


The Role of Regulatory T Cells in the Onset and Progression of Primary Sjögren’s Syndrome


Abstract
1. Introduction
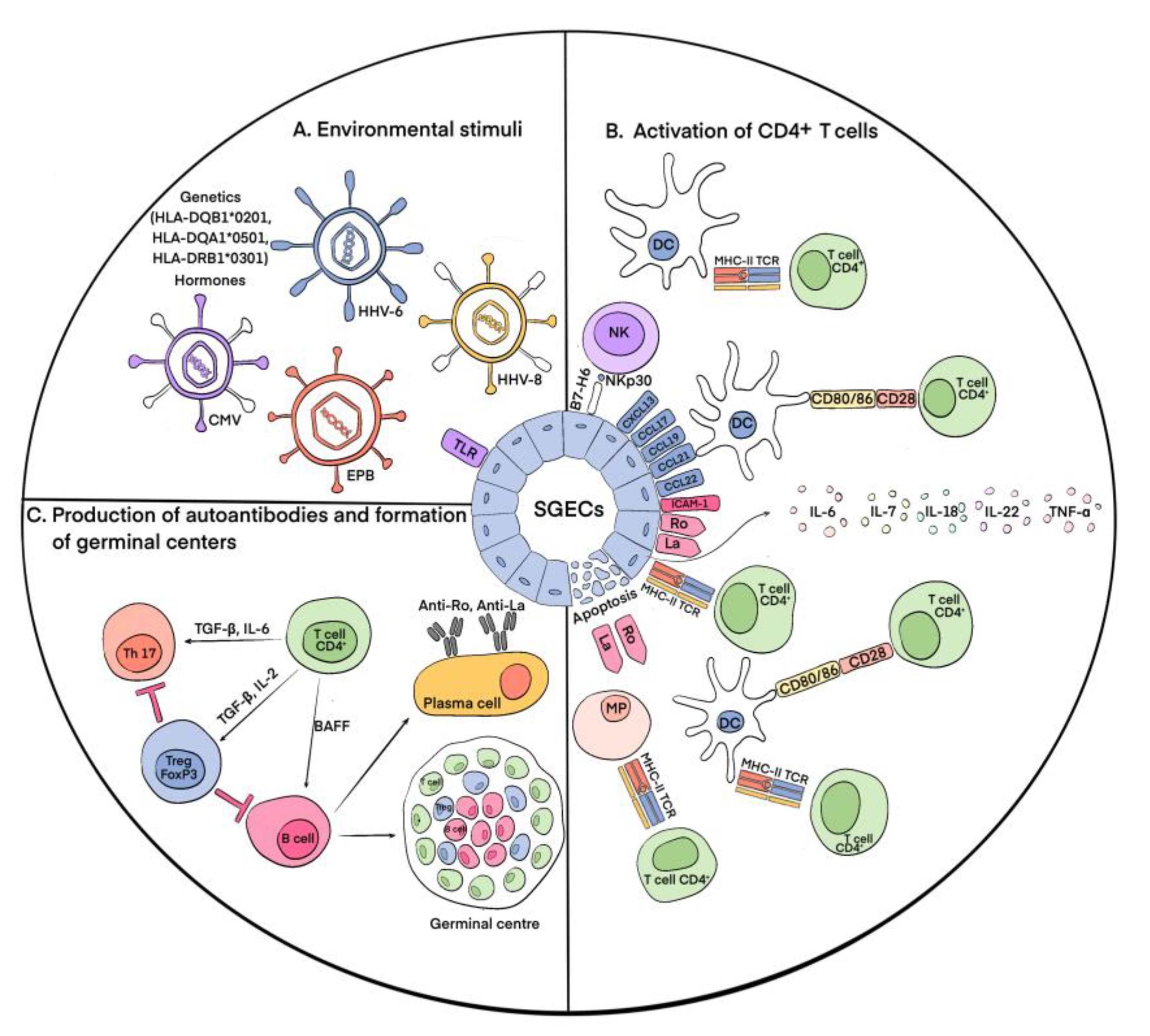
2. Immunopathogenesis of pSS
3. Regulatory T Cells (Tregs)
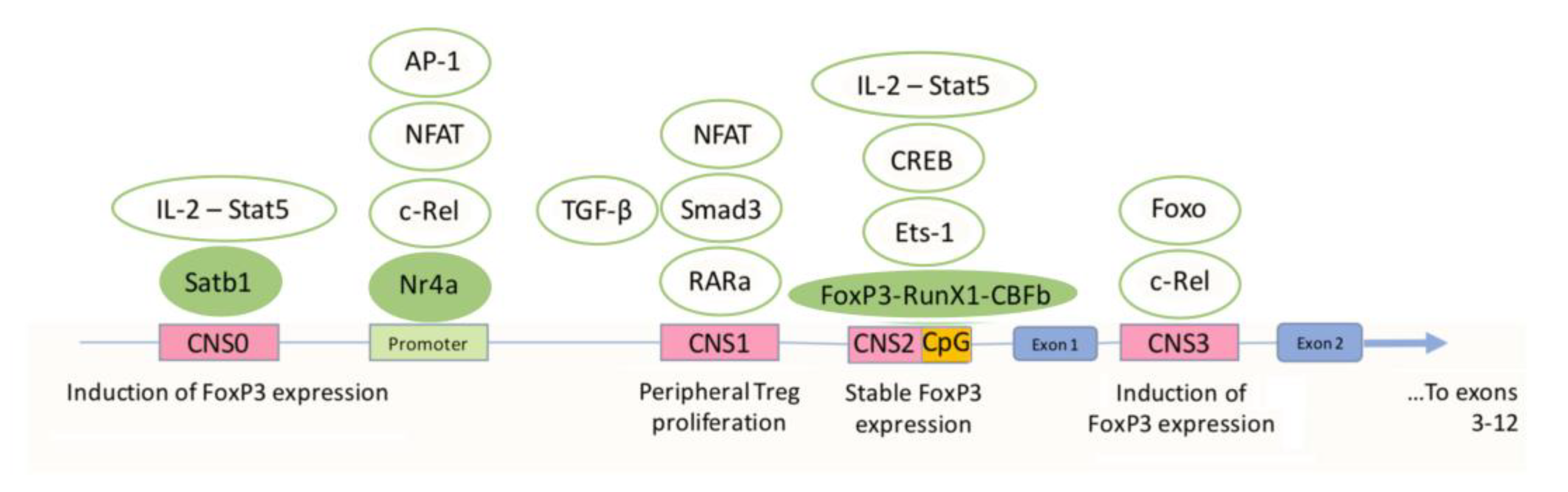
3.1. Differentiation and Activation of Tregs and the Role of FoxP3
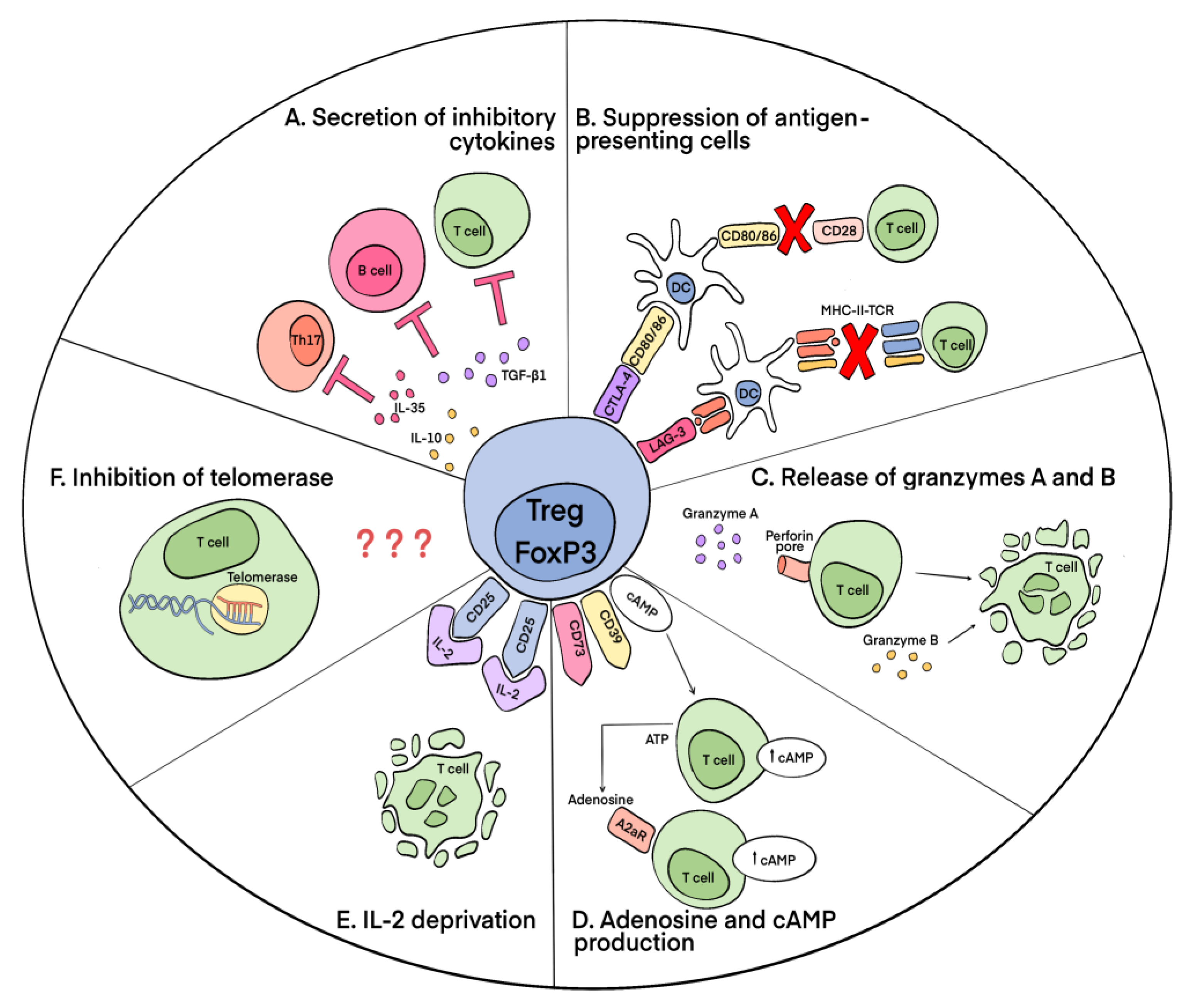
3.2. Mechanisms of Suppressive Action of Tregs
3.3. The Role of FoxP3 Protein in the Regulation of the Functional Activity of Tregs
4. Involvement of Tregs in pSS
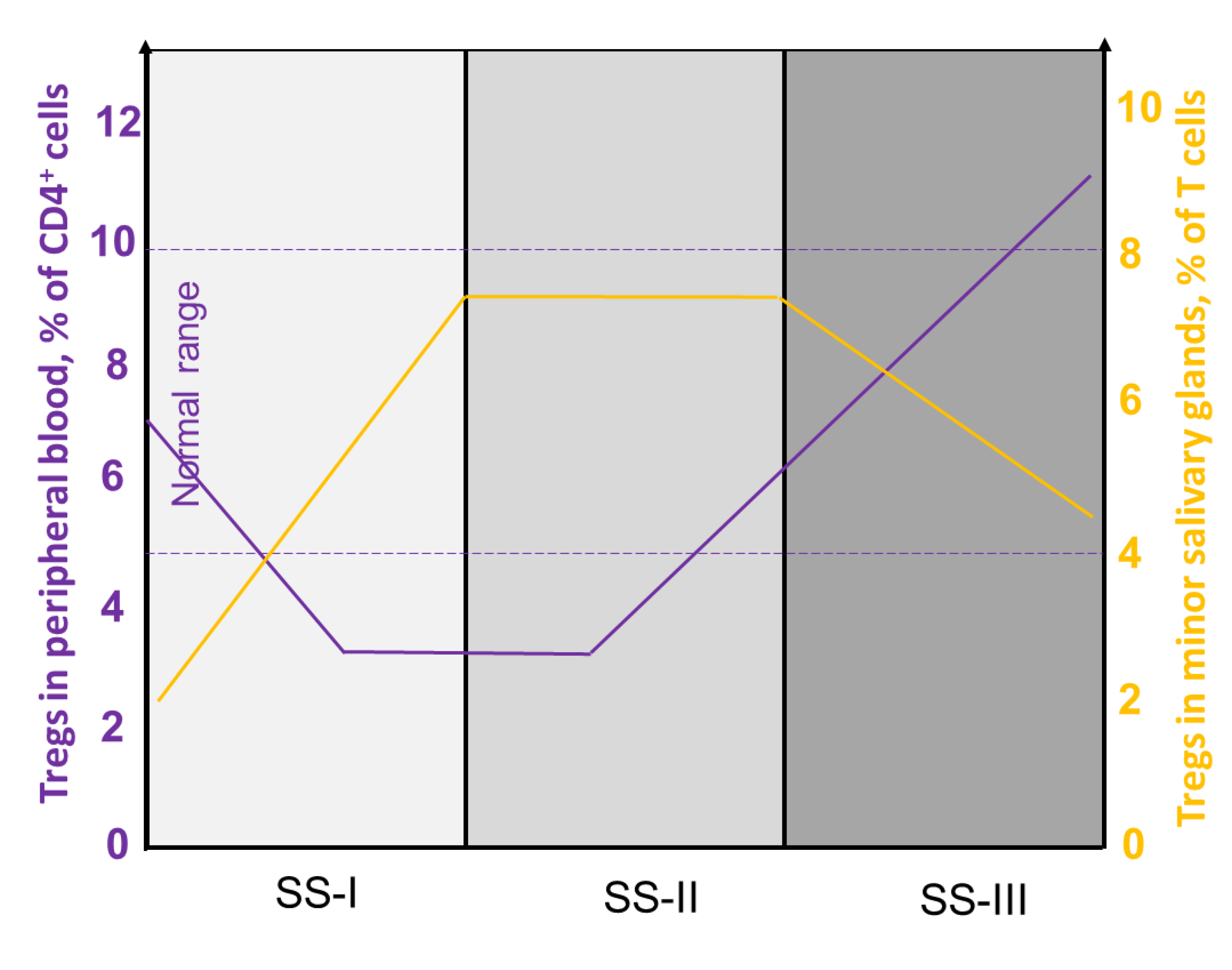
4.1. Heterogeneity of Tregs in pSS
4.2. Tregs and Ectopic Germinal Centers
5. Immunotherapy of pSS
5.1. Pharmaceutical B Cell Therapy
5.2. T Cell Therapy
5.2.1. Pharmaceutical Drug Therapy
5.2.2. Treg-Based Therapy
6. Future Perspectives on Treg Studies in pSS
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, Y.J.; Chih, P.L.; Huang, T.H.; Yu, H.S.; Hsieh, Y.L.; Yu, S. Skin Ultrastructural Findings in Acquired Generalized Hypohidrosis/Anhidrosis in a Patient with Subclinical Sjögren Syndrome. Acta Derm. Venereol. 2017, 97, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Hatta, K. Acquired Generalized Anhidrosis: Review of the Literature and Report of a Case with Lymphocytic Hidradenitis and Sialadenitis Successfully Treated with Cyclosporine. Dermatology 2013, 227, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Vivino, F.; Bunya, V.Y.; Massaro-Giordano, G.; Johr, C.R.; Giattino, S.L.; Schorpion, A.; Shafer, B.; Peck, A.; Sivils, K.; Rasmussen, A.; et al. Sjogren’s Syndrome: An Update on Disease Pathogenesis, Clinical Manifestations and Treatment. Clin. Immunol. 2019, 203, 81–121. [Google Scholar] [CrossRef]
- Fox, R.I.; Fox, C.M. Sjögren’s Syndrome: Practical Guidelines to Diagnosis and Therapy; Springer: Berlin/Heidelberg, Germany, 2011; p. 506. [Google Scholar]
- Negrini, S.; Emmi, G.; Greco, M.; Borro, M.; Sardanelli, F.; Murdaca, G.; Indiveri, F.; Puppo, F. Sjögren’s Syndrome: A Systemic Autoimmune Disease. Clin. Exp. Med. 2022, 22, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.E.; Priori, R.; Valesini, G.; Fairweather, D. Sex Differences in Sjögren’s Syndrome: A Comprehensive Review of Immune Mechanisms. Biol. Sex Differ. 2015, 6, 19. [Google Scholar] [CrossRef]
- Recalde, G.; Moreno-Sosa, T.; Yúdica, F.; Quintero, C.A.; Sánchez, M.B.; Jahn, G.A.; Kalergis, A.M.; Mackern-Oberti, J.P. Contribution of Sex Steroids and Prolactin to the Modulation of T and B Cells during Autoimmunity. Autoimmun. Rev. 2018, 17, 504–512. [Google Scholar] [CrossRef]
- Angum, F.; Khan, T.; Kaler, J.; Siddiqui, L.; Hussain, A. The Prevalence of Autoimmune Disorders in Women: A Narrative Review. Cureus 2020, 12, e8094. [Google Scholar] [CrossRef]
- Moulton, V.R. Sex Hormones in Acquired Immunity and Autoimmune Disease. Front. Immunol. 2018, 9, 2279. [Google Scholar] [CrossRef]
- Straub, R.H. The Complex Role of Estrogens in Inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef]
- Konttinen, Y.T.; Fuellen, G.; Bing, Y.; Porola, P.; Stegaev, V.; Trokovic, N.; Falk, S.S.I.; Liu, Y.; Szodoray, P.; Takakubo, Y. Sex Steroids in Sjögren’s Syndrome. J. Autoimmun. 2012, 39, 49–56. [Google Scholar] [CrossRef]
- Manoussakis, M.N.; Tsinti, M.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. The Salivary Gland Epithelial Cells of Patients with Primary Sjögren’s Syndrome Manifest Significantly Reduced Responsiveness to 17β-Estradiol. J. Autoimmun. 2012, 39, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, R.; Cheung, A.S.; Pang, K.; Saffery, R.; Novakovic, B. Sexual Dimorphism in Innate Immunity: The Role of Sex Hormones and Epigenetics. Front. Immunol. 2021, 11, 604000. [Google Scholar] [CrossRef] [PubMed]
- Bupp, M.R.G.; Jorgensen, T.N. Androgen-Induced Immunosuppression. Front. Immunol. 2018, 9, 794. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, M.I.; Kapsogeorgou, E.K.; Moutsopoulos, N.M.; Moutsopoulos, H.M. Foxp3+ T-Regulatory Cells in Sjögren’s Syndrome: Correlation with the Grade of the Autoimmune Lesion and Certain Adverse Prognostic Factors. Am. J. Pathol. 2008, 173, 1389–1396. [Google Scholar] [CrossRef]
