


Phylogenomic Analysis of micro-RNA Involved in Juvenile to Flowering-Stage Transition in Photophilic Rice and Its Sister Species

 , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval of MIR172 Homologs
2.2. MIRNA Precursor Sequence Analysis
2.3. Microsynteny Analysis
2.4. Plant Material, PCR, and Sequencing of Precursor MIR172
2.5. Expression Analysis of Mature MIR172
2.6. Phylogenomic Analysis of Precursor Sequences
2.7. Test of Neutrality
3. Results
3.1. Identification of MIR172 Homologs
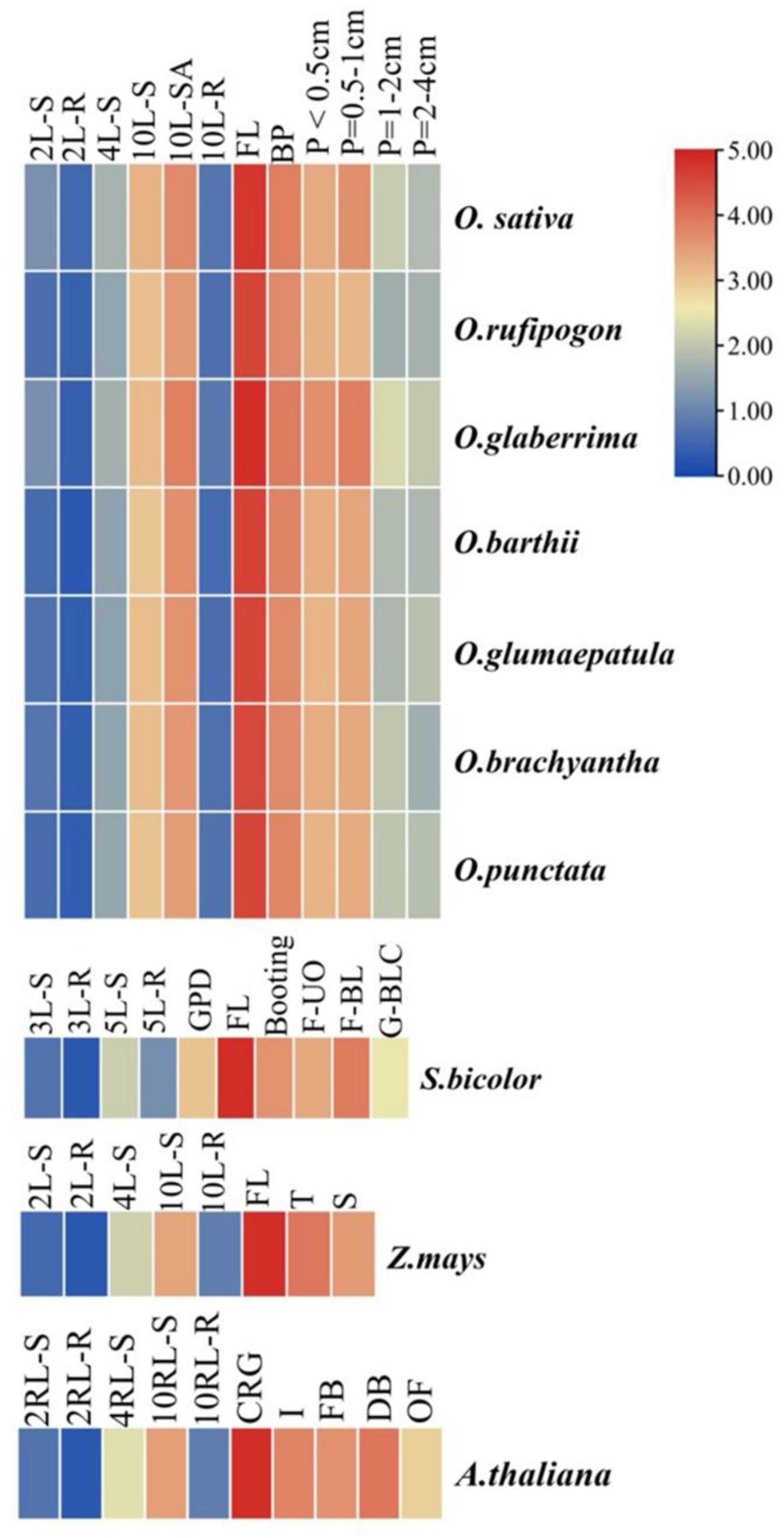
3.2. Expression Analysis of MIR172
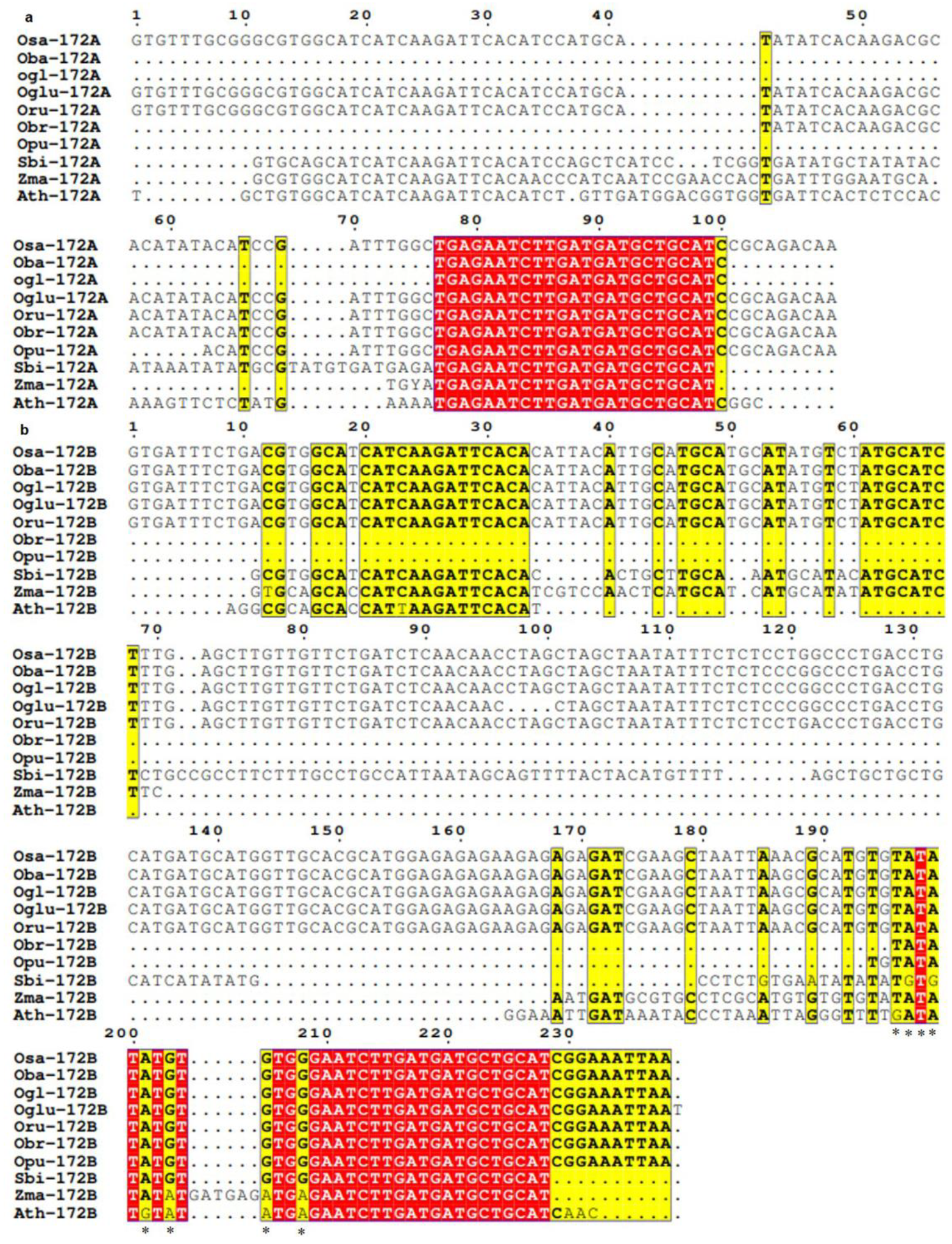
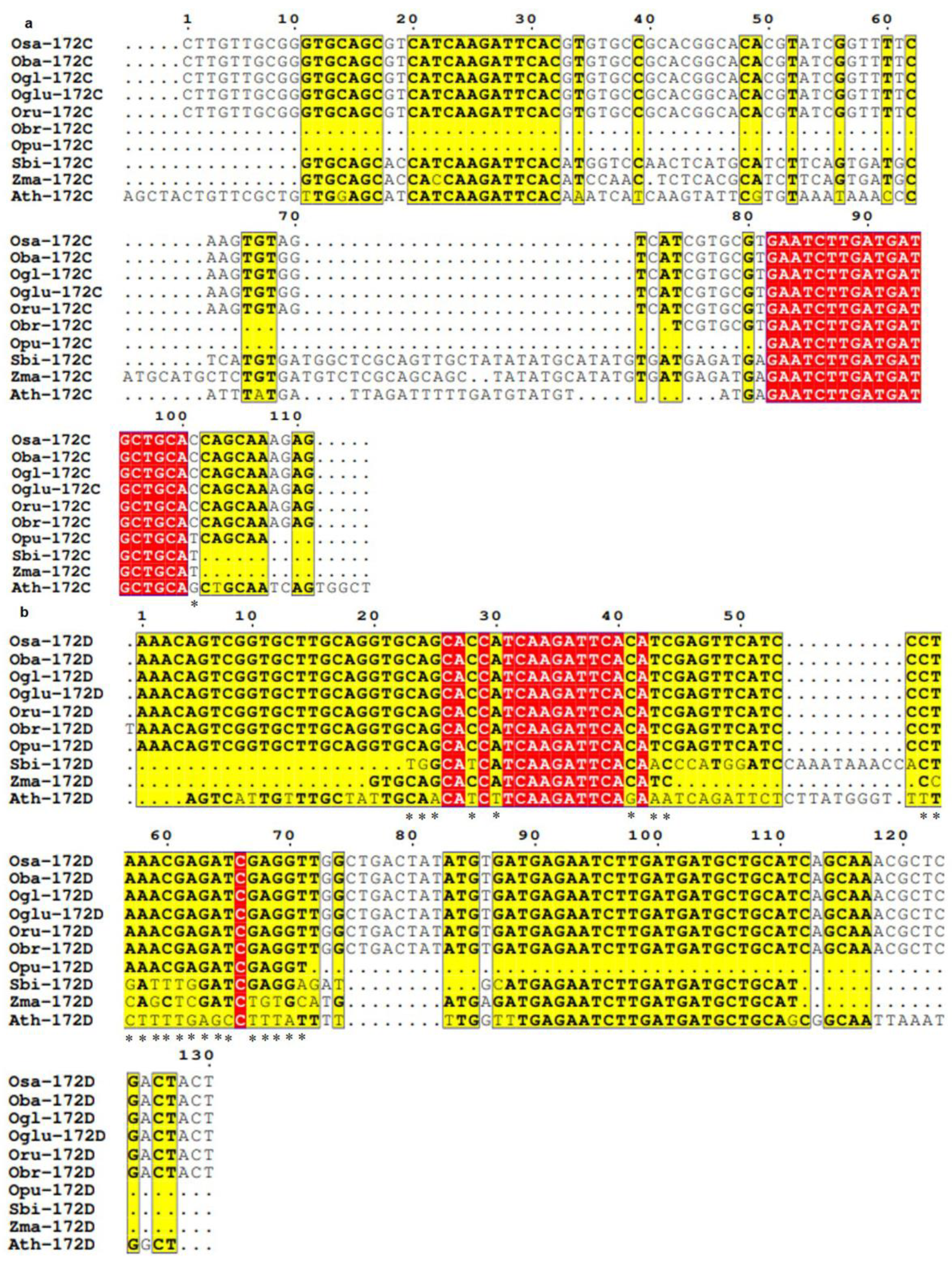
3.3. Conservation and Divergence in Mature and Precursor Sequence of MIR172
3.4. Spectrum of Sequence Variation in MIR172 Homologs
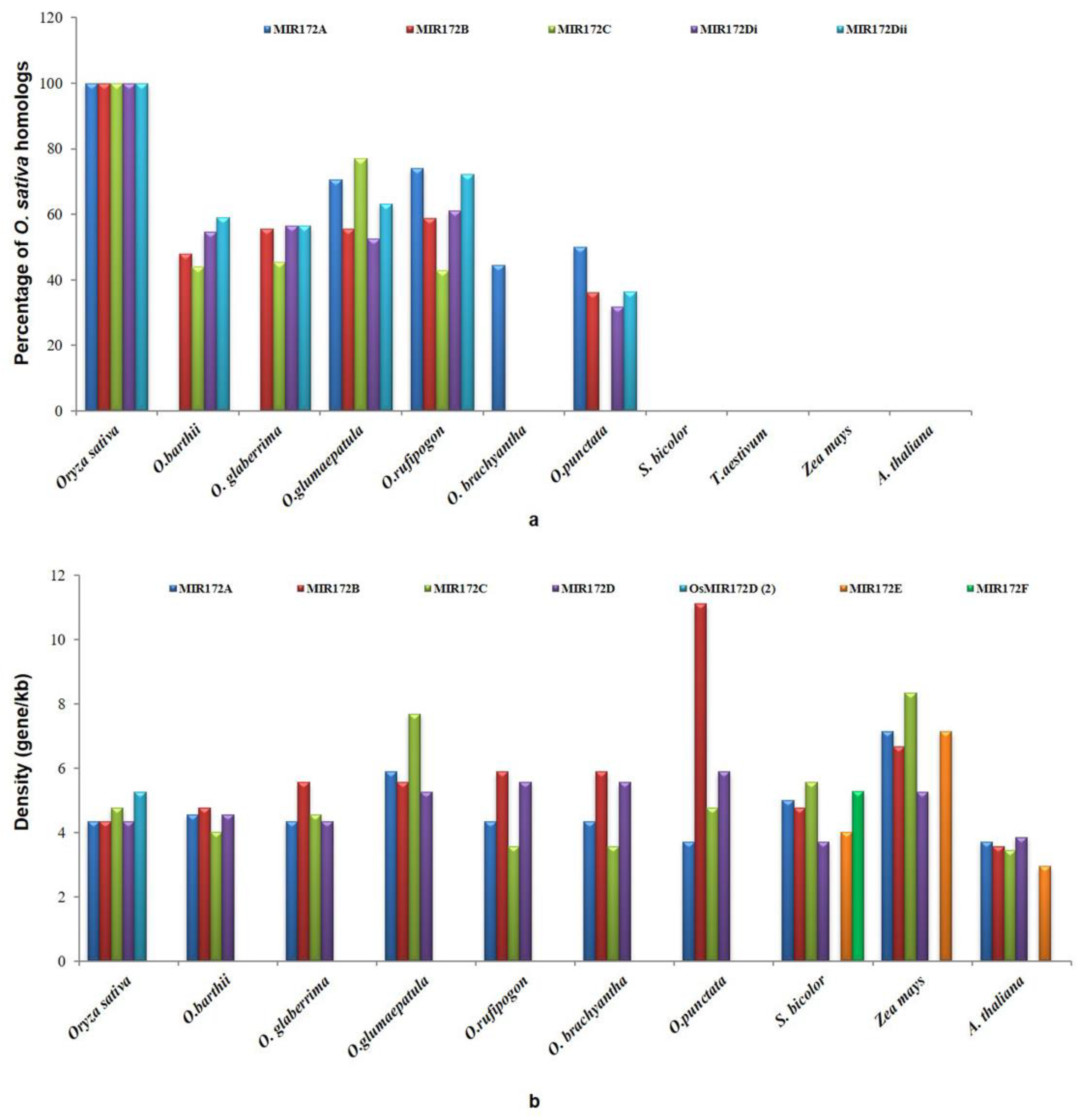
3.5. Gene Conservation and Gene Density Analysis
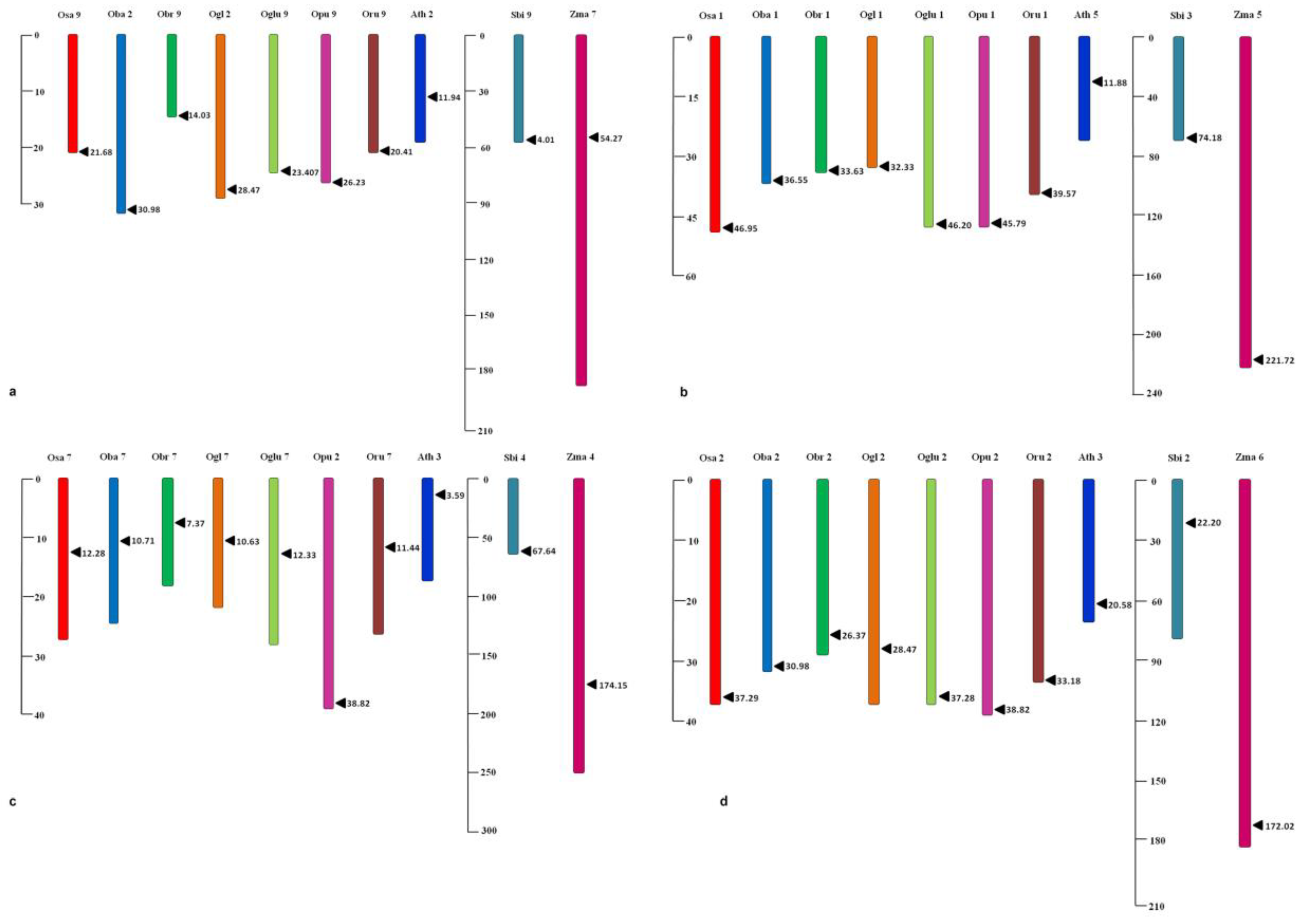
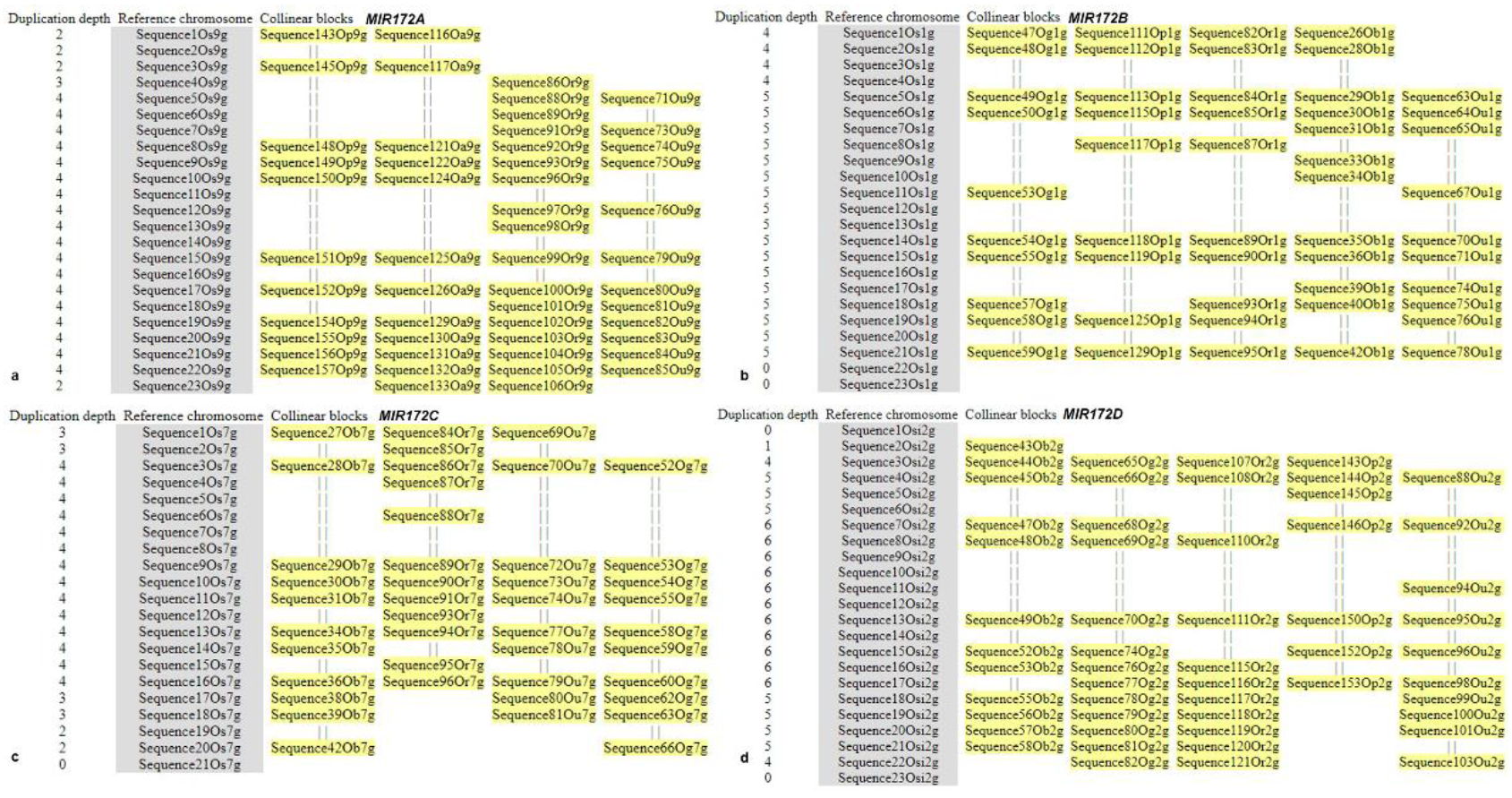
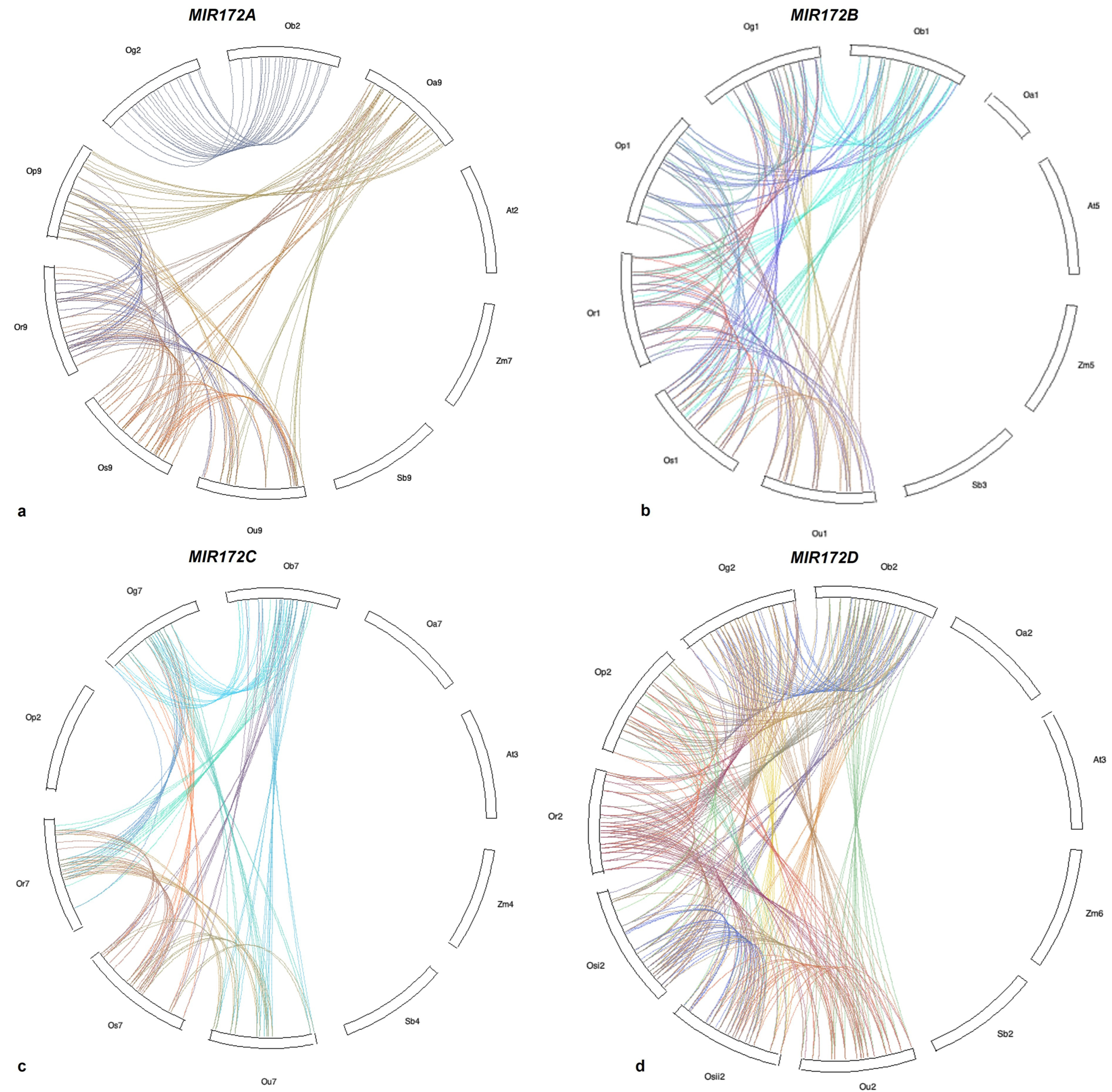
3.6. Microsynteny Analysis
- (i)
- MIR172A
- (ii)
- MIR172B
- (iii)
- MIR172C
- (iv)
- MIR172D
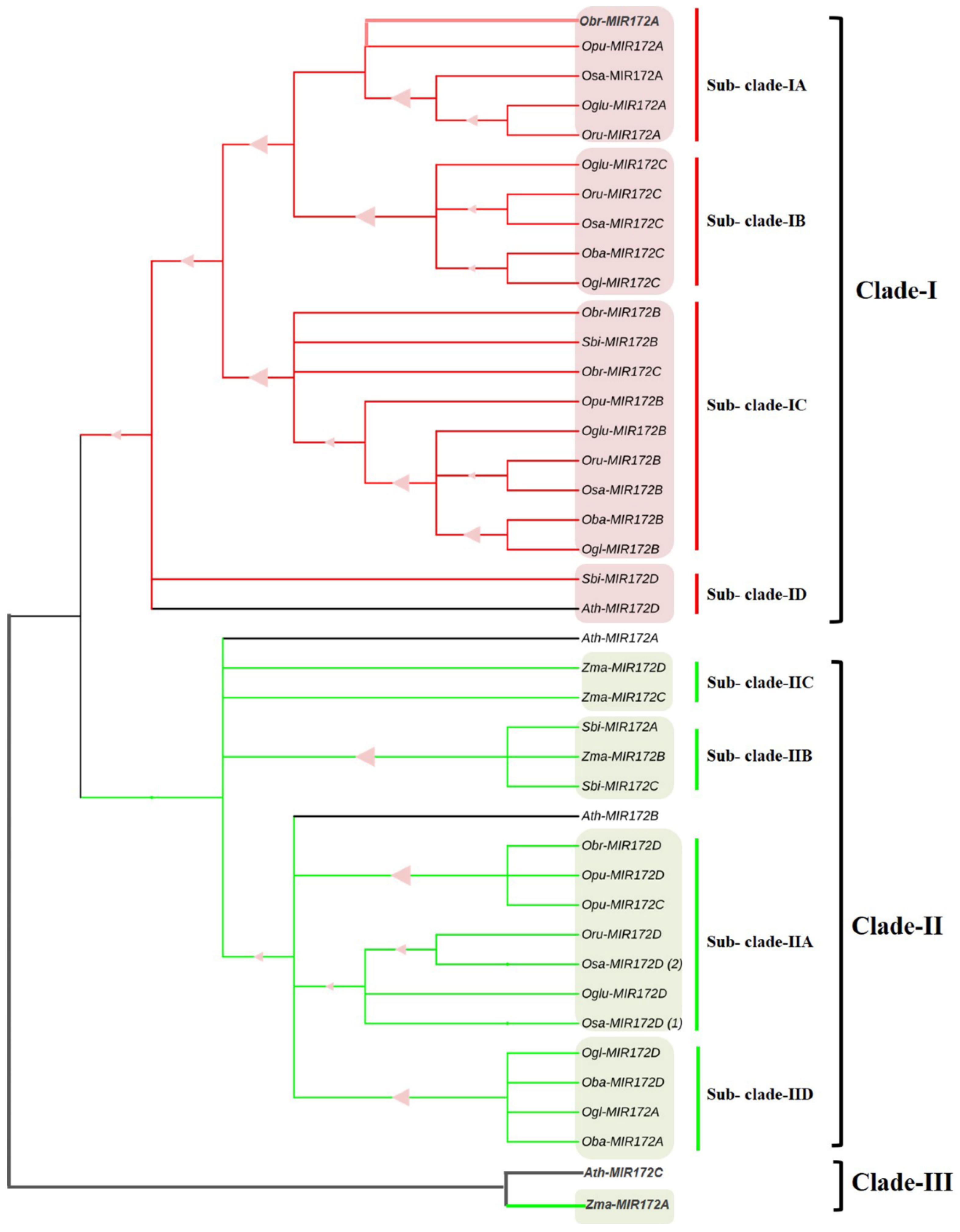
3.7. Phylogenomic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tang, M.; Bai, X.; Niu, L.-J.; Chai, X.; Chen, M.-S.; Xu, Z.-F. miR172 Regulates both Vegetative and Reproductive Development in the Perennial Woody Plant Jatropha curcas. Plant Cell Physiol. 2018, 59, 2549–2563. [Google Scholar] [CrossRef]
- Anwar, N.; Ohta, M.; Yazawa, T.; Sato, Y.; Li, C.; Tagiri, A.; Sakuma, M.; Nussbaumer, T.; Bregitzer, P.; Pourkheirandish, M.; et al. miR172 downregulates the translation of cleistogamy 1 in barley. Ann. Bot. 2018, 122, 251–265. [Google Scholar] [CrossRef]
- Dhaka, N.; Sharma, R. MicroRNA-mediated regulation of agronomically important seed traits: A treasure trove with shades of grey! Crit. Rev. Biotechnol. 2021, 41, 594–608. [Google Scholar] [CrossRef]
- Joshi, G.; Chauhan, C.; Das, S. Microsynteny analysis to understand evolution and impact of polyploidization on MIR319 family within Brassicaceae. Dev. Genes Evol. 2018, 228, 227–242. [Google Scholar] [CrossRef]
- Dash, P.K.; Gupta, P.; Pradhan, S.K.; Shasany, A.K.; Rai, R. Analysis of Homologous Regions of Small RNAs MIR397 and MIR408 Reveals the Conservation of Microsynteny among Rice Crop-Wild Relatives. Cells 2022, 11, 3461. [Google Scholar] [CrossRef]
- Moss, E.G. Heterochronic Genes and the Nature of Developmental Time. Curr. Biol. 2007, 17, R425–R434. [Google Scholar] [CrossRef]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.-W.; Weigel, D.; Poethig, R.S. The Sequential Action of miR156 and miR172 Regulates Developmental Timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef]
- Aukerman, M.J.; Sakai, H. Regulation of flowering time and floral organ identity by a microRNA and its APETALA2-like target genes. Plant Cell 2003, 15, 2730–2741. [Google Scholar] [CrossRef]
