
Proteomic Characterization of SAS Cell-Derived Extracellular Vesicles in Relation to Both BPA and Neutron Irradiation Doses

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Neutron Irradiation of SAS Cells
2.2. Cell Growth Assay
2.3. EVs Isolation from Cell Medium
2.4. LC-MS/MS Analysis of SAS Cells Medium-Derived EVs
2.4.1. In-Solution Digestion
2.4.2. Liquid Chromatography
2.4.3. Mass Spectrometry
2.5. Proteomic Data Processing and Data Mining
2.6. Network Analysis
3. Results
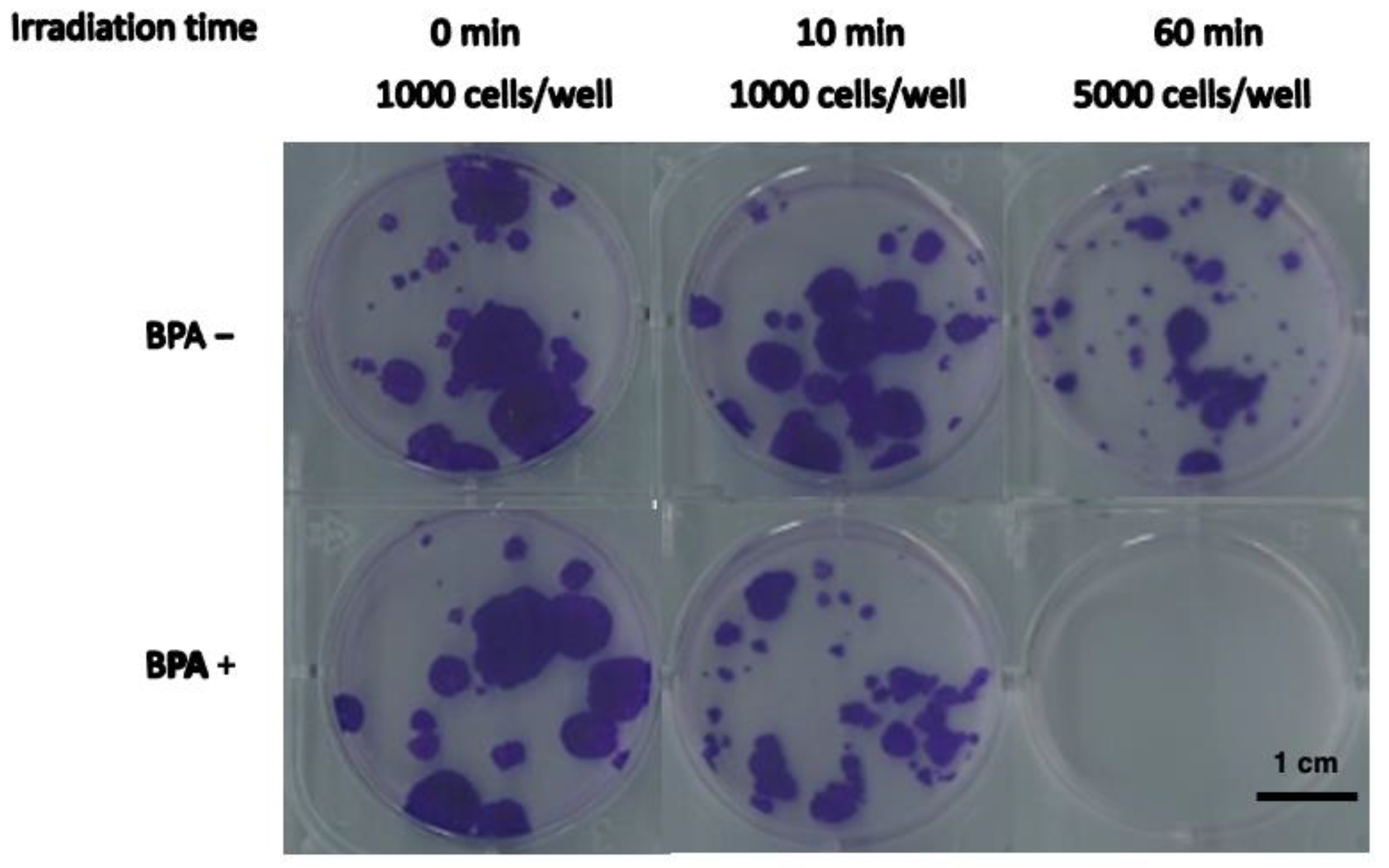
3.1. Evaluation of Cellular Growth and Viability after BNCT Treatment
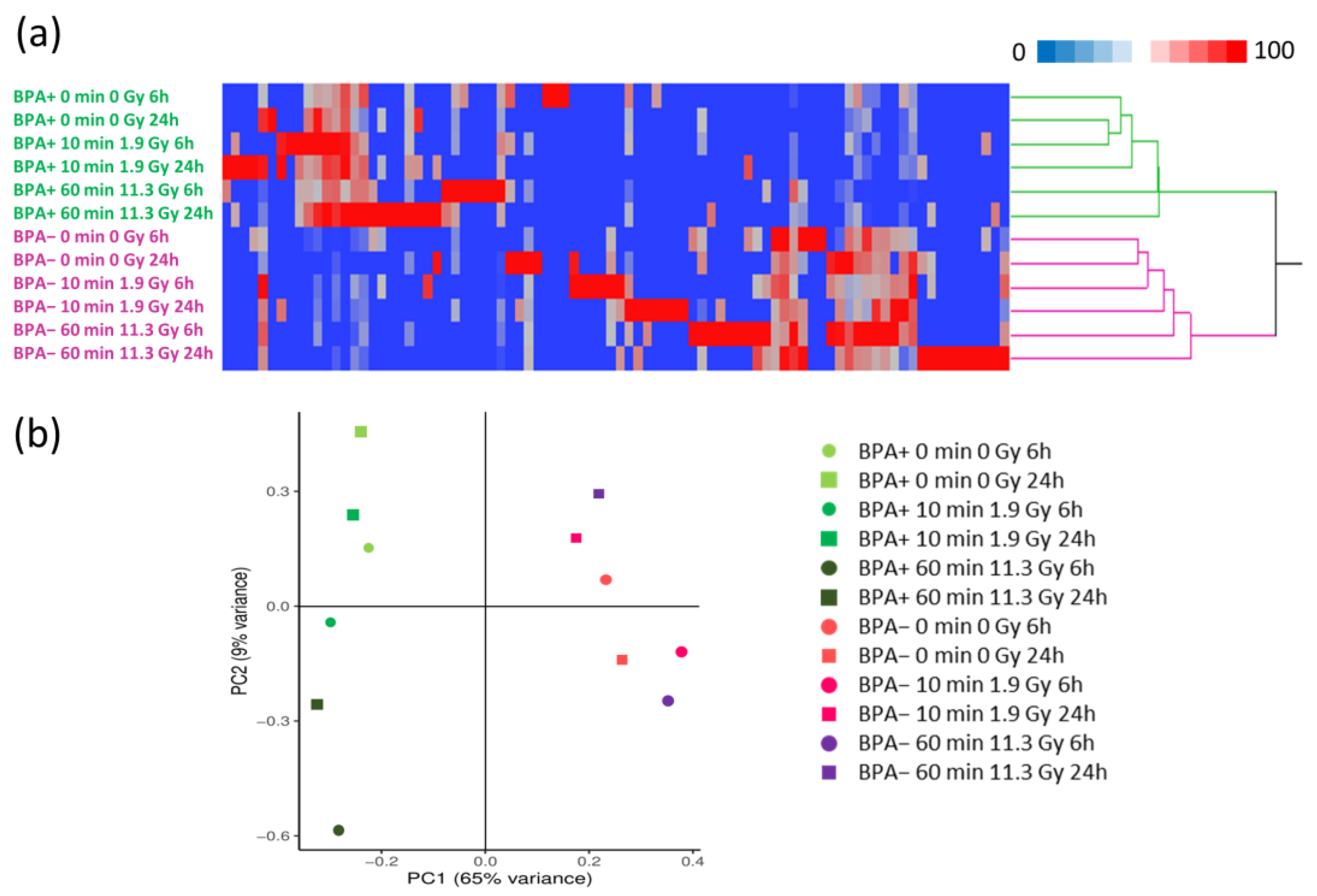
3.2. Proteomics Profiles of EVs under Different Conditions
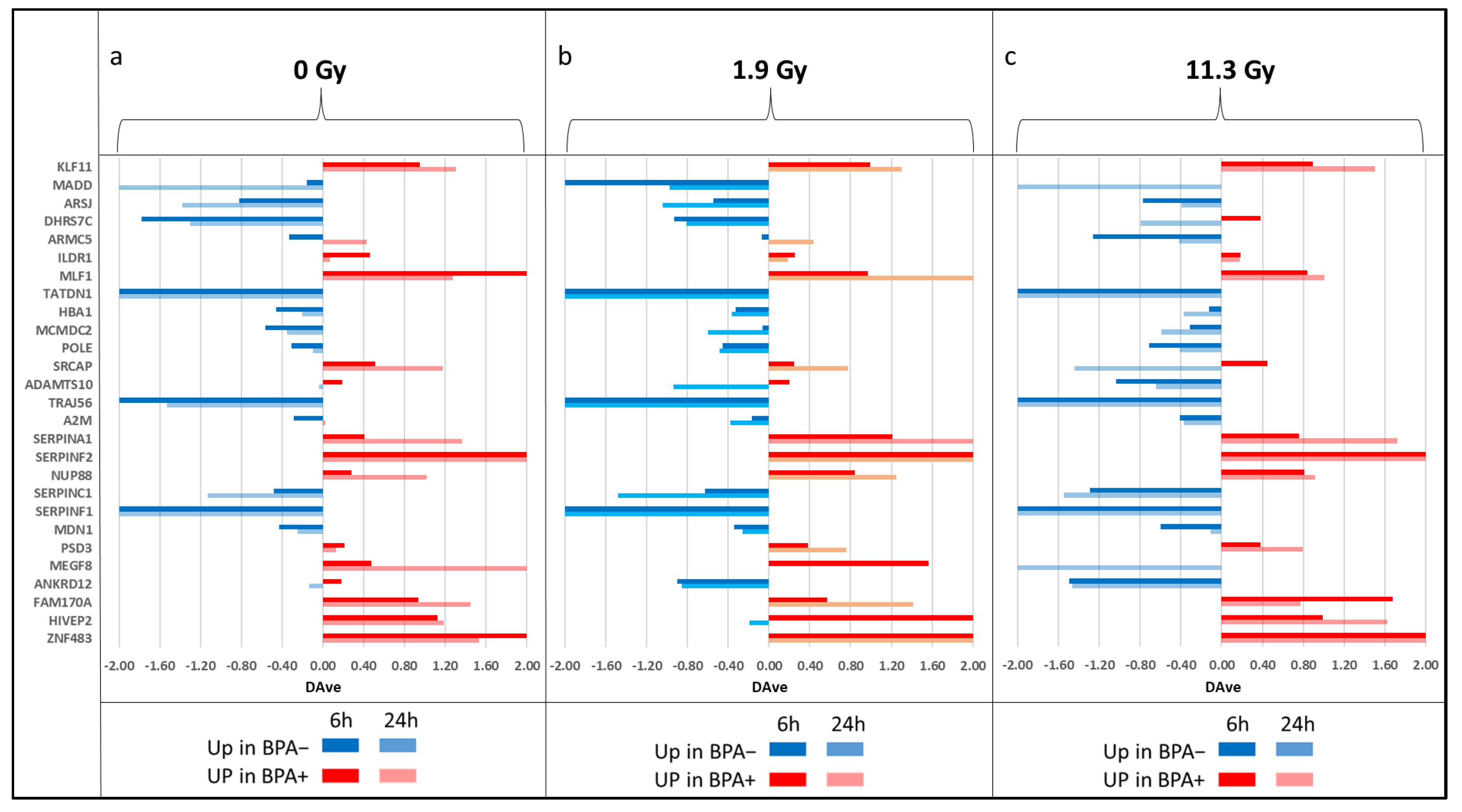
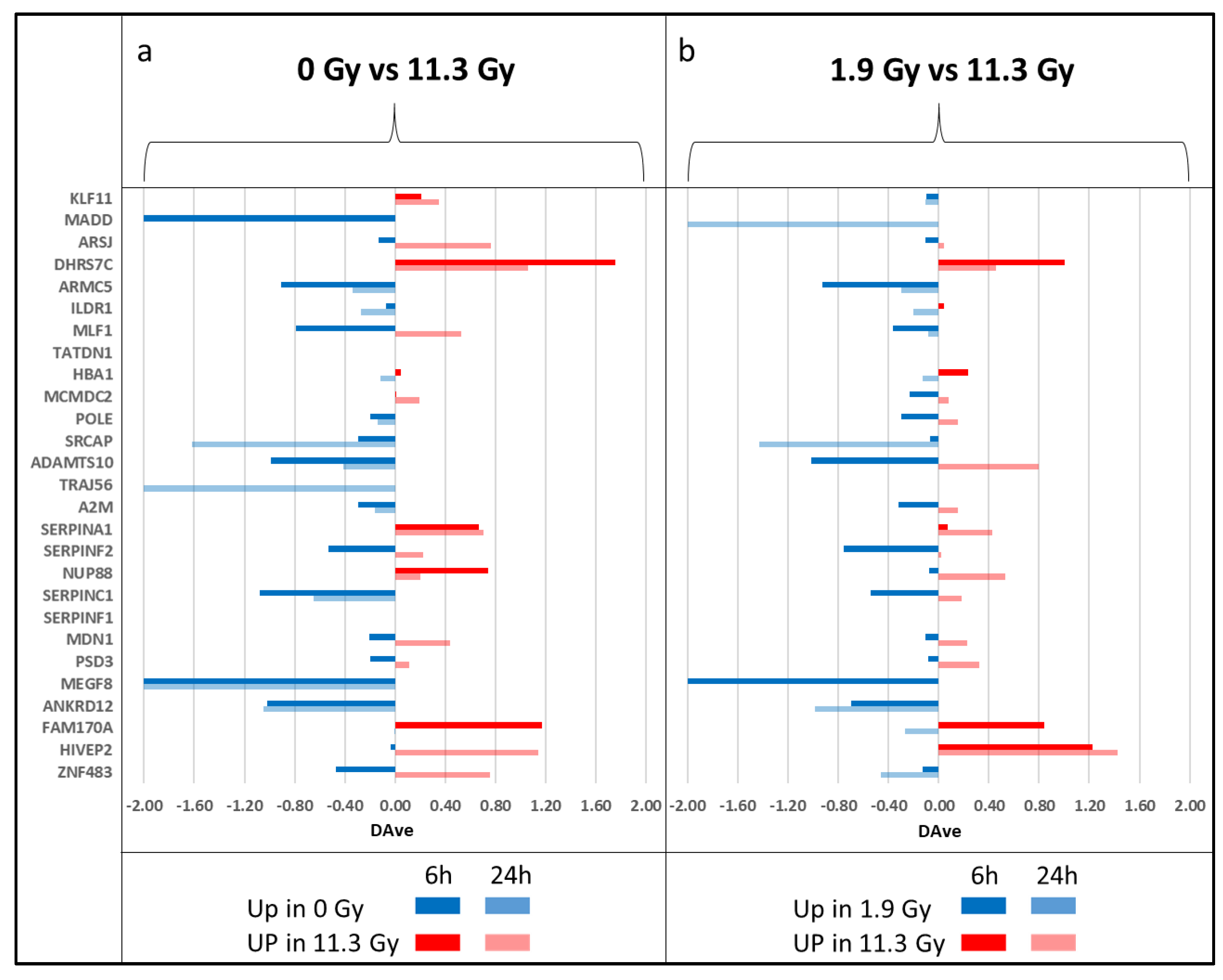
3.3. Differential Analysis of EVs between BPA+ and BPA− Conditions and within BPA+ Conditions between Different Times and Doses of Neutron Irradiation
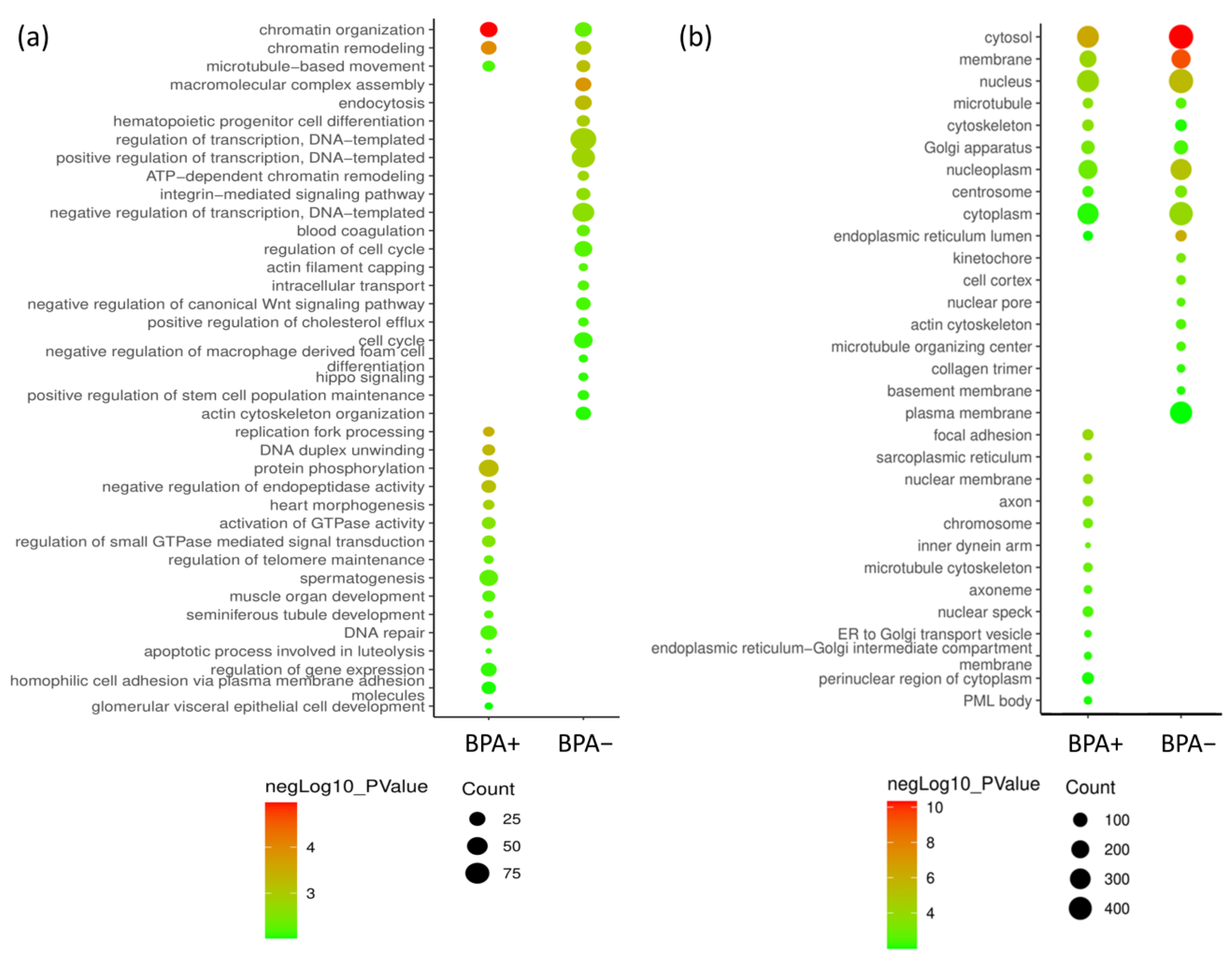
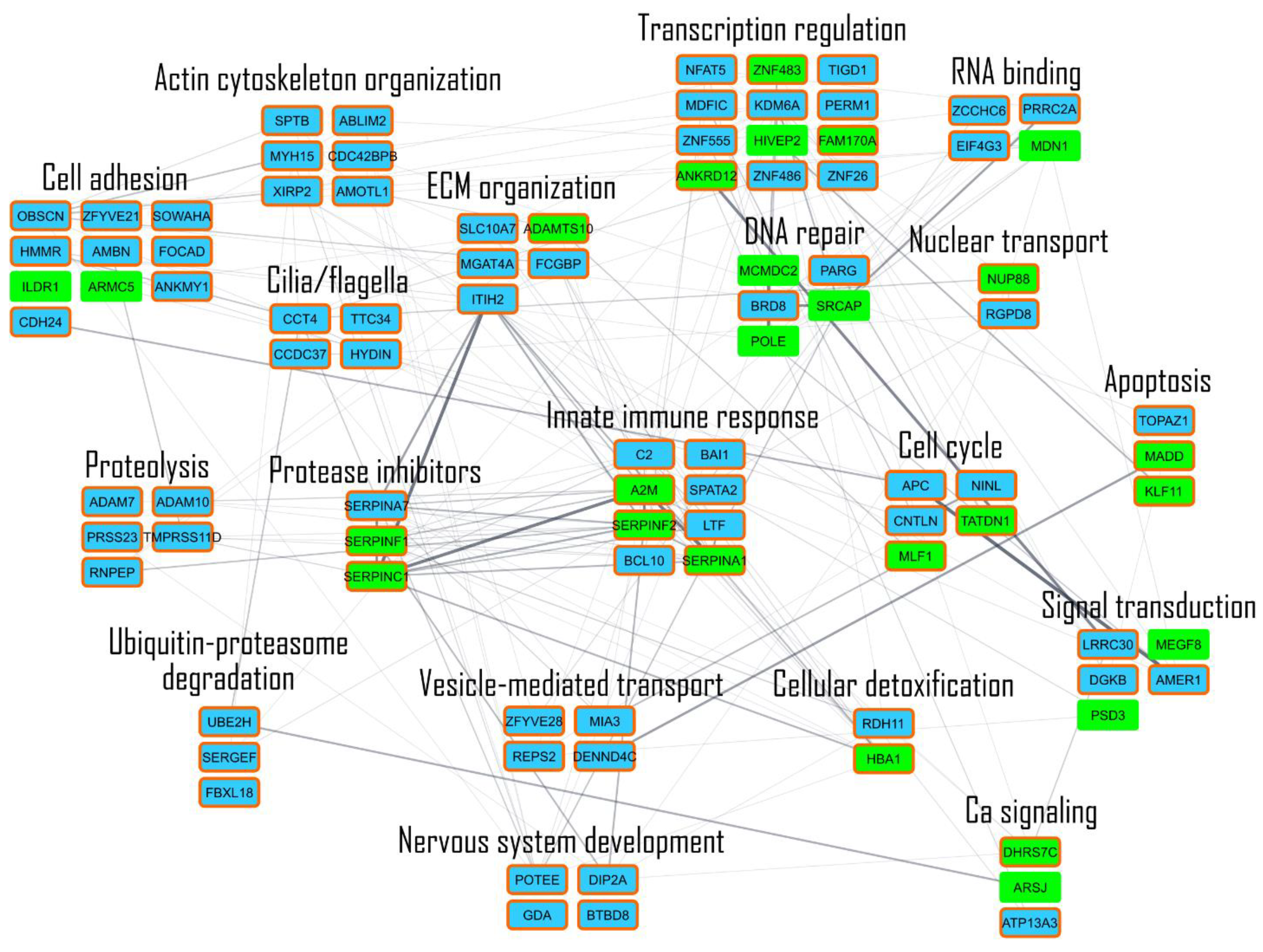
3.4. Network Analysis Shows the Major Pathways Involved in BNCT Effect and How They Are Connected