- Pijpe, J.; Kalk, W.W.I.; van der Wal, J.E.; Vissink, A.; Kluin, P.M.; Roodenburg, J.L.N.; Bootsma, H.; Kallenberg, C.G.M.; Spijkervet, F.K.L. Parotid Gland Biopsy Compared with Labial Biopsy in the Diagnosis of Patients with Primary Sjogren’s Syndrome. Rheumatology 2007, 46, 335–341. [Google Scholar] [CrossRef]
- Vasaitis, L.; Nordmark, G.; Theander, E.; Backlin, C.; Smedby, K.E.; Askling, J.; Rönnblom, L.; Sundström, C.; Baecklund, E. Population-Based Study of Patients with Primary Sjögren’s Syndrome and Lymphoma: Lymphoma Subtypes, Clinical Characteristics, and Gender Differences. Scand. J. Rheumatol. 2020, 49, 225–232. [Google Scholar] [CrossRef]
- Gorodetskiy, V.R.; Probatova, N.A.; Radenska-Lopovok, S.G.; Ryzhikova, N.V.; Sidorova, Y.V.; Sudarikov, A.B. Clonal Relationship of Marginal Zone Lymphoma and Diffuse Large B-Cell Lymphoma in Sjogren’s Syndrome Patients: Case Series Study and Review of the Literature. Rheumatol. Int. 2020, 40, 499–506. [Google Scholar] [CrossRef]
- Chivasso, C.; Sarrand, J.; Perret, J.; Delporte, C.; Soyfoo, M.S. The Involvement of Innate and Adaptive Immunity in the Initiation and Perpetuation of Sjögren’s Syndrome. Int. J. Mol. Sci. 2021, 22, 658. [Google Scholar] [CrossRef]
- Goules, A.V.; Tzioufas, A.G. Primary Sjӧgren’s Syndrome: Clinical Phenotypes, Outcome and the Development of Biomarkers. Autoimmun. Rev. 2016, 15, 695–703. [Google Scholar] [CrossRef]
- Goules, A.V.; Kapsogeorgou, E.K.; Tzioufas, A.G. Insight into Pathogenesis of Sjögren’s Syndrome: Dissection on Autoimmune Infiltrates and Epithelial Cells. Clin. Immunol. 2017, 182, 30–40. [Google Scholar] [CrossRef]
- Liu, Z.; Chu, A. Sjögren’s Syndrome and Viral Infections. Rheumatol. Ther. 2021, 8, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Manganelli, P.; Fietta, P. Apoptosis and Sjögren Syndrome. Semin. Arthritis Rheum. 2003, 33, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L. Nucleic Acid-Sensing Toll-like Receptors: Important Players in Sjögren’s Syndrome. Front. Immunol. 2022, 13, 980400. [Google Scholar] [CrossRef] [PubMed]
- Manoussakis, M.N.; Spachidou, M.P.; Maratheftis, C.I. Salivary Epithelial Cells from Sjogren’s Syndrome Patients Are Highly Sensitive to Anoikis Induced by TLR-3 Ligation. J. Autoimmun. 2010, 35, 212–218. [Google Scholar] [CrossRef]
- Ittah, M.; Miceli-Richard, C.; Gottenberg, J.E.; Sellam, J.; Eid, P.; Lebon, P.; Pallier, C.; Lepajolec, C.; Mariette, X. Viruses Induce High Expression of BAFF by Salivary Gland Epithelial Cells through TLR- and Type-I IFN-Dependent and -Independent Pathways. Eur. J. Immunol. 2008, 38, 1058–1064. [Google Scholar] [CrossRef]
- Kyriakidis, N.C.; Kapsogeorgou, E.K.; Gourzi, V.C.; Konsta, O.D.; Baltatzis, G.E.; Tzioufas, A.G. Toll-like Receptor 3 Stimulation Promotes Ro52/TRIM21 Synthesis and Nuclear Redistribution in Salivary Gland Epithelial Cells, Partially via Type I Interferon Pathway. Clin. Exp. Immunol. 2014, 178, 548–560. [Google Scholar] [CrossRef]
- Verstappen, G.M.; Pringle, S.; Bootsma, H.; Kroese, F.G.M. Epithelial-Immune Cell Interplay in Primary Sjögren Syndrome Salivary Gland Pathogenesis. Nat. Rev. Rheumatol. 2021, 17, 333–348. [Google Scholar] [CrossRef]
- Kiripolsky, J.; Kramer, J.M. Current and Emerging Evidence for Toll-Like Receptor Activation in Sjögren’s Syndrome. J. Immunol. Res. 2018, 2018, 1246818. [Google Scholar] [CrossRef]
- Lee, G.R. The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci. 2018, 19, 730. [Google Scholar] [CrossRef]
- Konsta, O.D.; Le Dantec, C.; Charras, A.; Cornec, D.; Kapsogeorgou, E.K.; Tzioufas, A.G.; Pers, J.O.; Renaudineau, Y. Defective DNA Methylation in Salivary Gland Epithelial Acini from Patients with Sjögren’s Syndrome Is Associated with SSB Gene Expression, Anti-SSB/LA Detection, and Lymphocyte Infiltration. J. Autoimmun. 2016, 68, 30–38. [Google Scholar] [CrossRef]