- Lauter, N.; Kampani, A.; Carlson, S.; Goebel, M.; Moose, S.P. microRNA172 down-regulates glossy15 to promote vegetative phase change in maize. Proc. Natl. Acad. Sci. USA 2005, 102, 9412–9417. [Google Scholar] [CrossRef]
- Wang, J.-W.; Park, M.Y.; Wang, L.-J.; Koo, Y.; Chen, X.-Y.; Weigel, D.; Poethig, R.S. MiRNA Control of Vegetative Phase Change in Trees. PLoS Genet. 2011, 7, e1002012. [Google Scholar] [CrossRef]
- Park, W.; Li, J.; Song, R.; Messing, J.; Chen, X. CARPEL FACTORY, a Dicer Homolog, and HEN1, a Novel Protein, Act in microRNA Metabolism in Arabidopsis thaliana. Curr. Biol. 2002, 12, 1484–1495. [Google Scholar] [CrossRef]
- Fornara, F.; Coupland, G. Plant Phase Transitions Make a SPLash. Cell 2009, 138, 625–627. [Google Scholar] [CrossRef]
- Zhu, Q.-H.; Helliwell, C.A. Regulation of flowering time and floral patterning by miR172. J. Exp. Bot. 2011, 62, 487–495. [Google Scholar] [CrossRef]
- Lee, D.-Y.; An, G. Two AP2 family genes, SUPERNUMERARY BRACT (SNB) and OsINDETERMINATE SPIKELET 1 (OsIDS1), synergistically control inflorescence architecture and floral meristem establishment in rice. Plant J. 2012, 69, 445–461. [Google Scholar] [CrossRef]
- Zeng, C.; Wang, W.; Zheng, Y.; Chen, X.; Bo, W.; Song, S.; Zhang, W.; Peng, M. Conservation and divergence of microRNAs and their functions in Euphorbiaceous plants. Nucleic Acids Res. 2010, 38, 981–995. [Google Scholar] [CrossRef]
- Zhao, L.; Kim, Y.; Dinh, T.T.; Chen, X. miR172 regulates stem cell fate and defines the inner boundary of APETALA3 and PISTILLATA expression domain in Arabidopsis floral meristems. Plant J. 2007, 51, 840–849. [Google Scholar] [CrossRef]
- Fouracre, J.P.; Poethig, R.S. The role of small RNAs in vegetative shoot development. Curr. Opin. Plant Biol. 2016, 29, 64–72. [Google Scholar] [CrossRef]
- Yu, S.; Lian, H.; Wang, J.-W. Plant developmental transitions: The role of microRNAs and sugars. Curr. Opin. Plant Biol. 2015, 27, 1–7. [Google Scholar] [CrossRef]
- Jung, J.-H.; Seo, P.J.; Kang, S.K.; Park, C.-M. miR172 signals are incorporated into the miR156 signaling pathway at the SPL3/4/5 genes in Arabidopsis developmental transitions. Plant Mol. Biol. 2011, 76, 35–45. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Lee, D.-Y.; Cho, L.-H.; An, G. Rice miR172 induces flowering by suppressing OsIDS1 and SNB, two AP2 genes that negatively regulate expression of Ehd1 and florigens. Rice 2014, 7, 31. [Google Scholar] [CrossRef]
- Tang, J.; Chu, C. MicroRNAs in crop improvement: Fine-tuners for complex traits. Nat. Plants 2017, 3, 17077. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, X. Secrets of the MIR172 family in plant development and flowering unveiled. PLoS Biol. 2021, 19, e3001099. [Google Scholar] [CrossRef]
- Gasser, C. Fruit development: miRNA pumps up the volume. Nat. Plants 2015, 1, 15037. [Google Scholar] [CrossRef]
