
Ketamine Reduces the Surface Density of the Astroglial Kir4.1 Channel and Inhibits Voltage-Activated Currents in a Manner Similar to the Action of Ba2+ on K+ Currents

 ,
,  , ,
, ,  and
and
Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Solutions
2.3. Plasmid and Cell Transfection
2.4. LysoTracker Labelling
2.5. Fixed and Live Cell Immunocytochemistry and Fluorescence Analysis
2.6. Analysis of Kir4.1 Vesicle Mobility
2.7. Electrophysiology
2.8. Statistical Analysis
3. Results
3.1. Kir4.1 Localizes to Aquaporin 4-Positive Vesicles and Vesicles Competent for SNARE- and Ca2+-Dependent Exocytosis
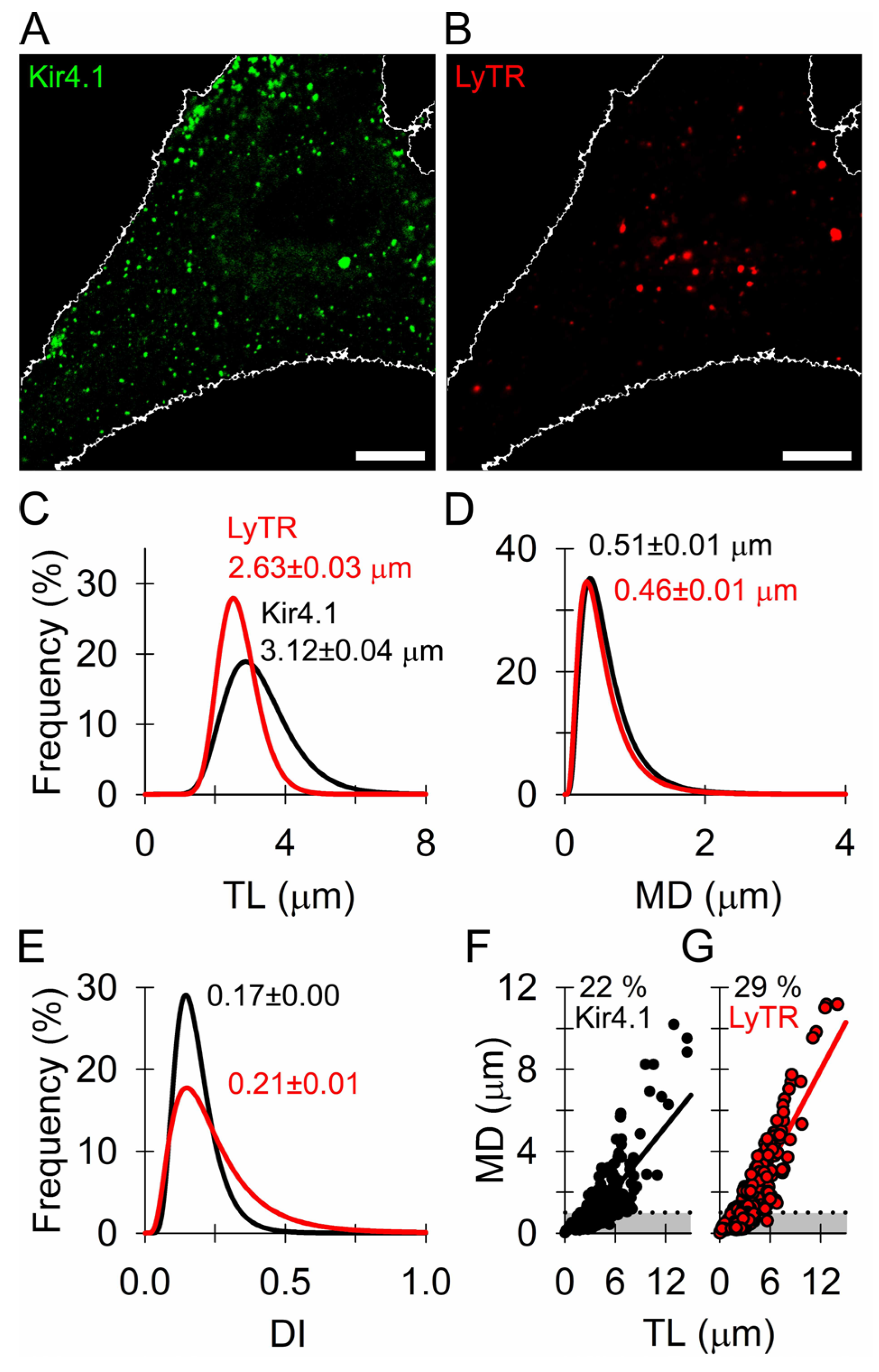
3.2. Similar Mobility of Kir4.1 Vesicles and Endo-Lysosomes
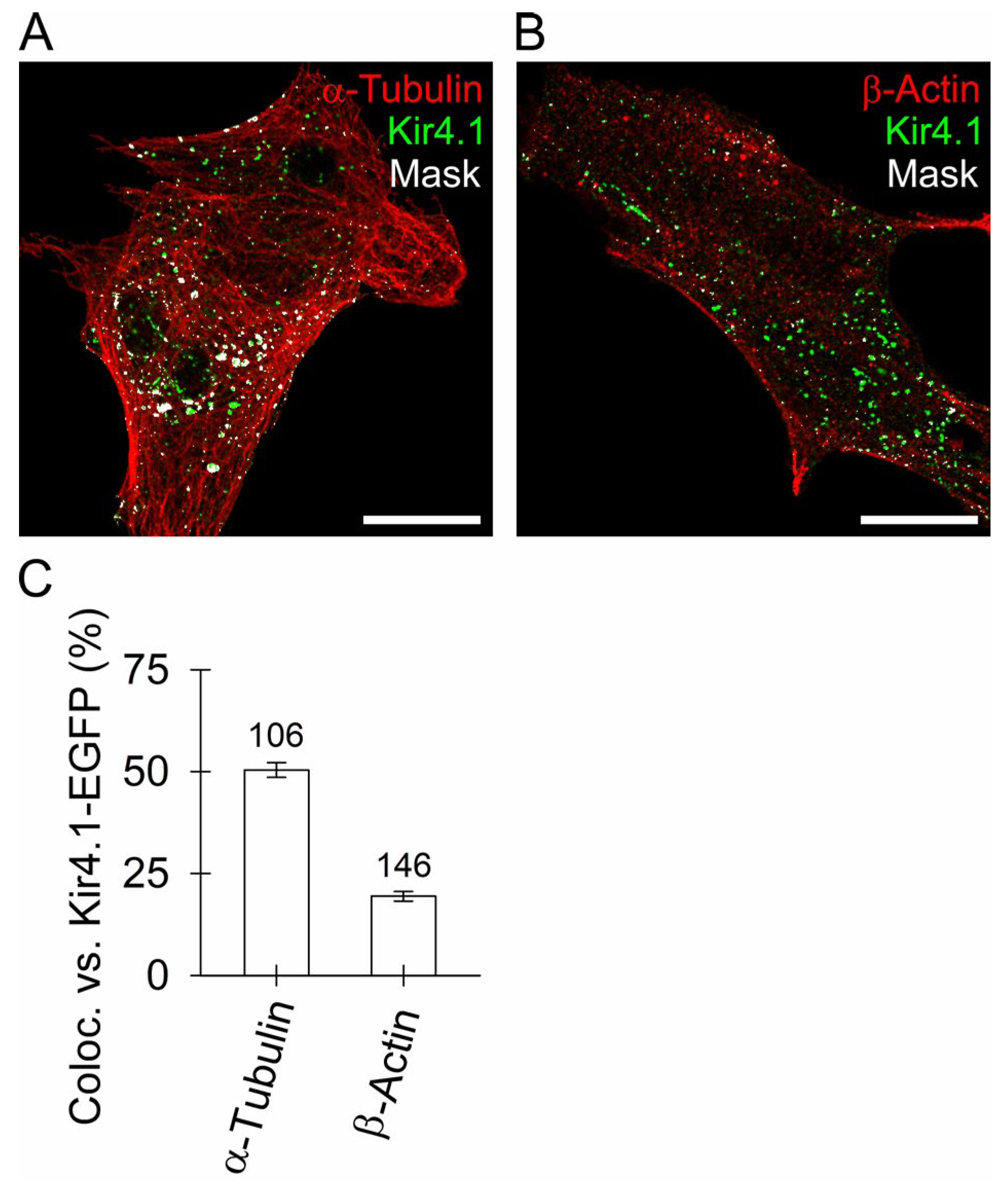
3.3. Kir4.1 Vesicles Associate More with Microtubules Than with Actin Filaments
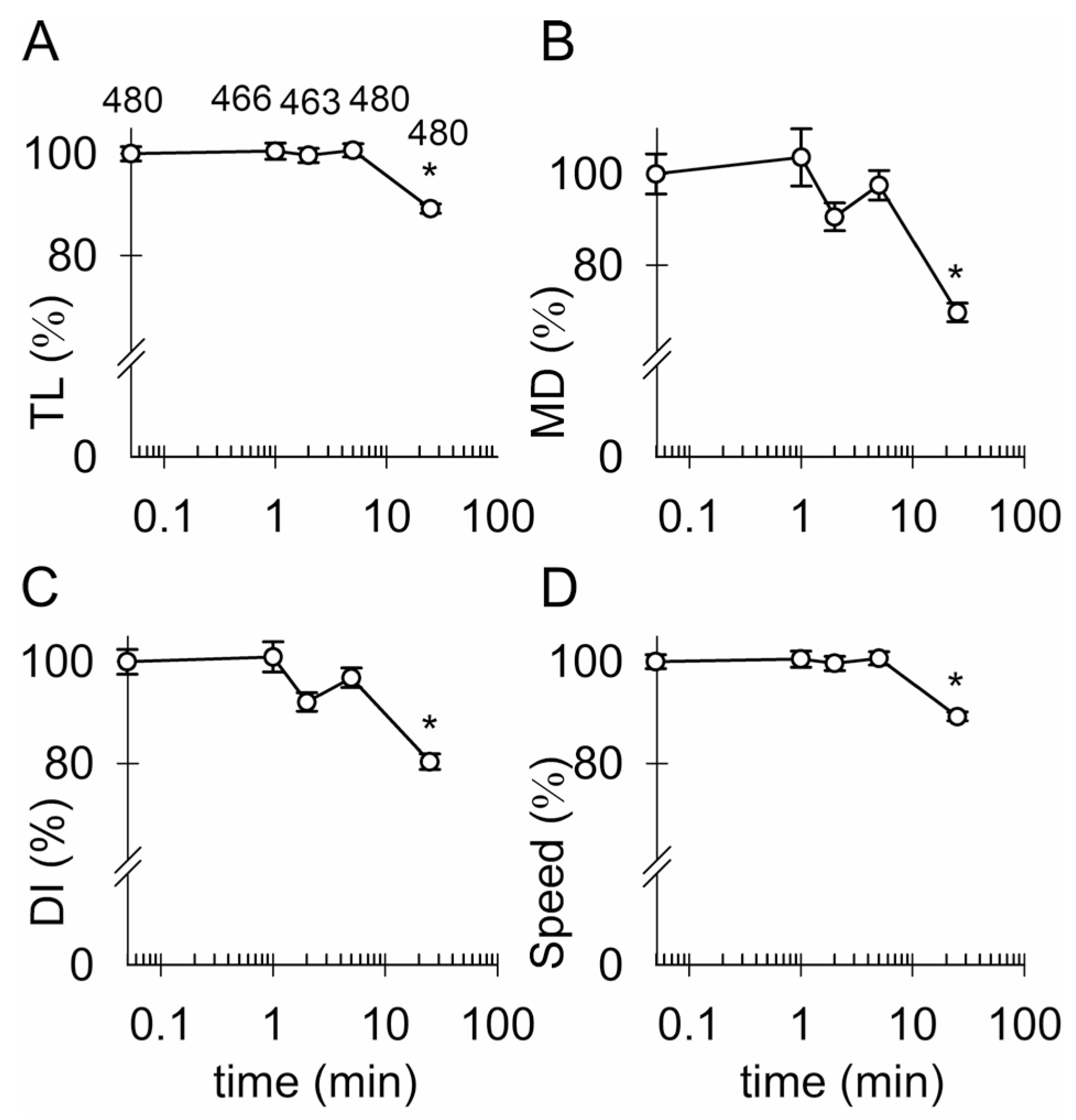
3.4. Ketamine and an Increase in Intracellular [cAMP] Attenuate the Mobility of Kir4.1-EGFP-Positive Vesicles
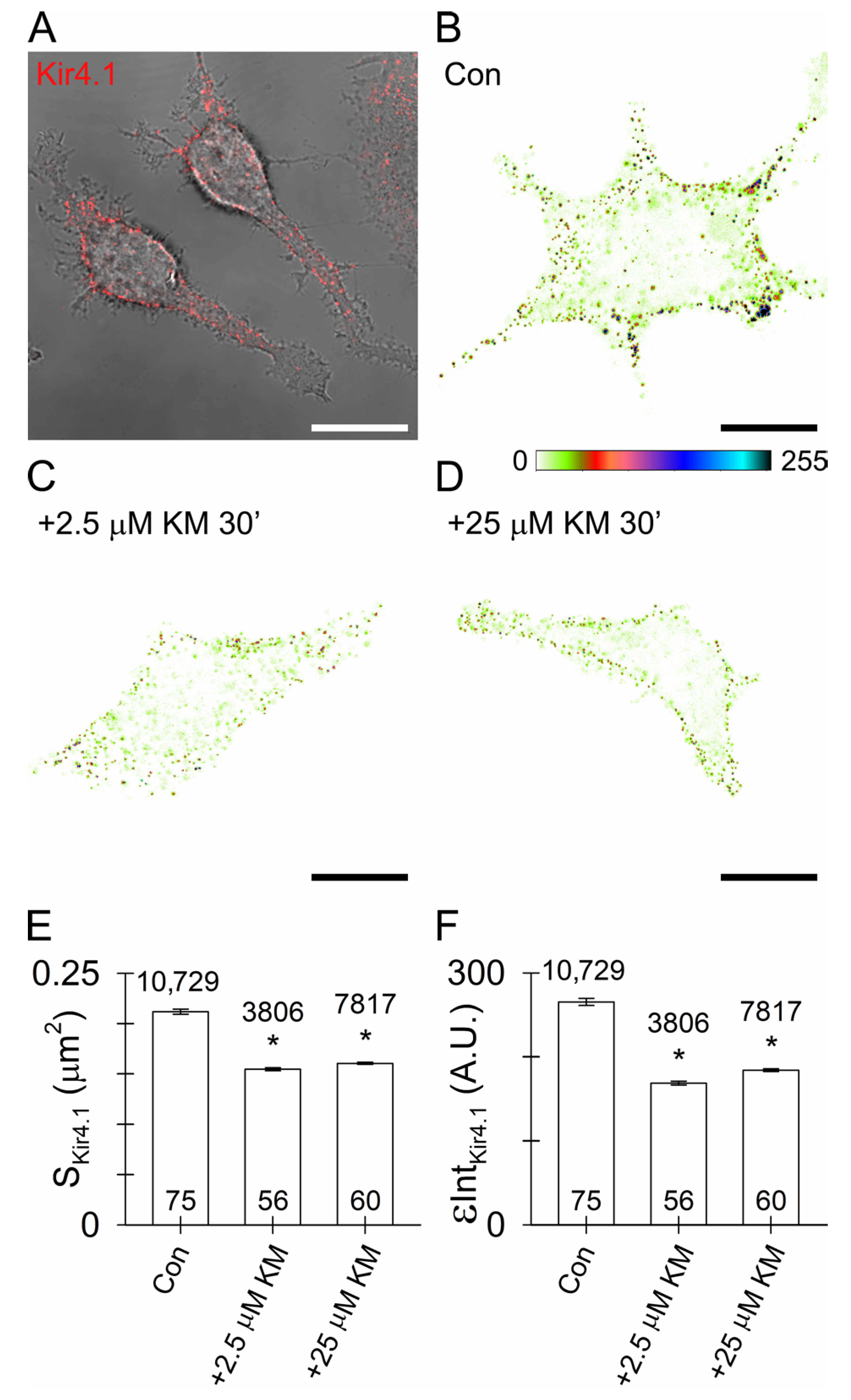
3.5. Reduced Surface Density of Astroglial Kir4.1 by Ketamine
3.6. Ketamine Inhibits Voltage-Activated Currents in Astroglia
4. Discussion
4.1. Kir4.1 and AQP4 Coalesce on Vesicles Competent for Regulated Exocytosis
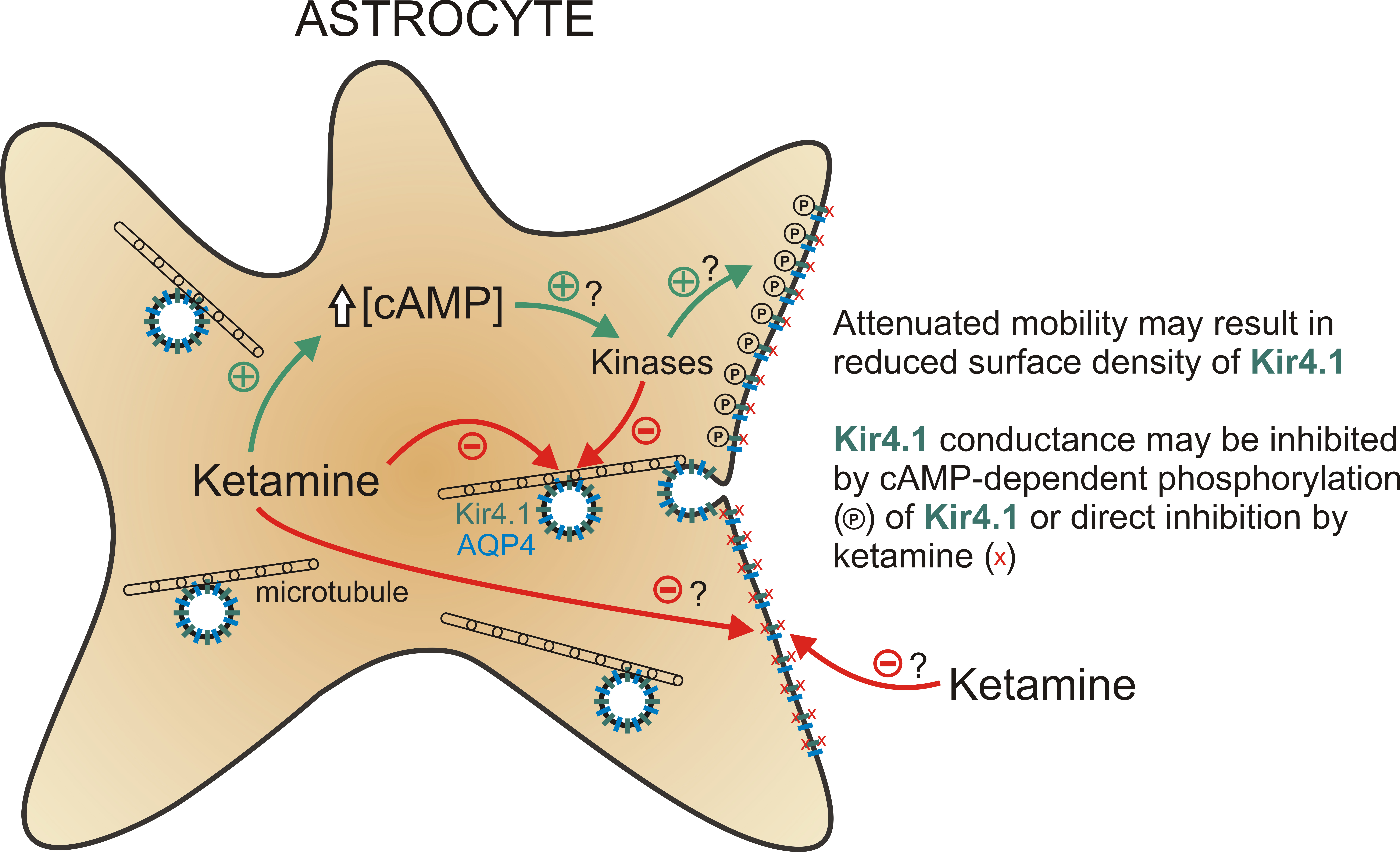
4.2. Ketamine Attenuates the Mobility of Kir4.1 Vesicles via a cAMP-Dependent Mechanism
4.3. Ketamine Reduces Astroglial Kir4.1 Surface Density
4.4. Ketamine Inhibits Astroglial K+ Conductance
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. An Acute Increase in [K+]o Requires a Delay to Alter the Mobility of Kir4.1 Vesicles

References
- Seri, B.; García-Verdugo, J.M.; McEwen, B.S.; Alvarez-Buylla, A. Astrocytes give rise to new neurons in the adult mammalian hippocampus. J. Neurosci. 2001, 21, 7153–7160. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiol. Rev. 2018, 98, 239–389. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Matteoli, M.; Parpura, V.; Mothet, J.-P.; Zorec, R. Astrocytes as secretory cells of the central nervous system: Idiosyncrasies of vesicular secretion. EMBO J. 2016, 35, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Zorec, R.; Verkhratsky, A.; Rodríguez, J.; Parpura, V. Astrocytic vesicles and gliotransmitters: Slowness of vesicular release and synaptobrevin2-laden vesicle nanoarchitecture. Neuroscience 2016, 323, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Barres, B.A. Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci. 2013, 14, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Zorec, R.; Horvat, A.; Vardjan, N.; Verkhratsky, A. Memory Formation Shaped by Astroglia. Front. Integr. Neurosci. 2015, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, P.; Newman, E.A. Potassium buffering in the central nervous system. Neuroscience 2004, 129, 1043–1056. [Google Scholar] [CrossRef]
- Orkand, R.K.; Nicholls, J.G.; Kuffler, S.W.; Verkhratsky, A.; Nedergaard, M.; Sawyer, J.E.R.; Hennebry, J.E.; Revill, A.; Brown, A.M.; Wanke, E.; et al. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J. Neurophysiol. 1966, 29, 788–806. [Google Scholar] [CrossRef]
- Kiyoshi, C.M.; Du, Y.; Zhong, S.; Wang, W.; Taylor, A.T.; Xiong, B.; Ma, B.; Terman, D.; Zhou, M. Syncytial isopotentiality: A system-wide electrical feature of astrocytic networks in the brain. Glia 2018, 66, 2756–2769. [Google Scholar] [CrossRef]