- Cruz-Tapias, P.; Rojas-Villarraga, A.; Maier-Moore, S.; Anaya, J.M. HLA and Sjögren’s Syndrome Susceptibility. A Meta-Analysis of Worldwide Studies. Autoimmun. Rev. 2012, 11, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Warner, B.M.; Odani, T.; Ji, Y.; Mo, Y.-Q.; Nakamura, H.; Jang, S.-I.; Yin, H.; Michael, D.G.; Hirata, N.; et al. LAMP3 Induces Apoptosis and Autoantigen Release in Sjögren’s Syndrome Patients. Sci. Rep. 2020, 10, 15169. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, S.; Seamon, V.; Azzarolo, A.M. Influence of Sex Hormones and Genetic Predisposition in Sjögren’s Syndrome: A New Clue to the Immunopathogenesis of Dry Eye Disease. Exp. Eye Res. 2012, 96, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, Y.; Ito, T.; Son, Y.; Amuro, H.; Shimamoto, K.; Sugimoto, H.; Katashiba, Y.; Ogata, M.; Miyamoto, R.; Murakami, N.; et al. Decrease of Blood Dendritic Cells and Increase of Tissue-Infiltrating Dendritic Cells Are Involved in the Induction of Sjögren’s Syndrome but Not in the Maintenance. Clin. Exp. Immunol. 2010, 159, 315. [Google Scholar] [CrossRef]
- Ainola, M.; Porola, P.; Takakubo, Y.; Przybyla, B.; Kouri, V.P.; Tolvanen, T.A.; Hänninen, A.; Nordström, D.C. Activation of Plasmacytoid Dendritic Cells by Apoptotic Particles—Mechanism for the Loss of Immunological Tolerance in Sjögren’s Syndrome. Clin. Exp. Immunol. 2018, 191, 301–310. [Google Scholar] [CrossRef]
- Aloisi, F.; Pujol-Borrell, R. Lymphoid Neogenesis in Chronic Inflammatory Diseases. Nat. Rev. Immunol. 2006, 6, 205–217. [Google Scholar] [CrossRef]
- Christodoulou, M.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. Characteristics of the Minor Salivary Gland Infiltrates in Sjögren’s Syndrome. J. Autoimmun. 2010, 34, 400–407. [Google Scholar] [CrossRef]
- Rusakiewicz, S.; Nocturne, G.; Lazure, T.; Semeraro, M.; Flament, C.; Caillat-Zucman, S.; Sène, D.; Delahaye, N.; Vivier, E.; Chaba1, K.; et al. NCR3/NKp30 Contributes to Pathogenesis in Primary Sjögren’s Syndrome. Sci. Transl. Med. 2013, 5, 195ra96. [Google Scholar] [CrossRef]
- Pontarini, E.; Sciacca, E.; Grigoriadou, S.; Rivellese, F.; Lucchesi, D.; Fossati-Jimack, L.; Coleby, R.; Chowdhury, F.; Calcaterra, F.; Tappuni, A.; et al. NKp30 Receptor Upregulation in Salivary Glands of Sjögren’s Syndrome Characterizes Ectopic Lymphoid Structures and Is Restricted by Rituximab Treatment. Front. Immunol. 2021, 12, 706737. [Google Scholar] [CrossRef]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic Lymphoid-like Structures in Infection, Cancer and Autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462. [Google Scholar] [CrossRef]
- Barr, J.Y.; Wang, X.; Meyerholz, D.K.; Lieberman, S.M. CD8 T Cells Contribute to Lacrimal Gland Pathology in the Nonobese Diabetic Mouse Model of Sjögren Syndrome. Immunol. Cell Biol. 2017, 95, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Marketos, N.; Cinoku, I.; Rapti, A.; Mavragani, C.P. Type I Interferon Signature in Sjögren’s Syndrome: Pathophysiological and Clinical Implications. Clin. Exp. Rheumatol. 2019, 37 (Suppl. S118), S185–S191. [Google Scholar]
- Youinou, P.; Pers, J.-O. Disturbance of Cytokine Networks in Sjögren’s Syndrome. Arthritis Res. Ther. 2011, 13, 227. [Google Scholar] [CrossRef]
- Katsifis, G.E.; Rekka, S.; Moutsopoulos, N.M.; Pillemer, S.; Wahl, S.M. Systemic and Local Interleukin-17 and Linked Cytokines Associated with Sjögren’s Syndrome Immunopathogenesis. Am. J. Pathol. 2009, 175, 1167. [Google Scholar] [CrossRef]
- Crotty, S. Follicular Helper CD4 T Cells (TFH). Annu. Rev. Immunol. 2011, 29, 621–663. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S. Naturally Arising Foxp3-Expressing CD25+CD4+ Regulatory T Cells in Immunological Tolerance to Self and Non-Self. Nat. Immunol. 2005, 6, 345–352. [Google Scholar] [CrossRef]
- Belkaid, Y.; Rouse, B.T. Natural Regulatory T Cells in Infectious Disease. Nat. Immunol. 2005, 6, 353–360. [Google Scholar] [CrossRef]
- Rocamora-Reverte, L.; Melzer, F.L.; Würzner, R.; Weinberger, B. The Complex Role of Regulatory T Cells in Immunity and Aging. Front. Immunol. 2021, 11, 3566. [Google Scholar] [CrossRef]
- Gottschalk, R.A.; Corse, E.; Allison, J.P. TCR Ligand Density and Affinity Determine Peripheral Induction of Foxp3 in Vivo. J. Exp. Med. 2010, 207, 1701–1711. [Google Scholar] [CrossRef]