- Ripoll, J.J.; Bailey, L.J.; Mai, Q.-A.; Wu, S.L.; Hon, C.T.; Chapman, E.J.; Ditta, G.S.; Estelle, M.; Yanofsky, M.F. microRNA regulation of fruit growth. Nat. Plants 2015, 1, 15036. [Google Scholar] [CrossRef]
- Debernardi, J.M.; Lin, H.; Chuck, G.; Faris, J.D.; Dubcovsky, J. microRNA172 plays a crucial role in wheat spike morphogen-esis and grain threshability. Development 2017, 144, 1966–1975. [Google Scholar] [CrossRef]
- Liu, P.; Liu, J.; Dong, H.; Sun, J. Functional regulation of Q by microRNA172 and transcriptional co-repressor TOPLESS in controlling bread wheat spikelet density. Plant Biotechnol. J. 2018, 16, 495–506. [Google Scholar] [CrossRef]
- D’ario, M.; Griffiths-Jones, S.; Kim, M. Small RNAs: Big Impact on Plant Development. Trends Plant Sci. 2017, 22, 1056–1068. [Google Scholar] [CrossRef]
- Yan, Z.; Hossain, S.; Wang, J.; Valdés-López, O.; Liang, Y.; Libault, M.; Qiu, L.; Stacey, G. miR172 Regulates Soybean Nodulation. Mol. Plant-Microbe Interact. 2013, 26, 1371–1377. [Google Scholar] [CrossRef]
- Li, W.; Wang, T.; Zhang, Y.; Li, Y. Overexpression of soybean miR172c confers tolerance to water deficit and salt stress, but increases ABA sensitivity in transgenic Arabidopsis thaliana. J. Exp. Bot. 2016, 67, 175–194. [Google Scholar] [CrossRef]
- Luan, Y.; Cui, J.; Li, J.; Jiang, N.; Liu, P.; Meng, J. Effective enhancement of resistance to Phytophthora infestans by overexpression of miR172a and b in Solanum lycopersicum. Planta 2018, 247, 127–138. [Google Scholar] [CrossRef]
- Dash, P.K.; Rai, R. Green revolution to grain revolution: Florigen in the frontiers. J. Biotechnol. 2021, 343, 38–46. [Google Scholar] [CrossRef]
- Dash, P.K.; Rai, R.; Mahato, A.K.; Gaikwad, K.; Singh, N.K. Transcriptome Landscape at Different Developmental Stages of a Drought Tolerant Cultivar of Flax (Linum usitatissimum). Front. Chem. 2017, 5, 82. [Google Scholar] [CrossRef]
- Shivaraj, S.M.; Deshmukh, R.K.; Rai, R.; Bélanger, R.; Agrawal, P.K.; Dash, P.K. Genome-wide identification, characterization, and expression profile of aquaporin gene family in flax (Linum usitatissimum). Sci. Rep. 2017, 7, srep46137. [Google Scholar] [CrossRef]
- Dash, P.K.; Cao, Y.; Jailani, A.K.; Gupta, P.; Venglat, P.; Xiang, D.; Rai, R.; Sharma, R.; Thirunavukkarasu, N.; Abdin, M.Z.; et al. Genome-wide analysis of drought induced gene expression changes in flax (Linum usitatissimum). GM Crop. Food 2014, 5, 106–119. [Google Scholar] [CrossRef]
- Wang, Z.; Hobson, N.; Galindo, L.; Zhu, S.; Shi, D.; McDill, J.; Yang, L.; Hawkins, S.; Neutelings, G.; Datla, R.; et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012, 72, 461–473. [Google Scholar] [CrossRef]
- Dash, P.K.; Gupta, P.; Rai, R. Hydroponic method of halophobic response elicitation in flax (Linum usitatissimum) for precise down-stream gene expression studies. Int. J. Tropic. Agric. 2015, 33, 1079–1085. [Google Scholar]
- Gupta, P.; Dash, P.K. Molecular details of secretory phospholipase A2 from flax (Linum usitatissimum L.) provide insight into its structure and function. Sci. Rep. 2017, 7, 11080. [Google Scholar] [CrossRef]