- Seifert, G.; Hüttmann, K.; Binder, D.K.; Hartmann, C.; Wyczynski, A.; Neusch, C.; Steinhäuser, C. Analysis of astroglial K+ channel expression in the developing hippocampus reveals a predominant role of the Kir4.1 subunit. J. Neurosci. 2009, 29, 7474–7488. [Google Scholar] [CrossRef]
- Tang, X.; Taniguchi, K.; Kofuji, P. Heterogeneity of Kir4.1 channel expression in glia revealed by mouse transgenesis. Glia 2009, 57, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Nwaobi, S.E.; Cuddapah, V.A.; Patterson, K.C.; Randolph, A.C.; Olsen, M.L. The role of glial-specific Kir4.1 in normal and pathological states of the CNS. Acta Neuropathol. 2016, 132, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Chever, O.; Djukic, B.; McCarthy, K.D.; Amzica, F. Implication of Kir4.1 channel in excess potassium clearance: An in vivo study on anesthetized glial-conditional Kir4.1 knock-out mice. J. Neurosci. 2010, 30, 15769–15777. [Google Scholar] [CrossRef] [PubMed]
- MacAulay, N. Molecular mechanisms of K+ clearance and extracellular space shrinkage—Glia cells as the stars. Glia 2020, 68, 2192–2211. [Google Scholar] [CrossRef]
- Dietz, A.G.; Goldman, S.A.; Nedergaard, M. Glial cells in schizophrenia: A unified hypothesis. Lancet Psychiatry 2019, 7, 272–281. [Google Scholar] [CrossRef]
- Peng, L.; Li, B.; Verkhratsky, A. Targeting astrocytes in bipolar disorder. Expert Rev. Neurother. 2016, 16, 649–657. [Google Scholar] [CrossRef]
- Rajkowska, G.; Hughes, J.; Stockmeier, C.A.; Miguel-Hidalgo, J.J.; Maciag, D. Coverage of blood vessels by astrocytic endfeet is reduced in major depressive disorder. Biol. Psychiatry 2013, 73, 613–621. [Google Scholar] [CrossRef]
- Stenovec, M.; Li, B.; Verkhratsky, A.; Zorec, R. Astrocytes in rapid ketamine antidepressant action. Neuropharmacology 2020, 173, 108158. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Rodríguez, J.J.; Steardo, L. Astrogliopathology: A central element of neuropsychiatric diseases? Neuropharmacology 2014, 20, 576–588. [Google Scholar] [CrossRef]
- Howe, W.M.; Kenny, P.J. Burst firing sets the stage for depression. Nature 2018, 554, 304–305. [Google Scholar] [CrossRef]
- Cui, Y.; Yang, Y.; Ni, Z.; Dong, Y.; Cai, G.; Foncelle, A.; Ma, S.; Sang, K.; Tang, S.; Li, Y.; et al. Astroglial Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature 2018, 554, 323–327. [Google Scholar] [CrossRef]
- Yang, Y.; Cui, Y.; Sang, K.; Dong, Y.; Ni, Z.; Ma, S.; Hu, H. Ketamine blocks bursting in the lateral habenula to rapidly relieve depression. Nature 2018, 554, 317–322. [Google Scholar] [CrossRef]
- Higashi, K.; Fujita, A.; Inanobe, A.; Tanemoto, M.; Doi, K.; Kubo, T.; Kurachi, Y. An inwardly rectifying K+ channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am. J. Physiol. Physiol. 2001, 281, C922–C931. [Google Scholar] [CrossRef]
- Domino, E.F.; Chodoff, P.; Corssen, G. Pharmacologic Effects of Ci-581, a New Dissociative Anesthetic, in Man. Clin. Pharmacol. Ther. 1965, 6, 279–291. [Google Scholar] [CrossRef]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Newport, D.J.; Carpenter, L.L.; McDonald, W.M.; Potash, J.B.; Tohen, M.; Nemeroff, C.B. The APA Council of Research Task Force on Novel Biomarkers and Treatments Ketamine and Other NMDA Antagonists: Early Clinical Trials and Possible Mechanisms in Depression. Am. J. Psychiatry 2015, 172, 950–966. [Google Scholar] [CrossRef]
- Zarate, C.A.; Singh, J.B.; Carlson, P.J.; Brutsche, N.E.; Ameli, R.; Luckenbaugh, D.A.; Charney, D.S.; Manji, H.K. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch. Gen. Psychiatry 2006, 63, 856–864. [Google Scholar] [CrossRef]