- Zheng, Y.; Josefowicz, S.; Chaudhry, A.; Peng, X.P.; Forbush, K.; Rudensky, A.Y. Role of Conserved Non-Coding DNA Elements in the Foxp3 Gene in Regulatory T-Cell Fate. Nature 2010, 463, 808. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Arvey, A.; Josefowicz, S.Z.; Peng, X.; Reynolds, A.; Sandstrom, R.; Neph, S.; Sabo, P.; Kim, J.M.; Liao, W.; et al. Foxp3 Exploits a Pre-Existent Enhancer Landscape for Regulatory T Cell Lineage Specification. Cell 2012, 151, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Rudra, D.; Deroos, P.; Chaudhry, A.; Niec, R.E.; Arvey, A.; Samstein, R.M.; Leslie, C.; Shaffer, S.A.; Goodlett, D.R.; Rudensky, A.Y. Transcription Factor Foxp3 and Its Protein Partners Form a Complex Regulatory Network. Nat. Immunol. 2012, 13, 1010–1019. [Google Scholar] [CrossRef]
- Arvey, A.; Van Der Veeken, J.; Samstein, R.M.; Feng, Y.; Stamatoyannopoulos, J.A.; Rudensky, A.Y. Inflammation-Induced Repression of Chromatin Bound by the Transcription Factor Foxp3 in Regulatory T Cells. Nat. Immunol. 2014, 15, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Liao, X.; Kang, Y. Tregs: Where We Are and What Comes Next? Front. Immunol. 2017, 8, 1578. [Google Scholar] [CrossRef] [PubMed]
- Fulford, T.S.; Grumont, R.; Wirasinha, R.C.; Ellis, D.; Barugahare, A.; Turner, S.J.; Naeem, H.; Powell, D.; Lyons, P.A.; Smith, K.G.C.; et al. C-Rel Employs Multiple Mechanisms to Promote the Thymic Development and Peripheral Function of Regulatory T Cells in Mice. Eur. J. Immunol. 2021, 51, 2006–2026. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Ohkura, N.; Kidani, Y.; Vandenbon, A.; Hirota, K.; Kawakami, R.; Yasuda, K.; Motooka, D.; Nakamura, S.; Kondo, M.; et al. Guidance of Regulatory T Cell Development by Satb1-Dependent Super-Enhancer Establishment. Nat. Immunol. 2017, 18, 173–183. [Google Scholar] [CrossRef]
- Dikiy, S.; Li, J.; Bai, L.; Jiang, M.; Janke, L.; Zong, X.; Hao, X.; Hoyos, B.; Wang, Z.M.; Xu, B.; et al. A Distal Foxp3 Enhancer Enables Interleukin-2 Dependent Thymic Treg Cell Lineage Commitment for Robust Immune Tolerance. Immunity 2021, 54, 931–946. [Google Scholar] [CrossRef]
- Rudensky, A.Y. Regulatory T Cells and Foxp3. Immunol. Rev. 2011, 241, 260. [Google Scholar] [CrossRef]
- Xiao, S.; Jin, H.; Korn, T.; Liu, S.M.; Oukka, M.; Lim, B.; Kuchroo, V.K. Retinoic Acid Increases Foxp3+ Regulatory T Cells and Inhibits Development of Th17 Cells by Enhancing TGF-β-Driven Smad3 Signaling and Inhibiting IL-6 and IL-23 Receptor Expression. J. Immunol. 2008, 181, 2277. [Google Scholar] [CrossRef]
- Ono, M. Control of Regulatory T-Cell Differentiation and Function by T-Cell Receptor Signalling and Foxp3 Transcription Factor Complexes. Immunology 2020, 160, 24–37. [Google Scholar] [CrossRef]
- Tone, Y.; Furuuchi, K.; Kojima, Y.; Tykocinski, M.L.; Greene, M.I.; Tone, M. Smad3 and NFAT Cooperate to Induce Foxp3 Expression through Its Enhancer. Nat. Immunol. 2008, 9, 194–202. [Google Scholar] [CrossRef]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of Treg and TH17 Cell Differentiation by the Aryl Hydrocarbon Receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Alunno, A.; Carubbi, F.; Bistoni, O.; Caterbi, S.; Bartoloni, E.; Mirabelli, G.; Cannarile, F.; Cipriani, P.; Giacomelli, R.; Gerli, R. T Regulatory and T Helper 17 Cells in Primary Sjögren’s Syndrome: Facts and Perspectives. Mediat. Inflamm. 2015, 2015, 243723. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 Control over Foxp3+ Regulatory T Cell Function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Legány, N.; Berta, L.; Kovács, L.; Balog, A.; Toldi, G. The Role of B7 Family Costimulatory Molecules and Indoleamine 2,3-Dioxygenase in Primary Sjögren’s Syndrome and Systemic Sclerosis. Immunol. Res. 2016, 65, 622–629. [Google Scholar] [CrossRef]
- Liang, B.; Workman, C.; Lee, J.; Chew, C.; Dale, B.M.; Colonna, L.; Flores, M.; Li, N.; Schweighoffer, E.; Greenberg, S.; et al. Regulatory T Cells Inhibit Dendritic Cells by Lymphocyte Activation Gene-3 Engagement of MHC Class II. J. Immunol. 2008, 180, 5916–5926. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-Inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Schmitt, E.G.; Williams, C.B. Generation and Function of Induced Regulatory T Cells. Front. Immunol. 2013, 4, 152. [Google Scholar] [CrossRef]
- Klein, M.; Bopp, T. Cyclic AMP Represents a Crucial Component of Treg Cell-Mediated Immune Regulation. Front. Immunol. 2016, 7, 315. [Google Scholar] [CrossRef]