- Gupta, P.; Dash, P.K. Precise method of in situ drought stress induction in flax (Linum usitatissimum) for RNA isolation towards down-stream analysis. An. Agric. Res. 2015, 36, 10–17. [Google Scholar]
- Dash, P.K.; Gupta, P.; Jailani, A.K.; Rai, R. Hydropenia induces expression of drought responsive genes (DRGs) erd1, hat, plD-δ, and zfa in Linum usitatissimum L. Indian J. Exp. Biol. 2018, 56, 743–749. [Google Scholar]
- Gupta, P.; Rai, R.; Vasudev, S.; Yadava, D.K.; Dash, P.K. Ex-foliar application of glycine betaine and its impact on protein, carbohydrates and induction of ROS scavenging system during drought stress in flax (Linum usitatissimum). J. Biotechnol. 2021, 337, 80–89. [Google Scholar] [CrossRef]
- Duong, H.V.; Repalli, S.K.; Gupta, P.; Sreevathsa, R.; Yadava, D.K.; Dash, P. Cloning and structural elucidation of a brassinosteroids biosynthetic gene (Atdwarf4) and genetic transformation of Indian mustard (Brassica juncea L.). Indian J. Biochem. Biophys. 2022, 59, 320–330. [Google Scholar] [CrossRef]
- Sahu, S.; Gupta, P.; Gowtham, T.; Yogesh, K.; Sanjay, T.; Singh, A.; Duong, H.V.; Pradhan, S.K.; Bisht, D.S.; Singh, N.K.; et al. Generation of High-Value Genomic Resource in Rice: A “Sub-Genomic Library” of Low-Light-Tolerant Rice Cultivar Swarnaprabha. Biology 2023, 12, 428. [Google Scholar] [CrossRef]
- Tyagi, S.; Rathinam, M.; Dokka, N.; Chaudhary, N.; Satish, L.; Dash, P.K.; Shasany, A.K.; Sreevathsa, R. Cajanus platycarpus Flavonoid 3′5′ Hydroxylase_2 (CpF3′5′H_2) Confers Resistance to Helicoverpa armigera by Modulating Total Polyphenols and Flavonoids in Transgenic Tobacco. Int. J. Mol. Sci. 2023, 24, 1755. [Google Scholar] [CrossRef]
- Gupta, P.; Dash, P.K.; Sanjay, T.D.; Pradhan, S.K.; Sreevathsa, R.; Rai, R. Cloning and Molecular Characterization of the phlD Gene Involved in the Biosynthesis of “Phloroglucinol”, a Compound with Antibiotic Properties from Plant Growth Promoting Bacteria Pseudomonas spp. Antibiotics 2023, 12, 260. [Google Scholar] [CrossRef]
- Gupta, P.; Karthik, K.; Sreevathsa, R.; Rai, R.; Dash, P.K. Cloning and characterization of phloroglucinol biosynthetic gene phlC from anIndian strain of Pseudomonas fluorescens. Experiment 2022, 60, 607–614. [Google Scholar] [CrossRef]
- Rathinam, M.; Tyagi, S.; Dokka, N.; Marimuthu, S.K.; Kumar, H.; Sagar, D.; Dash, P.K.; Shasany, A.K.; Sreevathsa, R. The plant specialized metabolite epicatechin-3-gallate (EC3G) perturbs lipid metabolism and attenuates fat accumulation in pigeonpea pod borer, Helicoverpa armigera. Int. J. Biol. Macromol. 2023, 231, 123325. [Google Scholar] [CrossRef]
- Pradhan, S.K.; Pandit, E.; Pawar, S.; Bharati, B.; Chatopadhyay, K.; Singh, S.; Dash, P.; Reddy, J.N. Association mapping reveals multiple QTLs for grain protein content in rice useful for biofortification. Mol. Genet. Genom. 2019, 294, 963–983. [Google Scholar] [CrossRef]
