cell migration and wound repair through regulation of lipocalin 2. Nat. Commun. 2014, 5, 4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathore, K.I.; Berard, J.L.; Redensek, A.; Chierzi, S.; Lopez-Vales, R.; Santos, M.; Akira, S.; David, S. Lipocalin 2 Plays an Immunomodulatory Role and Has Detrimental Effects after Spinal Cord Injury. J. Neurosci. 2011, 31, 13412–13419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhl, A.-L.; Wenzel, J. Interleukin-36 in Infectious and Inflammatory Skin Diseases. Front. Immunol. 2019, 10, 1162. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Me, R.; Gao, N.; Su, G.; Wu, X.; Yu, F.X. Role of IL-36gamma/IL-36R Signaling in Corneal Innate Defense Against Candida albicans Keratitis. Investig. Ophthalmol. Vis. Sci. 2021, 62, 10. [Google Scholar] [CrossRef]
- Luotola, K. IL-1 Receptor Antagonist (IL-1Ra) Levels and Management of Metabolic Disorders. Nutrients 2022, 14, 3422. [Google Scholar] [CrossRef]
- Bui, T.M.; Wiesolek, H.L.; Sumagin, R. ICAM-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J. Leukoc. Biol. 2020, 108, 787–799. [Google Scholar] [CrossRef]
- Queen, D.; Ediriweera, C.; Liu, L. Function and Regulation of IL-36 Signaling in Inflammatory Diseases and Cancer Development. Front. Cell Dev. Biol. 2019, 7, 317. [Google Scholar] [CrossRef]
- Yuan, Z.-C.; Xu, W.-D.; Liu, X.-Y.; Liu, X.-Y.; Huang, A.-F.; Su, L.-C. Biology of IL-36 Signaling and Its Role in Systemic Inflammatory Diseases. Front. Immunol. 2019, 10, 2532. [Google Scholar] [CrossRef] [Green Version]
- Gresnigt, M.S.; van de Veerdonk, F.L. Biology of IL-36 cytokines and their role in disease. Semin. Immunol. 2013, 25, 458–465. [Google Scholar] [CrossRef]
- Dong, H.; Hao, Y.; Li, W.; Yang, W.; Gao, P. IL-36 Cytokines: Their Roles in Asthma and Potential as a Therapeutic. Front. Immunol. 2022, 13, 921275. [Google Scholar] [CrossRef]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.-Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M.; et al. Interleukin-36–Receptor Antagonist Deficiency and Generalized Pustular Psoriasis. N. Engl. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Rossi-Semerano, L.; Piram, M.; Chiaverini, C.; De Ricaud, D.; Smahi, A.; Koné-Paut, I. First Clinical Description of an Infant With Interleukin-36-Receptor Antagonist Deficiency Successfully Treated with Anakinra. Pediatrics 2013, 132, e1043–e1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, D.W.; Kim, R.H. Epithelial cells release IL-36α in extracellular vesicles following mechanical damage. Biochem. Biophys. Res. Commun. 2022, 605, 56–62. [Google Scholar] [CrossRef]
- Bhusal, A.; Rahman, M.H.; Lee, I.K.; Suk, K. Role of Hippocampal Lipocalin-2 in Experimental Diabetic Encephalopathy. Front. Endocrinol. 2019, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Zhou, P.; Qi, Y. Down-regulation of LCN2 attenuates retinal vascular dysfunction and caspase-1-mediated pyroptosis in diabetes mellitus. Ann. Transl. Med. 2022, 10, 695. [Google Scholar] [CrossRef]
- Al Jaberi, S.; Cohen, A.; D’souza, C.; Abdulrazzaq, Y.M.; Ojha, S.; Bastaki, S.; Adeghate, E.A. Lipocalin-2: Structure, function, distribution and role in metabolic disorders. Biomed. Pharmacother. 2021, 142, 112002. [Google Scholar] [CrossRef]
- Foster, A.M.; Baliwag, J.; Chen, C.S.; Guzman, A.M.; Stoll, S.W.; Gudjonsson, J.E.; Ward, N.L.; Johnston, A. IL-36 Promotes Myeloid Cell Infiltration, Activation, and Inflammatory Activity in Skin. J. Immunol. 2014, 192, 6053–6061. [Google Scholar] [CrossRef] [Green Version]
- Moschen, A.R.; Adolph, T.E.; Gerner, R.R.; Wieser, V.; Tilg, H. Lipocalin-2: A Master Mediator of Intestinal and Metabolic Inflammation. Trends Endocrinol. Metab. 2017, 28, 388–397. [Google Scholar] [CrossRef]
- Kaji, Y.; Usui, T.; Oshika, T.; Matsubara, M.; Yamashita, H.; Araie, M.; Murata, T.; Ishibashi, T.; Nagai, R.; Horiuchi, S.; et al. Advanced glycation end products in diabetic corneas. Investig. Opthalmol. Vis. Sci. 2000, 41, 362–368. [Google Scholar]
- Gabay, C.; Lamacchia, C.; Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Yin, J.; Yoon, G.S.; Mi, Q.-S.; Yu, F.-S.X. Dendritic Cell–Epithelium Interplay Is a Determinant Factor for Corneal Epithelial Wound Repair. Am. J. Pathol. 2011, 179, 2243–2253. [Google Scholar] [CrossRef]
- Cavalli, G.; Dinarello, C.A. Anakinra Therapy for Non-cancer Inflammatory Diseases. Front. Pharmacol. 2018, 9, 1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachen, K.L.; Greving, C.N.A.; Towne, J.E. Role of IL-36 cytokines in psoriasis and other inflammatory skin conditions. Cytokine 2022, 156, 155897. [Google Scholar] [CrossRef] [PubMed]
- Carrier, Y.; Ma, H.-L.; Ramon, H.E.; Napierata, L.; Small, C.; O’Toole, M.; Young, D.A.; Fouser, L.A.; Nickerson-Nutter, C.; Collins, M.; et al. Inter-Regulation of Th17 Cytokines and the IL-36 Cytokines In Vitro and In Vivo: Implications in Psoriasis Pathogenesis. J. Investig. Dermatol. 2011, 131, 2428–2437. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, S.; Zhao, T.; Li, M. Serum IL-36 cytokines levels in type 2 diabetes mellitus patients and their association with obesity, insulin resistance, and inflammation. J Clin Lab Anal 2021, 35, e23611. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Q.; Gao, N.; Yu, F.-S. Interleukin-36 Receptor Signaling Attenuates Epithelial Wound Healing in C57BL/6 Mouse Corneas. Cells 2023, 12, 1587. https://doi.org/10.3390/cells12121587
Chen Q, Gao N, Yu F-S. Interleukin-36 Receptor Signaling Attenuates Epithelial Wound Healing in C57BL/6 Mouse Corneas. Cells. 2023; 12(12):1587. https://doi.org/10.3390/cells12121587
Chicago/Turabian StyleChen, Qi, Nan Gao, and Fu-Shin Yu. 2023. "Interleukin-36 Receptor Signaling Attenuates Epithelial Wound Healing in C57BL/6 Mouse Corneas" Cells 12, no. 12: 1587. https://doi.org/10.3390/cells12121587
APA StyleChen, Q., Gao, N., & Yu, F.-S. (2023). Interleukin-36 Receptor Signaling Attenuates Epithelial Wound Healing in C57BL/6 Mouse Corneas. Cells, 12(12), 1587. https://doi.org/10.3390/cells12121587