- Pandiyan, P.; Zheng, L.; Ishihara, S.; Reed, J.; Lenardo, M.J. CD4 + CD25 + Foxp3 + Regulatory T Cells Induce Cytokine Deprivation-Mediated Apoptosis of Effector CD4 + T Cells. Nat. Immunol. 2007, 8, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lei, L. Interleukin-35 Regulates the Balance of Th17 and Treg Responses during the Pathogenesis of Connective Tissue Diseases. Int. J. Rheum. Dis. 2021, 24, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.H.S.; Britton, G.J.; Hill, E.V.; Verhagen, J.; Burton, B.R.; Wraith, D.C. Regulation of Adaptive Immunity; The Role of Interleukin-10. Front. Immunol. 2013, 4, 129. [Google Scholar] [CrossRef]
- Mason, G.I.; Hamburger, J.; Bowman, S.; Matthews, J.B. Salivary Gland Expression of Transforming Growth Factor β Isoforms in Sjogren’s Syndrome and Benign Lymphoepithelial Lesions. Mol. Pathol. 2003, 56, 52. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular Mechanisms Oftreg-Mediatedt Cell Suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef] [PubMed]
- Zhdanov, D.D.; Gladilina, Y.A.; Grishin, D.V.; Grachev, V.A.; Orlova, V.S.; Pokrovskaya, M.V.; Alexandrova, S.S.; Pokrovsky, V.S.; Sokolov, N.N. Contact-Independent Suppressive Activity of Regulatory T Cells Is Associated with Telomerase Inhibition, Telomere Shortening and Target Lymphocyte Apoptosis. Mol. Immunol. 2018, 101, 229–244. [Google Scholar] [CrossRef]
- Zhdanov, D.D.; Gladilina, Y.A.; Pokrovsky, V.S.; Grishin, D.V.; Grachev, V.A.; Orlova, V.S.; Pokrovskaya, M.V.; Alexandrova, S.S.; Sokolov, N.N. Murine Regulatory T Cells Induce Death of Effector T, B, and NK Lymphocytes through a Contact-Independent Mechanism Involving Telomerase Suppression and Telomere-Associated Senescence. Cell. Immunol. 2018, 331, 146–160. [Google Scholar] [CrossRef]
- Wan, Y.Y.; Flavell, R.A. Regulatory T-Cell Functions Are Subverted and Converted Owing to Attenuated Foxp3 Expression. Nature 2007, 445, 766–770. [Google Scholar] [CrossRef]
- Gavin, M.A.; Rasmussen, J.P.; Fontenot, J.D.; Vasta, V.; Manganiello, V.C.; Beavo, J.A.; Rudensky, A.Y. Foxp3-Dependent Programme of Regulatory T-Cell Differentiation. Nature 2007, 445, 771–775. [Google Scholar] [CrossRef]
- Joly, A.L.; Liu, S.; Dahlberg, C.I.M.; Mailer, R.K.W.; Westerberg, L.S.; Andersson, J. Foxp3 Lacking Exons 2 and 7 Is Unable to Confer Suppressive Ability to Regulatory T Cells in Vivo. J. Autoimmun. 2015, 63, 23–30. [Google Scholar] [CrossRef]
- Blinova, V.G.; Novachly, N.S.; Gippius, S.N.; Hilal, A.; Gladilina, Y.A.; Eliseeva, D.D.; Zhdanov, D.D. Phenotypical and Functional Characteristics of Human Regulatory t Cells during Ex Vivo Maturation from Cd4+ t Lymphocytes. Appl. Sci. 2021, 11, 5776. [Google Scholar] [CrossRef]
- Du, J.; Wang, Q.; Yang, S.; Chen, S.; Fu, Y.; Spath, S.; Domeier, P.; Hagin, D.; Anover-Sombke, S.; Haouili, M.; et al. FOXP3 Exon 2 Controls Treg Stability and Autoimmunity. Sci. Immunol. 2022, 7, eabo5407. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Kozhaya, L.; Mercer, F.; Khaitan, A.; Fujii, H.; Unutmaz, D. Expression of GARP Selectively Identifies Activated Human FOXP3+ Regulatory T Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 13439–13444. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Liu, J.; Lee, E.; Perriman, R.; Roncarolo, M.G.; Bacchetta, R. Co-Expression of FOXP3FL and FOXP3Δ2 Isoforms Is Required for Optimal Treg-Like Cell Phenotypes and Suppressive Function. Front. Immunol. 2021, 12, 4184. [Google Scholar] [CrossRef] [PubMed]
- Szodoray, P.; Papp, G.; Horvath, I.F.; Barath, S.; Sipka, S.; Nakken, B.; Zeher, M. Cells with Regulatory Function of the Innate and Adaptive Immune System in Primary Sjögren’s Syndrome: ORIGINAL ARTICLE. Clin. Exp. Immunol. 2009, 157, 343–349. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Qian, L.; Wang, G.; Zhang, H.; Wang, X.; Chen, K.; Zhai, Z.; Li, Q.; Wang, Y.; et al. T Regulatory Cells Are Markedly Diminished in Diseased Salivary Glands of Patients with Primary Sjögren’s Syndrome. J. Rheumatol. 2007, 34, 2438–2445. [Google Scholar]
- Liu, M.F.; Lin, L.H.; Weng, C.T.; Weng, M.Y. Decreased CD4+CD25+bright T Cells in Peripheral Blood of Patients with Primary Sjögren’s Syndrome. Lupus 2008, 17, 34–39. [Google Scholar] [CrossRef]