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suzuki, M. Boron neutron capture therapy (BNCT): A unique role in radiotherapy with a view to entering the accelerator-based BNCT era. Int. J. Clin. Oncol. 2020, 25, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.I.; Wanibuchi, M. Boron neutron capture therapy for malignant brain tumors. J. Neurooncol. 2020, 149, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Sauerwein, W.; Wittig, A. Neutron Capture Therapy Principles and Applications; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2012. [Google Scholar]
- Ferrari, E.; Wittig, A. Urinary Proteomics Profiles Are Useful for Detection of Cancer Biomarkers and Changes Induced by Therapeutic Procedures. Molecules 2019, 24, 794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, A.; Itoh, T. Proteomic analysis of cellular response induced by boron neutron capture reaction in human squamous cell carcinoma SAS cells. Appl. Radiat. Isot. 2015, 106, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Imamichi, S.; Chen, L. Extracellular Release of HMGB1 as an Early Potential Biomarker for the Therapeutic Response in a Xenograft Model of Boron Neutron Capture Therapy. Biology 2022, 11, 420. [Google Scholar] [CrossRef]
- Perico, D.; Di Silvestre, D. Systems Biology Approach to Investigate Biomarkers, Boron-10 Carriers, and Mechanisms Useful for Improving Boron Neutron Capture Therapy. Cancer Biother. Radiopharm. 2022, 38, 152–159. [Google Scholar] [CrossRef]
- Qian, W.J.; Jacobs, J.M. Advances and challenges in liquid chromatography-mass spectrometry-based proteomics profiling for clinical applications. Mol. Cell. Proteom. 2006, 5, 1727–1744. [Google Scholar] [CrossRef] [Green Version]
- Kowalczyk, T.; Ciborowski, M. Mass spectrometry based proteomics and metabolomics in personalized oncology. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165690. [Google Scholar] [CrossRef]
- Simpson, K.L.; Whetton, A.D. Quantitative mass spectrometry-based techniques for clinical use: Biomarker identification and quantification. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 1240–1249. [Google Scholar] [CrossRef]