- Bastia, R.; Pandit, E.; Sanghamitra, P.; Barik, S.R.; Nayak, D.K.; Sahoo, A.; Moharana, A.; Meher, J.; Dash, P.K.; Raj, R.; et al. Association Mapping for Quantitative Trait Loci Controlling Superoxide Dismutase, Flavonoids, Anthocyanins, Carotenoids, γ-Oryzanol and Antioxidant Activity in Rice. Agronomy 2022, 12, 3036. [Google Scholar] [CrossRef]
- Pradhan, K.C.; Pandit, E.; Mohanty, S.P.; Moharana, A.; Sanghamitra, P.; Meher, J.; Jena, B.K.; Dash, P.K.; Behera, L.; Mohapatra, P.M.; et al. Development of Broad Spectrum and Durable Bacterial Blight Resistant Variety through Pyramiding of Four Resistance Genes in Rice. Agronomy 2022, 12, 1903. [Google Scholar] [CrossRef]
- Mohapatra, S.; Barik, S.R.; Dash, P.K.; Lenka, D.; Pradhan, K.C.; Raj, R.R.K.; Mohanty, S.P.; Mohanty, M.R.; Sahoo, A.; Jena, B.K.; et al. Molecular Breeding for Incorporation of Submergence Tolerance and Durable Bacterial Blight Resistance into the Popular Rice Variety ‘Ranidhan’. Biomolecules 2023, 13, 198. [Google Scholar] [CrossRef]
- Shenton, M.; Kobayashi, M.; Terashima, S.; Ohyanagi, H.; Copetti, D.; Hernández-Hernández, T.; Zhang, J.; Ohmido, N.; Fujita, M.; Toyoda, A.; et al. Evolution and diversity of the wild rice Oryza officinalis complex, across continents genome types, and ploidy levels. Genome Biol. Evol. 2020, 12, 413–428. [Google Scholar] [CrossRef]
- Ray, D.K.; Mueller, N.D.; West, P.C.; Foley, J.A. Yield trends are insufficient to double global crop production by 2050. PLoS ONE 2013, 8, e66428. [Google Scholar] [CrossRef]
- Mammadov, J.; Buyyarapu, R.; Guttikonda, S.K.; Parliament, K.; Abdurakhmonov, I.Y.; Kumpatla, S.P. Wild Relatives of Maize, Rice, Cotton, and Soybean: Treasure Troves for Tolerance to Biotic and Abiotic Stresses. Front. Plant Sci. 2018, 9, 886. [Google Scholar] [CrossRef]
- Zhang, Q.; Wing, R.A. (Eds.) Genetics and Genomics of Rice; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar] [CrossRef]
- Atwell, B.J.; Wang, H.; Scafaro, A.P. Could abiotic stress tolerance in wild relatives of rice be used to improve Oryza sativa? Plant Sci. 2014, 215, 48–58. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Solovyev, V.; Fokin, O.; Seledtsov, I.; Salamov, A.; Molodtsov, V.; Okhalin, N.; Bachinskii, A.; Kosarev, P.; Bakulina, N.; Genaev, M.; et al. MolQuest–Bioinformatics Toolbox for Analysis of Biomedical Data. 2021. Available online: https://molquest.com/molquest.phtml?topic=downloads (accessed on 12 March 2023).
- Salamov, A.A.; Solovyev, V.V. Ab initio Gene Finding in Drosophila Genomic DNA. Genome Res. 2000, 10, 516–522. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 3, 607–612. [Google Scholar] [CrossRef]
- Fu, Y.X.; Li, W.H. Statistical tests of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, M.; Miura, S.; Nei, M. Origins and Evolution of MicroRNA Genes in Plant Species. Genome Biol. Evol. 2012, 4, 230–239. [Google Scholar] [CrossRef]