- Banica, L.; Besliu, A.; Pistol, G.; Stavaru, C.; Ionescu, R.; Forsea, A.M.; Tanaseanu, C.; Dumitrache, S.; Otelea, D.; Tamsulea, I.; et al. Quantification and Molecular Characterization of Regulatory T Cells in Connective Tissue Diseases. Autoimmunity 2009, 42, 41–49. [Google Scholar] [CrossRef]
- Alunno, A.; Petrillo, M.G.; Nocentini, G.; Bistoni, O.; Bartoloni, E.; Caterbi, S.; Bianchini, R.; Baldini, C.; Nicoletti, I.; Riccardi, C.; et al. Characterization of a New Regulatory CD4+ T Cell Subset in Primary Sjögren’s Syndrome. Rheumatology 2013, 52, 1387–1396. [Google Scholar] [CrossRef]
- Sarigul, M.; Yazisiz, V.; Başsorgun, C.I.; Ulker, M.; Avci, A.B.; Erbasan, F.; Gelen, T.; Gorczynski, R.M.; Terzioǧlu, E. The Numbers of Foxp3+ Treg Cells Are Positively Correlated with Higher Grade of Infiltration at the Salivary Glands in Primary Sjögren’s Syndrome. Lupus 2010, 19, 138–145. [Google Scholar] [CrossRef]
- Gottenberg, J.E.; Lavie, F.; Abbed, K.; Gasnault, J.; Le Nevot, E.; Delfraissy, J.F.; Taoufik, Y.; Mariette, X. CD4 CD25high Regulatory T Cells Are Not Impaired in Patients with Primary Sjögren’s Syndrome. J. Autoimmun. 2005, 24, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Getnet, D.; Grosso, J.F.; Goldberg, M.V.; Harris, T.J.; Yen, H.R.; Bruno, T.C.; Durham, N.M.; Hipkiss, E.L.; Pyle, K.J.; Wada, S.; et al. A Role for the Transcription Factor Helios in Human CD4(+)CD25(+) Regulatory T Cells. Mol. Immunol. 2010, 47, 1595–1600. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Guan, Z.; Zhao, L.; Song, Y.; Wang, H. Elevated Level of Circulating CD4+Helios+FoxP3+ Cells in Primary Sjogren’s Syndrome Patients. Mod. Rheumatol. 2017, 27, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Qi, J.; Sun, L. SAT0016 Il-12 Suppress Tr1 Cells in the SjÖgren’s Syndrome. Ann. Rheum. Dis. 2018, 77, 876. [Google Scholar] [CrossRef]
- Risselada, A.P.; Looije, M.F.; Kruize, A.A.; Bijlsma, J.W.J.; Van Roon, J.A.G. The Role of Ectopic Germinal Centers in the Immunopathology of Primary Sjögren’s Syndrome: A Systematic Review. Semin. Arthritis Rheum. 2013, 42, 368–376. [Google Scholar] [CrossRef]
- Lee, K.-E.; Kang, J.-H.; Yim, Y.-R.; Kim, J.-E.; Lee, J.-W.; Wen, L.; Park, D.-J.; Kim, T.-J.; Park, Y.-W.; Yoon, K.C.; et al. The Significance of Ectopic Germinal Centers in the Minor Salivary Gland of Patients with Sjögren’s Syndrome. J. Korean Med. Sci. 2016, 31, 190. [Google Scholar] [CrossRef]
- Stebegg, M.; Kumar, S.D.; Silva-Cayetano, A.; Fonseca, V.R.; Linterman, M.A.; Graca, L. Regulation of the Germinal Center Response. Front. Immunol. 2018, 9, 2469. [Google Scholar] [CrossRef]
- Sage, P.T.; Sharpe, A.H. T Follicular Regulatory Cells in the Regulation of B Cell Responses. Trends Immunol. 2015, 36, 410–418. [Google Scholar] [CrossRef]
- Chung, Y.; Tanaka, S.; Chu, F.; Nurieva, R.I.; Martinez, G.J.; Rawal, S.; Wang, Y.H.; Lim, H.; Reynolds, J.M.; Zhou, X.H.; et al. Follicular Regulatory T Cells Expressing Foxp3 and Bcl-6 Suppress Germinal Center Reactions. Nat. Med. 2011, 17, 983–988. [Google Scholar] [CrossRef]
- Yao, Y.; Ma, J.F.; Chang, C.; Xu, T.; Gao, C.Y.; Gershwin, M.E.; Lian, Z.X. Immunobiology of T Cells in Sjögren’s Syndrome. Clin. Rev. Allergy Immunol. 2021, 60, 111–131. [Google Scholar] [CrossRef]
- Verstappen, G.M.; Kroese, F.G.M.; Bootsma, H. T Cells in Primary Sjögren’s Syndrome: Targets for Early Intervention. Rheumatology 2019, 60, 3088–3098. [Google Scholar] [CrossRef] [PubMed]
- Verstappen, G.M.; Nakshbandi, U.; Mossel, E.; Haacke, E.A.; van der Vegt, B.; Vissink, A.; Bootsma, H.; Kroese, F.G.M. Is the T Follicular Regulatory:Follicular Helper T Cell Ratio in Blood a Biomarker for Ectopic Lymphoid Structure Formation in Sjögren’s Syndrome? Comment on the Article by Fonseca et Al. Arthritis Rheumatol. 2018, 70, 1354–1355. [Google Scholar] [CrossRef]