- Zarate, C.A.; Brutsche, N.E.; Ibrahim, L.; Franco-Chaves, J.; Diazgranados, N.; Cravchik, A.; Selter, J.; Marquardt, C.A.; Victoria Liberty; Luckenbaugh, D.A. Replication of ketamine’s antidepressant efficacy in bipolar depression: A randomized controlled add-on trial. Biol. Psychiatry 2012, 71, 939–946. [Google Scholar] [CrossRef]
- Gideons, E.S.; Kavalali, E.T.; Monteggia, L.M. Mechanisms underlying differential effectiveness of memantine and ketamine in rapid antidepressant responses. Proc. Natl. Acad. Sci. USA 2014, 111, 8649–8654. [Google Scholar] [CrossRef]
- Stenovec, M.; Lasič, E.; Božić, M.; Bobnar, S.T.; Stout, R.F.; Grubišić, V.; Parpura, V.; Zorec, R. Ketamine Inhibits ATP-Evoked Exocytotic Release of Brain-Derived Neurotrophic Factor from Vesicles in Cultured Rat Astrocytes. Mol. Neurobiol. 2016, 53, 6882–6896. [Google Scholar] [CrossRef]
- Lasič, E.; Rituper, B.; Jorgačevski, J.; Kreft, M.; Stenovec, M.; Zorec, R. Subanesthetic doses of ketamine stabilize the fusion pore in a narrow flickering state in astrocytes. J. Neurochem. 2016, 138, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Lasič, E.; Lisjak, M.; Horvat, A.; Božić, M.; Šakanović, A.; Anderluh, G.; Verkhratsky, A.; Vardjan, N.; Jorgačevski, J.; Stenovec, M.; et al. Astrocyte Specific Remodeling of Plasmalemmal Cholesterol Composition by Ketamine Indicates a New Mechanism of Antidepressant Action. Sci. Rep. 2019, 9, 10957. [Google Scholar] [CrossRef] [PubMed]
- Senese, N.B.; Rasenick, M.M. Antidepressants Produce Persistent Gαs-Associated Signaling Changes in Lipid Rafts after Drug Withdrawal. Mol. Pharmacol. 2021, 100, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Schwarts, J.P.; Wilson, D.J. Preparation and characterization of type 1 astrocytes cultured from adult rat cortex, cerebellum, and striatum. Glia 1992, 5, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Méndez-González, M.P.; Kucheryavykh, Y.V.; Zayas-Santiago, A.; Vélez-Carrasco, W.; Maldonado-Martínez, G.; Cubano, L.A.; Nichols, C.G.; Skatchkov, S.N.; Eaton, M.J. Novel KCNJ10 Gene Variations Compromise Function of Inwardly Rectifying Potassium Channel 4.1. J. Biol. Chem. 2016, 291, 7716–7726. [Google Scholar] [CrossRef]
- Sala-Rabanal, M.; Kucheryavykh, L.Y.; Skatchkov, S.N.; Eaton, M.J.; Nichols, C.G. Molecular mechanisms of EAST/SeSAME syndrome mutations in Kir4.1 (KCNJ10). J. Biol. Chem. 2010, 285, 36040–36048. [Google Scholar] [CrossRef]
- Kreft, M.; Milisav, I.; Potokar, M.; Zorec, R. Automated high through-put colocalization analysis of multichannel confocal images. Comput. Methods Programs Biomed. 2004, 74, 63–67. [Google Scholar] [CrossRef]
- Božić, M.; Verkhratsky, A.; Zorec, R.; Stenovec, M. Exocytosis of large-diameter lysosomes mediates interferon γ-induced relocation of MHC class II molecules toward the surface of astrocytes. Cell. Mol. Life Sci. 2020, 77, 3245–3264. [Google Scholar] [CrossRef]
- Potokar, M.; Kreft, M.; Pangršič, T.; Zorec, R. Vesicle mobility studied in cultured astrocytes. Biochem. Biophys. Res. Commun. 2005, 329, 678–683. [Google Scholar] [CrossRef]
- Stenovec, M.; Božić, M.; Pirnat, S.; Zorec, R. Astroglial Mechanisms of Ketamine Action Include Reduced Mobility of Kir4.1-Carrying Vesicles. Neurochem. Res. 2019, 45, 109–121. [Google Scholar] [CrossRef]