- Freeling, M. Bias in Plant Gene Content Following Different Sorts of Duplication: Tandem, Whole-Genome, Segmental, or by Transposition. Annu. Rev. Plant Biol. 2009, 60, 433–453. [Google Scholar] [CrossRef]
- Goicoechea, J.L.; Ammiraju, J.S.S.; Marri, P.R.; Chen, M.; Jackson, S.; Yu, Y.; Rounsley, S.; Wing, R.A. The Future of Rice Genomics: Sequencing the Collective Oryza Genome. Rice 2010, 3, 89–97. [Google Scholar] [CrossRef]
- Hilu, K.W. Phylogenetics and chromosomal evolution in the Poaceae (grasses). Aust. J. Bot. 2004, 52, 13–22. [Google Scholar] [CrossRef]
- Salse, J. In silico archeogenomics unveils modern plant genome organisation, regulation and evolution. Curr. Opin. Plant Biol. 2012, 15, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chia, J.-M.; Kumari, S.; Stein, J.C.; Liu, Z.; Narechania, A.; Maher, C.A.; Guill, K.; McMullen, M.D.; Ware, D. A Genome-Wide Characterization of MicroRNA Genes in Maize. PLoS Genet. 2009, 5, e1000716. [Google Scholar] [CrossRef]
- Sun, S.; Wang, J.; Yu, J.; Meng, F.; Xia, R.; Wang, L.; Wang, Z.; Ge, W.; Liu, X.; Li, Y.; et al. Alignment of Common Wheat and Other Grass Genomes Establishes a Comparative Genomics Research Platform. Front. Plant Sci. 2017, 8, 1480. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Zheng, Y. Comprehensive Analysis of Single Nucleotide Polymorphisms in Human MicroRNAs. PLoS ONE 2013, 8, e78028. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Tong, Y.; Zhang, H.-M.; Wang, K.; Hu, T.; Shan, G.; Sun, J.; Guo, A.-Y. Genome-wide identification of SNPs in microRNA genes and the SNP effects on microRNA target binding and biogenesis. Hum. Mutat. 2012, 33, 254–263. [Google Scholar] [CrossRef]
- Ehrenreich, I.M.; Purugganan, M.D. Sequence Variation of MicroRNAs and Their Binding Sites in Arabidopsis. Plant Physiol. 2008, 146, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Hu, B.; Zhang, C. microRNAs and Their Roles in Plant Development. Front. Plant Sci. 2022, 13, 824240. [Google Scholar] [CrossRef]
- Lian, H.; Wang, L.; Ma, N.; Zhou, C.-M.; Han, L.; Zhang, T.-Q.; Wang, J.-W. Redundant and specific roles of individual MIR172 genes in plant development. PLoS Biol. 2021, 19, e3001044. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zwaenepoel, A.; Xue, J.-Y.; Kao, S.-M.; Li, Z.; Schranz, M.E.; Van de Peer, Y. Whole-genome microsynteny-based phylogeny of angiosperms. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- de Meaux, J.; Hu, J.-Y.; Tartler, U.; Goebel, U. Structurally different alleles of the ath-MIR824 microRNA precursor are maintained at high frequency in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2008, 105, 8994–8999. [Google Scholar] [CrossRef]