- Huang, Z.; Ma, L. Proteomic profiling of human plasma for cancer biomarker discovery. Proteomics 2017, 17, 1600240. [Google Scholar] [CrossRef]
- Uluisik, I.; Karakaya, H.C. The importance of boron in biological systems. J. Trace Elem. Med. Biol. 2018, 45, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Hakki, S.S.; Bozkurt, B.S. Boron regulates mineralized tissue-associated proteins in osteoblasts (MC3T3-E1). J. Trace Elem. Med. Biol. 2010, 24, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Rico, P.; Rodrigo-Navarro, A. Borax induces osteogenesis by stimulating NaBC1 transporter via activation of BMP pathway. Commun. Biol. 2020, 3, 717. [Google Scholar] [CrossRef] [PubMed]
- Spielberg, S.P.; Butler, J.D. Treatment of glutathione synthetase deficient fibroblasts by inhibiting gamma-glutamyl transpeptidase activity with serine and borate. Biochem. Biophys. Res. Commun. 1979, 89, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Routray, I.; Ali, S. Boron Induces Lymphocyte Proliferation and Modulates the Priming Effects of Lipopolysaccharide on Macrophages. PLoS ONE 2016, 11, e0150607. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.; Ceresa, D. Comprehensive Profiling of Secretome Formulations from Fetal- and Perinatal Human Amniotic Fluid Stem Cells. Int. J. Mol. Sci. 2021, 22, 3713. [Google Scholar] [CrossRef]
- Barrachina, M.N.; García, Á. Clinical Proteomics for the Analysis of Circulating Extracellular Vesicles. Methods Mol. Biol. 2021, 2259, 13–23. [Google Scholar] [CrossRef]
- Zhang, W.; Ou, X. Proteomics profiling of plasma exosomes in epithelial ovarian cancer: A potential role in the coagulation cascade, diagnosis and prognosis. Int. J. Oncol. 2019, 54, 1719–1733. [Google Scholar] [CrossRef] [Green Version]
- Moreira-Costa, L.; Barros, A.S. Exosome-Derived Mediators as Potential Biomarkers for Cardiovascular Diseases: A Network Approach. Proteomes 2021, 9, 8. [Google Scholar] [CrossRef]
- Rollet-Cohen, V.; Bourderioux, M. Comparative proteomics of respiratory exosomes in cystic fibrosis, primary ciliary dyskinesia and asthma. J. Proteom. 2018, 185, 1–7. [Google Scholar] [CrossRef]
- Muraoka, S.; Jedrychowski, M.P. Proteomic Profiling of Extracellular Vesicles Derived from Cerebrospinal Fluid of Alzheimer’s Disease Patients: A Pilot Study. Cells 2020, 9, 1959. [Google Scholar] [CrossRef] [PubMed]
- Käll, L.; Canterbury, J.D. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 4, 923–925. [Google Scholar] [CrossRef] [PubMed]
- Mauri, P.; Dehò, G. A Proteomic Approach to the Analysis of RNA Degradosome Composition in Escherichia coli. Methods Enzymol. 2008, 447, 99–117. [Google Scholar] [CrossRef]
- De Palma, A.; Fanelli, G. Gcn5p and Ubp8p Affect Protein Ubiquitylation and Cell Proliferation by Altering the Fermentative/Respiratory Flux Balance in Saccharomyces cerevisiae. mBio 2020, 11, e01504-20. [Google Scholar] [CrossRef]
- Vesiclepedia: Home—Extracellular Vesicles Database. Available online: http://microvesicles.org/ (accessed on 1 December 2022).
- FunRich: Functional Enrichment Analysis Tool: Home. Available online: http://www.funrich.org/ (accessed on 1 December 2022).