- Rituper, B.; Guček, A.; Jorgačevski, J.; Flašker, A.; Kreft, M.; Zorec, R. High-resolution membrane capacitance measurements for the study of exocytosis and endocytosis. Nat. Protoc. 2013, 8, 1169–1183. [Google Scholar] [CrossRef]
- Hibino, H.; Inanobe, A.; Furutani, K.; Murakami, S.; Findlay, I.; Kurachi, Y.; Yamamura, H.; Suzuki, Y.; Yamamura, H.; Asai, K.; et al. Inwardly rectifying potassium channels: Their structure, function, and physiological roles. Physiol. Rev. 2010, 90, 291–366. [Google Scholar] [CrossRef]
- Kržan, M.; Stenovec, M.; Kreft, M.; Pangršič, T.; Grilc, S.; Haydon, P.G.; Zorec, R. Calcium-dependent exocytosis of atrial natriuretic peptide from astrocytes. J. Neurosci. 2003, 23, 1580–1583. [Google Scholar] [CrossRef]
- Nagelhus, E.; Mathiisen, T.; Ottersen, O. Aquaporin-4 in the central nervous system: Cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience 2004, 129, 905–913. [Google Scholar] [CrossRef]
- Montana, V.; Malarkey, E.B.; Verderio, C.; Matteoli, M.; Parpura, V. Vesicular transmitter release from astrocytes. Glia 2006, 54, 700–715. [Google Scholar] [CrossRef]
- Paco, S.; Margelí, M.A.; Olkkonen, V.M.; Imai, A.; Blasi, J.; Fischer-Colbrie, R.; Aguado, F. Regulation of exocytotic protein expression and Ca2+-dependent peptide secretion in astrocytes. J. Neurochem. 2009, 110, 143–156. [Google Scholar] [CrossRef]
- Zhang, Q.; Fukuda, M.; Van Bockstaele, E.; Pascual, O.; Haydon, P.G. Synaptotagmin IV regulates glial glutamate release. Proc. Natl. Acad. Sci. USA 2004, 101, 9441–9446. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Castonguay, J.; Orth, J.H.C.; Müller, T.; Sleman, F.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Mallmann, R.T.; Bildl, W.; Schulte, U.; et al. The two-pore channel TPC1 is required for efficient protein processing through early and recycling endosomes. Sci. Rep. 2017, 7, 10038. [Google Scholar] [CrossRef]
- Vanlandingham, P.A.; Ceresa, B.P. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J. Biol. Chem. 2009, 284, 12110–12124. [Google Scholar] [CrossRef]
- Wandinger-Ness, A.; Zerial, M. Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb. Perspect. Biol. 2014, 6, a022616. [Google Scholar] [CrossRef] [PubMed]
- Potokar, M.; Stenovec, M.; Gabrijel, M.; Li, L.; Kreft, M.; Grilc, S.; Pekny, M.; Zorec, R. Intermediate filaments attenuate stimulation-dependent mobility of endosomes/lysosomes in astrocytes. Glia 2010, 58, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Potokar, M.; Kreft, M.; Li, L.; Andersson, J.D.; Pangršič, T.; Chowdhury, H.H.; Pekny, M.; Zorec, R. Cytoskeleton and vesicle mobility in astrocytes. Traffic 2007, 8, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Stenovec, M.; Trkov, S.; Kreft, M.; Zorec, R. Alterations of calcium homoeostasis in cultured rat astrocytes evoked by bioactive sphingolipids. Acta Physiol. 2014, 212, 49–61. [Google Scholar] [CrossRef]
- Schultz, C.; Vajanaphanich, M.; Genieser, H.-G.; Jastorff, B.; Barrett, K.E.; Tsien, R.Y. Membrane-permeant derivatives of cyclic AMP optimized for high potency, prolonged activity, or rapid reversibility. Mol. Pharmacol. 1994, 46, 702–708. [Google Scholar]
- Zhang, J.; Ma, Y.; Taylor, S.S.; Tsien, R.Y. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc. Natl. Acad. Sci. USA 2001, 98, 14997–15002. [Google Scholar] [CrossRef]