- Sayin, I.; Radtke, A.J.; Vella, L.A.; Jin, W.; Wherry, E.J.; Buggert, M.; Betts, M.R.; Herati, R.S.; Germain, R.N.; Canaday, D.H. Spatial Distribution and Function of T Follicular Regulatory Cells in Human Lymph Nodes. J. Exp. Med. 2018, 215, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, V.R.; Agua-Doce, A.; Maceiras, A.R.; Pierson, W.; Ribeiro, F.; Romão, V.C.; Pires, A.R.; da Silva, S.L.; Fonseca, J.E.; Sousa, A.E.; et al. Human Blood Tfr Cells Are Indicators of Ongoing Humoral Activity Not Fully Licensed with Suppressive Function. Sci. Immunol. 2017, 2, eaan1487. [Google Scholar] [CrossRef] [PubMed]
- Sage, P.T.; Alvarez, D.; Godec, J.; Von Andrian, U.H.; Sharpe, A.H. Circulating T Follicular Regulatory and Helper Cells Have Memory-like Properties. J. Clin. Investig. 2014, 124, 5191–5204. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Makarenkova, H.P. Innate Immunity and Biological Therapies for the Treatment of Sjögren’s Syndrome. Int. J. Mol. Sci. 2020, 21, 9172. [Google Scholar] [CrossRef]
- Dass, S.; Bowman, S.J.; Vital, E.M.; Ikeda, K.; Pease, C.T.; Hamburger, J.; Richards, A.; Rauz, S.; Emery, P. Reduction of Fatigue in Sjögren Syndrome with Rituximab: Results of a Randomised, Double-Blind, Placebo-Controlled Pilot Study. Ann. Rheum. Dis. 2008, 67, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Meijer, J.M.; Meiners, P.M.; Vissink, A.; Spijkervet, F.K.L.; Abdulahad, W.; Kamminga, N.; Brouwer, E.; Kallenberg, C.G.M.; Bootsma, H. Effectiveness of Rituximab Treatment in Primary Sjögren’s Syndrome: A Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheum. 2010, 62, 960–968. [Google Scholar] [CrossRef]
- Gottenberg, J.E.; Cinquetti, G.; Larroche, C.; Combe, B.; Hachulla, E.; Meyer, O.; Pertuiset, E.; Kaplanski, G.; Chiche, L.; Berthelot, J.M.; et al. Efficacy of Rituximab in Systemic Manifestations of Primary Sjogren’s Syndrome: Results in 78 Patients of the AutoImmune and Rituximab Registry. Ann. Rheum. Dis. 2013, 72, 1026–1031. [Google Scholar] [CrossRef]
- Carubbi, F.; Cipriani, P.; Marrelli, A.; Benedetto, P.D.; Ruscitti, P.; Berardicurti, O.; Pantano, I.; Liakouli, V.; Alvaro, S.; Alunno, A.; et al. Efficacy and Safety of Rituximab Treatment in Early Primary Sjögren’s Syndrome: A Prospective, Multi-Center, Follow-up Study. Arthritis Res. 2013, 15, R172. [Google Scholar] [CrossRef]
- Meiners, P.M.; Arends, S.; Brouwer, E.; Spijkervet, F.K.L.; Vissink, A.; Bootsma, H. Responsiveness of Disease Activity Indices ESSPRI and ESSDAI in Patients with Primary Sjögren’s Syndrome Treated with Rituximab. Ann. Rheum. Dis. 2012, 71, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Devauchelle-Pensec, V.; Mariette, X.; Jousse-Joulin, S.; Berthelot, J.-M.; Perdriger, A.; Puéchal, X.; Le Guern, V.; Sibilia, J.; Gottenberg, J.-E.; Chiche, L.; et al. Treatment of Primary Sjögren Syndrome with Rituximab: A Randomized Trial. Ann. Intern. Med. 2014, 160, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Navarro Coy, N.; Pitzalis, C.; Emery, P.; Pavitt, S.; Gray, J.; Hulme, C.; Hall, F.; Busch, R.; Smith, P.; et al. The TRACTISS Protocol: A Randomised Double Blind Placebo Controlled Clinical TRial of Anti-B-Cell Therapy In Patients with Primary Sjögren’s Syndrome. BMC Musculoskelet. Disord. 2014, 15, 21. [Google Scholar] [CrossRef]
- Bowman, S.J.; Everett, C.C.; O’Dwyer, J.L.; Emery, P.; Pitzalis, C.; Ng, W.F.; Pease, C.T.; Price, E.J.; Sutcliffe, N.; Gendi, N.S.T.; et al. Randomized Controlled Trial of Rituximab and Cost-Effectiveness Analysis in Treating Fatigue and Oral Dryness in Primary Sjögren’s Syndrome. Arthritis Rheumatol. 2017, 69, 1440–1450. [Google Scholar] [CrossRef] [PubMed]
- Steinfeld, S.D.; Tant, L.; Burmester, G.R.; Teoh, N.K.W.; Wegener, W.A.; Goldenberg, D.M.; Pradier, O. Epratuzumab (Humanised Anti-CD22 Antibody) in Primary Sjögren’s Syndrome: An Open-Label Phase I/II Study. Arthritis Res. 2006, 8, R129. [Google Scholar] [CrossRef] [PubMed]