- Chen, T.; Liu, Y.-X. ImageGP: An easy-to-use data visualization web server for scientific researchers. iMeta 2022, 1, e5. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Morris, J.H. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [Green Version]
- Saito, R.; Smoot, M.E. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Sereni, L.; Castiello, M.C. Autonomous role of Wiskott-Aldrich syndrome platelet deficiency in inducing autoimmunity and inflammation. J. Allergy Clin. Immunol. 2018, 142, 1272–1284. [Google Scholar] [CrossRef] [Green Version]
- Gitaí, D.L.G.; Dos Santos, Y.D.R. Extracellular Vesicles in the Forebrain Display Reduced miR-346 and miR-331-3p in a Rat Model of Chronic Temporal Lobe Epilepsy. Mol. Neurobiol. 2020, 57, 1674–1687. [Google Scholar] [CrossRef]
- Hilario, M.; Kalousis, A. Approaches to dimensionality reduction in proteomic biomarker studies. Brief. Bioinform. 2007, 9, 102–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, B.T.; Hao, M. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Pietrowska, M.; Wlosowicz, A. MS-Based Proteomic Analysis of Serum and Plasma: Problem of High Abundant Components and Lights and Shadows of Albumin Removal. Adv. Exp. Med. Biol. 2019, 1073, 57–76. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Nakajima, D. A Simple Method for In-Depth Proteome Analysis of Mammalian Cell Culture Conditioned Media Containing Fetal Bovine Serum. Int. J. Mol. Sci. 2021, 22, 2565. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Lee, J. Comparative Proteomic Analysis of the Mesenchymal Stem Cells Secretome from Adipose, Bone Marrow, Placenta and Wharton’s Jelly. Int. J. Mol. Sci. 2021, 22, 845. [Google Scholar] [CrossRef]
- Cebeci, E.; Yüksel, B. Anti-cancer effect of boron derivatives on small-cell lung cancer. J. Trace Elem. Med. Biol. 2022, 70, 126923. [Google Scholar] [CrossRef]
- Leach, J.K.; Van Tuyle, G. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001, 61, 3894–3901. [Google Scholar]
- Bettinger, J.Q.; Simon, M. Accurate Proteomewide Measurement of Methionine Oxidation in Aging Mouse Brains. J. Proteome Res. 2022, 21, 1495–1509. [Google Scholar] [CrossRef]
- Dewey, W.C.; Ling, C.C. Radiation-induced apoptosis: Relevance to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 1995, 33, 781–796. [Google Scholar] [CrossRef]
- Veuger, S.J.; Curtin, N.J. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res. 2003, 63, 6008–6015. [Google Scholar] [PubMed]
- Liu, H.Y.; Liu, Y.Y. Acetylation of MORC2 by NAT10 regulates cell-cycle checkpoint control and resistance to DNA-damaging chemotherapy and radiotherapy in breast cancer. Nucleic Acids Res. 2020, 48, 3638–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, M.; Patin, E.C. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 2020, 20, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Lucas, A.; Yaron, J.R. Overview of Serpins and Their Roles in Biological Systems. Methods Mol. Biol. 2018, 1826, 1–7. [Google Scholar] [CrossRef]
- Zheng, D.; Chen, H. Serpins for diagnosis and therapy in cancer. Cardiovasc. Hematol. Disord. Drug Targets 2013, 13, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Kajiya, A. Aberrant expression of serpin squamous cell carcinoma antigen 2 in human tumor tissues and cell lines: Evidence of protection from tumor necrosis factor-mediated apoptosis. Biol. Chem. 2002, 383, 1231–1236. [Google Scholar] [CrossRef]
- Zhang, T.; Guan, M. Pigment epithelium-derived factor inhibits glioma cell growth in vitro and in vivo. Life Sci. 2007, 81, 1256–1263. [Google Scholar] [CrossRef]
- Miao, R.Q.; Agata, J. Kallistatin is a new inhibitor of angiogenesis and tumor growth. Blood 2002, 100, 3245–3252. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Li, H. SERPINB2 down-regulation contributes to chemoresistance in head and neck cancer. Mol. Carcinog. 2014, 53, 777–786. [Google Scholar] [CrossRef]
- Pavón, M.A.; Arroyo-Solera, I. uPA/uPAR and SERPINE1 in head and neck cancer: Role in tumor resistance, metastasis, prognosis and therapy. Oncotarget 2016, 7, 57351–57366. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.W.; Chang, K.P. Identification of Salivary Biomarkers for Oral Cancer Detection with Untargeted and Targeted Quantitative Proteomics Approaches. Mol. Cell. Proteom. 2019, 18, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, R.; Bollinger, J.G. A targeted proteomic strategy for the measurement of oral cancer candidate biomarkers in human saliva. Proteomics 2016, 16, 159–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strybel, U.; Marczak, L. Molecular Composition of Serum Exosomes Could Discriminate Rectal Cancer Patients with Different Responses to Neoadjuvant Radiotherapy. Cancers 2022, 14, 993. [Google Scholar] [CrossRef] [PubMed]