- Choi, H.B.; Gordon, G.R.J.; Zhou, N.; Tai, C.; Rungta, R.L.; Martinez, J.; Milner, T.A.; Ryu, J.K.; McLarnon, J.G.; Tresguerres, M.; et al. Metabolic communication between astrocytes and neurons via bicarbonate-responsive soluble adenylyl cyclase. Neuron 2012, 75, 1094–1104. [Google Scholar] [CrossRef]
- Zhou, Z.; Ikegaya, Y.; Koyama, R. The Astrocytic cAMP Pathway in Health and Disease. Int. J. Mol. Sci. 2019, 20, 779. [Google Scholar] [CrossRef]
- Duffy, S.; MacVicar, B. Potassium-dependent calcium influx in acutely isolated hippocampal astrocytes. Neuroscience 1994, 61, 51–61. [Google Scholar] [CrossRef]
- Hibino, H.; Kurachi, Y. Distinct detergent-resistant membrane microdomains (lipid rafts) respectively harvest K+ and water transport systems in brain astroglia. Eur. J. Neurosci. 2007, 26, 2539–2555. [Google Scholar] [CrossRef]
- Pangršič, T.; Potokar, M.; Haydon, P.G.; Zorec, R.; Kreft, M. Astrocyte swelling leads to membrane unfolding, not membrane insertion. J. Neurochem. 2006, 99, 514–523. [Google Scholar] [CrossRef]
- Su, S.; Ohno, Y.; Lossin, C.; Hibino, H.; Inanobe, A.; Kurachi, Y. Inhibition of astroglial inwardly rectifying Kir4.1 channels by a tricyclic antidepressant, nortriptyline. J. Pharmacol. Exp. Ther. 2007, 320, 573–580. [Google Scholar] [CrossRef]
- Ohno, Y.; Hibino, H.; Lossin, C.; Inanobe, A.; Kurachi, Y. Inhibition of astroglial Kir4.1 channels by selective serotonin reuptake inhibitors. Brain Res. 2007, 1178, 44–51. [Google Scholar] [CrossRef]
- Kucheryavykh, Y.V.; Kucheryavykh, L.Y.; Nichols, C.G.; Maldonado, H.M.; Baksi, K.; Reichenbach, A.; Skatchkov, S.N.; Eaton, M.J. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 2007, 55, 274–281. [Google Scholar] [CrossRef]
- Takumi, T.; Ishii, T.; Horio, Y.; Morishige, K.-I.; Takahashi, N.; Yamada, M.; Yamashita, T.; Kiyama, H.; Sohmiya, K.; Nakanishi, S.; et al. A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J. Biol. Chem. 1995, 270, 16339–16346. [Google Scholar] [CrossRef]
- Tanemoto, M.; Kittaka, N.; Inanobe, A.; Kurachi, Y. In vivo formation of a proton-sensitive K + channel by heteromeric subunit assembly of Kir5.1 with Kir4.1. J. Physiol. 2000, 525 Pt 3, 587–592. [Google Scholar] [CrossRef]
- Rushworth, M.F.; Noonan, M.P.; Boorman, E.D.; Walton, M.E.; Behrens, T.E. Frontal cortex and reward-guided learning and decision-making. Neuron 2021, 70, 1054–1069. [Google Scholar] [CrossRef]
- Ferenczi, E.A.; Zalocusky, K.A.; Liston, C.; Grosenick, L.; Warden, M.R.; Amatya, D.; Katovich, K.; Mehta, H.; Patenaude, B.; Ramakrishnan, C.; et al. Prefrontal cortical regulation of brainwide circuit dynamics and reward-related behavior. Science 2016, 351, aac9698. [Google Scholar] [CrossRef]
- Potokar, M.; Stenovec, M.; Jorgačevski, J.; Holen, T.; Kreft, M.; Ottersen, O.P.; Zorec, R. Regulation of AQP4 surface expression via vesicle mobility in astrocytes. Glia 2013, 61, 917–928. [Google Scholar] [CrossRef]
- Puwarawuttipanit, W.; Bragg, A.; Frydenlund, D.; Mylonakou, M.-N.; Nagelhus, E.; Peters, M.; Kotchabhakdi, N.; Adams, M.; Froehner, S.; Haug, F.-M.; et al. Differential effect of α-syntrophin knockout on aquaporin-4 and Kir4.1 expression in retinal macroglial cells in mice. Neuroscience 2006, 137, 165–175. [Google Scholar] [CrossRef]
- Li, D.; Hérault, K.; Zylbersztejn, K.; Lauterbach, M.A.; Guillon, M.; Oheim, M.; Ropert, N. Astrocyte VAMP3 vesicles undergo Ca2+-independent cycling and modulate glutamate transporter trafficking. J. Physiol. 2015, 593, 2807–2832. [Google Scholar] [CrossRef] [PubMed]