- Ammiraju, J.S.; Lu, F.; Sanyal, A.; Yu, Y.; Song, X.; Jiang, N.; Pontaroli, A.C.; Rambo, T.; Currie, J.; Collura, K.; et al. Dynamic Evolution of Oryza Genomes Is Revealed by Comparative Genomic Analysis of a Genus-Wide Vertical Data Set. Plant Cell 2008, 20, 3191–3209. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yu, Y.; Haberer, G.; Marri, P.R.; Fan, C.; Goicoechea, J.L.; Zuccolo, A.; Song, X.; Kudrna, D.; Ammiraju, J.S.S.; et al. The genome sequence of African rice (Oryza glaberrima) and evidence for independent domestication. Nat. Genet. 2014, 46, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Lye, Z.N.; Groen, S.C.; Dai, X.; Rughani, P.; Zaaijer, S.; Harrington, E.D.; Juul, S.; Purugganan, M.D. Nanopore sequencing-based genome assembly and evolutionary genomics of circum-basmati rice. Genome Biol. 2020, 21, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.K.; Rai, R.; Pradhan, S.; Shivaraj, S.; Deshmukh, R.; Sreevathsa, R.; Singh, N. Drought and oxidative stress in flax (Linum usitatissimum L.) entails harnessing non-canonical reference gene for precise quantification of qRT-PCR gene expression. Antioxidants 2023, 12, 950. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | MIRNA | Location | Precursor Coordinates | |
|---|---|---|---|---|
| Start | End | |||
| Oryza sativa | MIR172A | Chr 9 | 21,688,003 | 21,688,111 |
| MIR172B | Chr 1 | 46,953,572 | 46,953,809 | |
| MIR172C | Chr 7 | 12,288,035 | 12,288,145 | |
| MIR172D | Chr 2 | 37,298,091 | 37,298,220 | |
| Chr 2 | 37,328,665 | 37,328,794 | ||
| Oryza barthii | MIR172A | Chr 2 | 30,983,600 | 30,983,623 |
| MIR172B | Chr 1 | 36,558,471 | 36,558,708 | |
| MIR172C | Chr 7 | 10,711,579 | 10,711,689 | |
| MIR172D | Chr 2 | 30,983,582 | 30,983,711 | |
| Oryza glaberrima | MIR172A | Chr 2 | 28,473,275 | 28,473,298 |
| MIR172B | Chr 1 | 32,331,490 | 32,331,727 | |
| MIR172C | Chr 7 | 10,632,540 | 10,632,650 | |
| MIR172D | Chr 2 | 28,473,257 | 28,473,386 | |
| Oryza glumaepatula | MIR172A | Chr 9 | 23,407,188 | 23,407,296 |
| MIR172B | Chr 1 | 46,207,906 | 46,208,139 | |
| MIR172C | Chr 7 | 12,334,350 | 12,334,460 | |
| MIR172D | Chr 2 | 37,285,412 | 37,285,541 | |
| Oryza punctata | MIR172A | Ch9 | 26,230,640 | 26,230,686 |
| MIR172B | Chr1 | 45,791,747 | 45,791,791 | |
| MIR172C | Chr2 | 38,821,836 | 38,821,861 | |
| MIR172D | Chr2 | 38,821,883 | 38,821,953 | |
| Oryza rufipogon | MIR172A | Chr 9 | 20,417,006 | 20,417,114 |
| MIR172B | Chr 1 | 39,578,018 | 39,578,255 | |
| MIR172C | Chr 7 | 11,443,165 | 11,443,275 | |
| MIR172D | Chr 2 | 33,182,612 | 33,182,741 | |
| Oryza brachyantha | MIR172A | Chr9 | 14,032,760 | 14,032,800 |
| MIR172B | Chr1 | 33638435 | 33638477 | |
| MIR172C | Chr7 | 7,379,288 | 7,379,325 | |
| MIR172D | Chr2 | 26,379,138 | 26,429,195 | |
| Sorghum bicolor | MIR172A | Chr 9 | 58,774,558 | 58,774,659 |
| MIR172B | Chr 3 | 74,188,339 | 74,188,508 | |
| MIR172C | Chr 4 | 67,645,991 | 67,646,109 | |
| MIR172D | Chr 2 | 22,201,215 | 22,201,302 | |
| MIR172E | Chr 2 | 14,122,957 | 14,123,071 | |
| MIR172F | Chr 5 | 19,295,951 | 19,296,068 | |
| Zea mays | MIR172A | Chr 7 | 54,276,707 | 54,276,789 |
| MIR172B | Chr 5 | 221,721,349 | 221,721,474 | |