- Drobin, K.; Marczyk, M. Molecular Profiling for Predictors of Radiosensitivity in Patients with Breast or Head-and-Neck Cancer. Cancers 2020, 12, 753. [Google Scholar] [CrossRef] [Green Version]
- Dillon, M.T.; Bergerhoff, K.F. ATR Inhibition Potentiates the Radiation-induced Inflammatory Tumor Microenvironment. Clin. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Riaz, N. Functional landscapes of POLE and POLD1 mutations in checkpoint blockade-dependent antitumor immunity. Nat. Genet. 2022, 54, 996–1012. [Google Scholar] [CrossRef]
- Dong, S.; Han, J. The human SRCAP chromatin remodeling complex promotes DNA-end resection. Curr. Biol. 2014, 24, 2097–2110. [Google Scholar] [CrossRef] [Green Version]
- Kondo, N. DNA damage and biological responses induced by Boron Neutron Capture Therapy (BNCT). Enzymes 2022, 51, 65–78. [Google Scholar] [CrossRef]
- Huang, C.Y.; Lai, Z.Y. Boron Neutron Capture Therapy Eliminates Radioresistant Liver Cancer Cells by Targeting DNA Damage and Repair Responses. J. Hepatocell. Carcinoma 2022, 9, 1385–1401. [Google Scholar] [CrossRef]
- Fujita, Y.; Kato, I. Role of p53 mutation in the effect of boron neutron capture therapy on oral squamous cell carcinoma. Radiat. Oncol. 2009, 4, 63. [Google Scholar] [CrossRef] [Green Version]
- Faião-Flores, F.; Coelho, P.R. Apoptosis through Bcl-2/Bax and cleaved caspase up-regulation in melanoma treated by boron neutron capture therapy. PLoS ONE 2013, 8, e59639. [Google Scholar] [CrossRef]
- Wang, P.; Zhen, H. Boron neutron capture therapy induces apoptosis of glioma cells through Bcl-2/Bax. BMC Cancer 2010, 10, 661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, B.; Jeon, J. Radiotherapy in combination with hyperthermia suppresses lung cancer progression via increased NR4A3 and KLF11 expression. Int. J. Radiat. Biol. 2019, 95, 1696–1707. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, H. Use of exosome transcriptome-based analysis to identify novel biomarkers in patients with locally advanced esophageal squamous cell carcinoma undergoing neoadjuvant chemoradiotherapy. Ann. Transl. Med. 2023, 11, 182. [Google Scholar] [CrossRef] [PubMed]
- Kononikhin, A.S.; Brzhozovskiy, A.G. Targeted MRM Quantification of Urinary Proteins in Chronic Kidney Disease Caused by Glomerulopathies. Molecules 2023, 28, 3323. [Google Scholar] [CrossRef] [PubMed]
- Canetti, D.; Brambilla, F. Clinical Amyloid Typing by Proteomics: Performance Evaluation and Data Sharing between Two Centres. Molecules 2021, 26, 1913. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Irradiated Time (min) | Inoculated Cell Numbers | Cell Growth Ratio * | |
|---|---|---|---|
| BPA+ | BPA− | ||
| 0 | 1000 | 1.00 ± 0.04 | 1.00 ± 0.11 |
| 10 | 1000 | 0.40 ± 0.20 | 1.11 ± 0.15 |
| 60 | 5000 | <6.5 × 10−4 | 0.10 ± 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perico, D.; Tong, Y.; Chen, L.; Imamichi, S.; Sanada, Y.; Ishiai, M.; Suzuki, M.; Masutani, M.; Mauri, P. Proteomic Characterization of SAS Cell-Derived Extracellular Vesicles in Relation to Both BPA and Neutron Irradiation Doses. Cells 2023, 12, 1562. https://doi.org/10.3390/cells12121562
Perico D, Tong Y, Chen L, Imamichi S, Sanada Y, Ishiai M, Suzuki M, Masutani M, Mauri P. Proteomic Characterization of SAS Cell-Derived Extracellular Vesicles in Relation to Both BPA and Neutron Irradiation Doses. Cells. 2023; 12(12):1562. https://doi.org/10.3390/cells12121562
Chicago/Turabian StylePerico, Davide, Ying Tong, Lichao Chen, Shoji Imamichi, Yu Sanada, Masamichi Ishiai, Minoru Suzuki, Mitsuko Masutani, and Pierluigi Mauri. 2023. "Proteomic Characterization of SAS Cell-Derived Extracellular Vesicles in Relation to Both BPA and Neutron Irradiation Doses" Cells 12, no. 12: 1562. https://doi.org/10.3390/cells12121562
APA StylePerico, D., Tong, Y., Chen, L., Imamichi, S., Sanada, Y., Ishiai, M., Suzuki, M., Masutani, M., & Mauri, P. (2023). Proteomic Characterization of SAS Cell-Derived Extracellular Vesicles in Relation to Both BPA and Neutron Irradiation Doses. Cells, 12(12), 1562. https://doi.org/10.3390/cells12121562