- Ropert, N.; Jalil, A.; Li, D. Expression and cellular function of vSNARE proteins in brain astrocytes. Neuroscience 2016, 323, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Denovan-Wright, E.M.; Newton, R.A.; Armstrong, J.N.; Babity, J.M.; Robertson, H.A. Acute administration of cocaine, but not amphetamine, increases the level of synaptotagmin IV mRNA in the dorsal striatum of rat. Mol. Brain Res. 1998, 55, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Premkumar, A.; Mossner, R.; Fukuda, M.; Lesch, K.-P.; Simantov, R. Synaptotagmin I and IV are differentially regulated in the brain by the recreational drug 3,4-methylenedioxymethamphetamine (MDMA). Mol. Brain Res. 2002, 108, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-T.; Grishanin, R.; Earles, C.A.; Chang, P.Y.; Martin, T.F.J.; Chapman, E.R.; Jackson, M.B. Synaptotagmin modulation of fusion pore kinetics in regulated exocytosis of dense-core vesicles. Science 2001, 294, 1111–1115. [Google Scholar] [CrossRef]
- Wang, C.-T.; Lu, J.-C.; Bai, J.; Chang, P.Y.; Martin, T.F.J.; Chapman, E.R.; Jackson, M.B. Different domains of synaptotagmin control the choice between kiss-and-run and full fusion. Nature 2003, 424, 943–947. [Google Scholar] [CrossRef]
- Song, Y.; Gunnarson, E. Potassium dependent regulation of astrocyte water permeability is mediated by cAMP signaling. PLoS ONE 2012, 7, e34936. [Google Scholar] [CrossRef]
- Chesler, M. The regulation and modulation of pH in the nervous system. Prog. Neurobiol. 1990, 34, 401–427. [Google Scholar] [CrossRef]
- Ransom, B.R. Chapter 3: Glial modulation of neural excitability mediated by extracellular pH: A hypothesis. Prog. Brain. Res. 1992, 94, 37–46. [Google Scholar] [CrossRef]
- Wray, N.H.; Schappi, J.M.; Singh, H.; Senese, N.B.; Rasenick, M.M. NMDAR-independent, cAMP-dependent antidepressant actions of ketamine. Mol. Psychiatry 2018, 24, 1833–1843. [Google Scholar] [CrossRef]
- Shin, C.; Kim, Y.-K. Ketamine in Major Depressive Disorder: Mechanisms and Future Perspectives. Psychiatry Investig. 2020, 17, 181–192. [Google Scholar] [CrossRef]
- Sos, P.; Klirova, M.; Novak, T.; Kohutova, B.; Horacek, J.; Palenicek, T. Relationship of ketamine’s antidepressant and psychotomimetic effects in unipolar depression. Neuro. Endocrinol. Lett. 2013, 34, 287–293. [Google Scholar]
- Keil, M.F.; Briassoulis, G.; Stratakis, C.A.; Wu, T.J. Protein Kinase A and Anxiety-Related Behaviors: A Mini-Review. Front. Endocrinol. 2016, 7, 83. [Google Scholar] [CrossRef]
- Khaled, M.; Larribere, L.; Bille, K.; Aberdam, E.; Ortonne, J.P.; Ballotti, R.; Bertolotto, C. Glycogen synthase kinase 3beta is activated by cAMP and plays an active role in the regulation of melanogenesis. J. Biol. Chem. 2002, 277, 33690–33697. [Google Scholar] [CrossRef]
- Gunawardena, S.; Yang, G.; Goldstein, L.S. Presenilin controls kinesin-1 and dynein function during APP-vesicle transport in vivo. Hum. Mol. Genet. 2013, 22, 3828–3843. [Google Scholar] [CrossRef]
- Pigino, G.; Morfini, G.; Pelsman, A.; Mattson, M.P.; Brady, S.T.; Busciglio, J. Alzheimer’s presenilin 1 mutations impair kinesin-based axonal transport. J. Neurosci. 2003, 23, 4499–4508. [Google Scholar] [CrossRef]
- Morfini, G.; Szebenyi, G.; Elluru, R.; Ratner, N.; Brady, S.T. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002, 21, 281–293. [Google Scholar] [CrossRef]