| MIR172C | Chr 4 | 174,154,928 | 174,155,050 | |
| MIR172D | Chr 6 | 172,029,786 | 172,029,859 | |
| MIR172E | Chr 3 | 145,801,592 | 145,801,765 | |
| Arabidopsis thaliana | MIR172A | Chr2 | 11,942,914 | 11,943,015 |
| MIR172B | Chr5 | 1,188,207 | 1,188,301 | |
| MIR172C | Chr3 | 3,599,776 | 3,599,908 | |
| MIR172D | Chr3 | 20,587,904 | 20,588,027 | |
| MIR172E | Chr5 | 23,988,472 | 23,988,596 | |
| MIRNA | Plant Species | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Oryza sativa | Oryza barthii | Oryza glaberrima | Oryza glumaepatula | Oryza rufipogon | Oryza brachyantha | Oryza punctata | Sorghum bicolor | Triticum aestivum | Zea mays | Arabidopsis thaliana | |||
| No. of genes predicted | MIR172 | MIR172A | 23 | 22 | 23 | 17 | 23 | 27 | 22 | 20 | 0 | 14 | 27 |
| MIR172B | 23 | 21 | 18 | 18 | 17 | 9 | 25 | 21 | 0 | 15 | 28 | ||
| MIR172C | 21 | 25 | 22 | 13 | 28 | 21 | 22 | 18 | 0 | 12 | 29 | ||
| MIR172D | 1–23, 2–19 | 22 | 23 | 19 | 18 | 17 | 22 | 27 | 0 | 19 | 26 | ||
| MIR172E | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 25 | 0 | 14 | 34 | ||
| MIR172F | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 19 | 0 | 0 | 0 | ||
| No. of homologs predicted | MIR172A | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | |
| MIR172B | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | ||
| MIR172C | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | ||
| MIR172D | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0 | 1 | 1 | ||
| MIR172E | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | ||
| MIR172F | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dash, P.K.; Gupta, P.; Sreevathsa, R.; Pradhan, S.K.; Sanjay, T.D.; Mohanty, M.R.; Roul, P.K.; Singh, N.K.; Rai, R. Phylogenomic Analysis of micro-RNA Involved in Juvenile to Flowering-Stage Transition in Photophilic Rice and Its Sister Species. Cells 2023, 12, 1370. https://doi.org/10.3390/cells12101370
Dash PK, Gupta P, Sreevathsa R, Pradhan SK, Sanjay TD, Mohanty MR, Roul PK, Singh NK, Rai R. Phylogenomic Analysis of micro-RNA Involved in Juvenile to Flowering-Stage Transition in Photophilic Rice and Its Sister Species. Cells. 2023; 12(10):1370. https://doi.org/10.3390/cells12101370
Chicago/Turabian StyleDash, Prasanta K., Payal Gupta, Rohini Sreevathsa, Sharat Kumar Pradhan, Tenkabailu Dharmanna Sanjay, Mihir Ranjan Mohanty, Pravat K. Roul, Nagendra K. Singh, and Rhitu Rai. 2023. "Phylogenomic Analysis of micro-RNA Involved in Juvenile to Flowering-Stage Transition in Photophilic Rice and Its Sister Species" Cells 12, no. 10: 1370. https://doi.org/10.3390/cells12101370
APA StyleDash, P. K., Gupta, P., Sreevathsa, R., Pradhan, S. K., Sanjay, T. D., Mohanty, M. R., Roul, P. K., Singh, N. K., & Rai, R. (2023). Phylogenomic Analysis of micro-RNA Involved in Juvenile to Flowering-Stage Transition in Photophilic Rice and Its Sister Species. Cells, 12(10), 1370. https://doi.org/10.3390/cells12101370