- Numakawa, T.; Odaka, H.; Adachi, N. Actions of Brain-Derived Neurotrophin Factor in the Neurogenesis and Neuronal Function, and Its Involvement in the Pathophysiology of Brain Diseases. Int. J. Mol. Sci. 2018, 19, 3650. [Google Scholar] [CrossRef]
- Paco, S.; Hummel, M.; Plá, V.; Sumoy, L.; Aguado, F. Cyclic AMP signaling restricts activation and promotes maturation and antioxidant defenses in astrocytes. BMC Genom. 2016, 17, 304. [Google Scholar] [CrossRef]
- Bensel, B.M.; Guzik-Lendrum, S.; Masucci, E.M.; Woll, K.A.; Eckenhoff, R.G.; Gilbert, S.P. Common general anesthetic propofol impairs kinesin processivity. Proc. Natl. Acad. Sci. USA 2017, 114, E4281–E4287. [Google Scholar] [CrossRef]
- Connors, N.C.; Adams, M.E.; Froehner, S.C.; Kofuji, P. The potassium channel Kir4.1 associates with the dystrophin-glycoprotein complex via alpha-syntrophin in glia. J. Biol. Chem. 2004, 279, 28387–28392. [Google Scholar] [CrossRef] [PubMed]
- Tanemoto, M.; Toyohara, T.; Abe, T.; Ito, S. MAGI-1a functions as a scaffolding protein for the distal renal tubular basolateral K+ channels. J. Biol. Chem. 2008, 283, 12241–12247. [Google Scholar] [CrossRef] [PubMed]
- Welling, P.A. Regulation of potassium channel trafficking in the distal nephron. Curr. Opin. Nephrol. Hypertens. 2013, 22, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Pirnat, S.; Božić, M.; Dolanc, D.; Horvat, A.; Tavčar, P.; Vardjan, N.; Verkhratsky, A.; Zorec, R.; Stenovec, M. Astrocyte arborization enhances Ca 2+ but not cAMP signaling plasticity. Glia 2021, 69, 2899–2916. [Google Scholar] [CrossRef]
- Schachtrup, C.; Ryu, J.K.; Helmrick, M.J.; Vagena, E.; Galanakis, D.K.; Degen, J.L.; Margolis, R.U.; Akassoglou, K. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-β after vascular damage. J. Neurosci. 2010, 30, 5843–5854. [Google Scholar] [CrossRef]
- Escartin, C.; Galea, E.; Lakatos, A.; O’callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhäuser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat. Neurosci. 2021, 24, 312–325. [Google Scholar] [CrossRef]
- McKhann, G.M.; D’ambrosio, R.; Janigro, D. Heterogeneity of astrocyte resting membrane potentials and intercellular coupling revealed by whole-cell and gramicidin-perforated patch recordings from cultured neocortical and hippocampal slice astrocytes. J. Neurosci. 1997, 17, 6850–6863. [Google Scholar] [CrossRef]
- Zhou, M.; Kimelberg, H.K. Freshly isolated astrocytes from rat hippocampus show two distinct current patterns and different [K+]o uptake capabilities. J. Neurophysiol. 2000, 84, 2746–2757. [Google Scholar] [CrossRef]
- Furutani, K.; Ohno, Y.; Inanobe, A.; Hibino, H.; Kurachi, Y. Mutational and in silico analyses for antidepressant block of astroglial inward-rectifier Kir4.1 channel. Mol. Pharmacol. 2009, 75, 1287–1295. [Google Scholar] [CrossRef]
- Ohno, Y.; Kinboshi, M.; Shimizu, S. Inwardly Rectifying Potassium Channel Kir4.1 as a Novel Modulator of BDNF Expression in Astrocytes. Int. J. Mol. Sci. 2018, 19, 3313. [Google Scholar] [CrossRef]
- Ohno, Y.; Kunisawa, N.; Shimizu, S. Emerging Roles of Astrocyte Kir4.1 Channels in the Pathogenesis and Treatment of Brain Diseases. Int. J. Mol. Sci. 2021, 22, 10236. [Google Scholar] [CrossRef]
- Kaye, A.; Ross, D.A. The Habenula: Darkness, Disappointment, and Depression. Biol. Psychiatry 2017, 81, e27–e28. [Google Scholar] [CrossRef]
- Levitan, I.B. Modulation of ion channels by protein phosphorylation. How the brain works. Adv. Second. Messenger Phosphoprot. Res. 1999, 33, 3–22. [Google Scholar] [CrossRef]
- Gao, F.; Yang, S.; Wang, J.; Zhu, G. cAMP-PKA cascade: An outdated topic for depression? Biomed. Pharmacother. 2022, 150, 113030. [Google Scholar] [CrossRef]
- Beguin, P.; Nagashima, K.; Nishimura, M.; Gonoi, T.; Seino, S. PKA-mediated phosphorylation of the human K(ATP) channel: Separate roles of Kir6.2 and SUR1 subunit phosphorylation. EMBO J. 1999, 18, 4722–4732. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, L.; Thomas, S.; Wang, K.; Lin, D.-H.; Rinehart, J.; Wang, W.-H. Src family protein tyrosine kinase regulates the basolateral K channel in the distal convoluted tubule (DCT) by phosphorylation of KCNJ10 protein. J. Biol. Chem. 2013, 288, 26135–26146. [Google Scholar] [CrossRef]
- Rojas, A.; Cui, N.; Su, J.; Yang, L.; Muhumuza, J.-P.; Jiang, C. Protein kinase C dependent inhibition of the heteromeric Kir4.1–Kir5.1 channel. Biochim. Biophys. Acta 2007, 1768, 2030–2042. [Google Scholar] [CrossRef]
- Kohrs, R.; Durieux, M.E. Ketamine: Teaching an old drug new tricks. Anesth. Analg. 1998, 87, 1186–1193. [Google Scholar] [CrossRef]
- Mion, G.; Villevieille, T. Ketamine pharmacology: An update (pharmacodynamics and molecular aspects, recent findings). CNS Neurosci. Ther. 2013, 19, 370–380. [Google Scholar] [CrossRef]
- Lester, H.A.; Lavis, L.D.; Dougherty, D.A. Ketamine inside neurons? Am. J. Psychiatry 2015, 172, 1064–1066. [Google Scholar] [CrossRef]
- Rohaim, A.; Gong, L.; Li, J.; Rui, H.; Blachowicz, L.; Roux, B. Open and Closed Structures of a Barium-Blocked Potassium Channel. J. Mol. Biol. 2020, 432, 4783–4798. [Google Scholar] [CrossRef] [PubMed]
- Hakey, P.; Ouellette, W.; Zubieta, J.; Korter, T. (S)-(+)-Ketamine hydrochloride. Acta Crystallogr. Sect. E Struct. Rep. Online 2008, 64, o1487. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Inhibited (All) Cells | Erev (mV)− | Erev (mV)+ | p-Value |
|---|---|---|---|---|
| ECS (control) | 8 (8) | −64 ± 3 | −57 ± 3 | 0.130 |
| 300 µM Ba2+ | 4 (8) | −53 ± 7 | −53 ± 5 | 0.886 |
| 2.5 µM KM | 6 (8) | −65 ± 7 | −63 ± 7 | 0.589 |
| KM + Ba2+ | 4 (10) | −73 ± 0 | −60 ± 10 | 0.029 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Božić, M.; Pirnat, S.; Fink, K.; Potokar, M.; Kreft, M.; Zorec, R.; Stenovec, M. Ketamine Reduces the Surface Density of the Astroglial Kir4.1 Channel and Inhibits Voltage-Activated Currents in a Manner Similar to the Action of Ba2+ on K+ Currents. Cells 2023, 12, 1360. https://doi.org/10.3390/cells12101360
Božić M, Pirnat S, Fink K, Potokar M, Kreft M, Zorec R, Stenovec M. Ketamine Reduces the Surface Density of the Astroglial Kir4.1 Channel and Inhibits Voltage-Activated Currents in a Manner Similar to the Action of Ba2+ on K+ Currents. Cells. 2023; 12(10):1360. https://doi.org/10.3390/cells12101360
Chicago/Turabian StyleBožić, Mićo, Samo Pirnat, Katja Fink, Maja Potokar, Marko Kreft, Robert Zorec, and Matjaž Stenovec. 2023. "Ketamine Reduces the Surface Density of the Astroglial Kir4.1 Channel and Inhibits Voltage-Activated Currents in a Manner Similar to the Action of Ba2+ on K+ Currents" Cells 12, no. 10: 1360. https://doi.org/10.3390/cells12101360
APA StyleBožić, M., Pirnat, S., Fink, K., Potokar, M., Kreft, M., Zorec, R., & Stenovec, M. (2023). Ketamine Reduces the Surface Density of the Astroglial Kir4.1 Channel and Inhibits Voltage-Activated Currents in a Manner Similar to the Action of Ba2+ on K+ Currents. Cells, 12(10), 1360. https://doi.org/10.3390/cells12101360