- Gottenberg, J.E.; Dörner, T.; Bootsma, H.; Devauchelle-Pensec, V.; Bowman, S.J.; Mariette, X.; Bartz, H.; Oortgiesen, M.; Shock, A.; Koetse, W.; et al. Efficacy of Epratuzumab, an Anti-CD22 Monoclonal IgG Antibody, in Systemic Lupus Erythematosus Patients With Associated Sjögren’s Syndrome: Post Hoc Analyses From the EMBODY Trials. Arthritis Rheumatol. 2018, 70, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Mariette, X.; Seror, R.; Quartuccio, L.; Baron, G.; Salvin, S.; Fabris, M.; Desmoulins, F.; Nocturne, G.; Ravaud, P.; De Vita, S. Efficacy and Safety of Belimumab in Primary Sjögren’s Syndrome: Results of the BELISS Open-Label Phase II Study. Ann. Rheum. Dis. 2015, 74, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Dörner, T.; Posch, M.G.; Li, Y.; Petricoul, O.; Cabanski, M.; Milojevic, J.M.; Kamphausen, E.; Valentin, M.A.; Simonett, C.; Mooney, L.; et al. Treatment of Primary Sjögren’s Syndrome with Ianalumab (VAY736) Targeting B Cells by BAFF Receptor Blockade Coupled with Enhanced, Antibody-Dependent Cellular Cytotoxicity. Ann. Rheum. Dis. 2019, 78, 641–647. [Google Scholar] [CrossRef]
- Remibrutinib (LOU064) in Sjögren’s Syndrome: Safety and Efficacy Results from a 24-Week Placebo-Controlled Proof-of-Concept Study—ACR Meeting Abstracts. Available online: https://acrabstracts.org/abstract/remibrutinib-lou064-in-sjogrens-syndrome-safety-and-efficacy-results-from-a-24%E2%80%91week-placebo-controlled-proof-of-concept-study/ (accessed on 18 April 2023).
- Bowman, S.J.; Fox, R.; Dörner, T.; Mariette, X.; Papas, A.; Grader-Beck, T.; Fisher, B.A.; Barcelos, F.; De Vita, S.; Schulze-Koops, H.; et al. Safety and Efficacy of Subcutaneous Ianalumab (VAY736) in Patients with Primary Sjögren’s Syndrome: A Randomised, Double-Blind, Placebo-Controlled, Phase 2b Dose-Finding Trial. Lancet 2022, 399, 161–171. [Google Scholar] [CrossRef]
- St. Clair, E.W.; Baer, A.N.; Wei, C.; Noaiseh, G.; Parke, A.; Coca, A.; Utset, T.O.; Genovese, M.C.; Wallace, D.J.; McNamara, j. Clinical Efficacy and Safety of Baminercept, a Lymphotoxin β Receptor Fusion Protein, in Primary Sjögren’s Syndrome: Results From a Phase II Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2018, 70, 1470–1480. [Google Scholar] [CrossRef]
- Adler, S.; Körner, M.; Förger, F.; Huscher, D.; Caversaccio, M.D.; Villiger, P.M. Evaluation of Histologic, Serologic, and Clinical Changes in Response to Abatacept Treatment of Primary Sjögren’s Syndrome: A Pilot Study. Arthritis Care Res. 2013, 65, 1862–1868. [Google Scholar] [CrossRef] [PubMed]
- Meiners, P.M.; Vissink, A.; Kroese, F.G.M.; Spijkervet, F.K.L.; Smitt-Kamminga, N.S.; Abdulahad, W.H.; Bulthuis-Kuiper, J.; Brouwer, E.; Arends, S.; Bootsma, H. Abatacept Treatment Reduces Disease Activity in Early Primary Sjögren’s Syndrome (Open-Label Proof of Concept ASAP Study). Ann. Rheum. Dis. 2014, 73, 1393–1396. [Google Scholar] [CrossRef] [PubMed]
- Verstappen, G.M.; Meiners, P.M.; Corneth, O.B.J.; Visser, A.; Arends, S.; Abdulahad, W.H.; Hendriks, R.W.; Vissink, A.; Kroese, F.G.M.; Bootsma, H. Attenuation of Follicular Helper T Cell-Dependent B Cell Hyperactivity by Abatacept Treatment in Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2017, 69, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Spijkervet, F.K.L.; Onno Teng, Y.K.; Arends, E.J.; van Nimwegen, J.F.; van Zuiden, G.S.; Arends, S.; Mossel, E.; Wijnsma, R.F.; Stel, A.J.; Delli, K.; et al. Abatacept Treatment of Patients with Early Active Primary Sjogren’s Syndrome—A Randomized, Double-Blind Placebo-Controlled Phase Iii-Study (ASAP-III). Ann. Rheum. Dis. 2019, 78, 93. [Google Scholar] [CrossRef]
- Papp, K.A. The Long-Term Efficacy and Safety of New Biological Therapies for Psoriasis. Arch. Dermatol. Res. 2006, 298, 7–15. [Google Scholar] [CrossRef] [PubMed]
- A Phase 2a Study of MEDI5872 (AMG557), a Fully Human Anti-ICOS Ligand Monoclonal Antibody in Patients with Primary Sjögren’s Syndrome—ACR Meeting Abstracts. Available online: https://acrabstracts.org/abstract/a-phase-2a-study-of-medi5872-amg557-a-fully-human-anti-icos-ligand-monoclonal-antibody-in-patients-with-primary-sjogrens-syndrome/ (accessed on 21 April 2023).
- Pilat, N.; Sprent, J. Treg Therapies Revisited: Tolerance Beyond Deletion. Front. Immunol. 2021, 11, 3663. [Google Scholar] [CrossRef]
- Desreumaux, P.; Foussat, A.; Allez, M.; Beaugerie, L.; Hébuterne, X.; Bouhnik, Y.; Nachury, M.; Brun, V.; Bastian, H.; Belmonte, N.; et al. Safety and Efficacy of Antigen-Specific Regulatory T-Cell Therapy for Patients with Refractory Crohn’s Disease. Gastroenterology 2012, 143, 1207–1217. [Google Scholar] [CrossRef]
- Canavan, J.B.; Scottà, C.; Vossenkämper, A.; Goldberg, R.; Elder, M.J.; Shoval, I.; Marks, E.; Stolarczyk, E.; Lo, J.W.; Powell, N.; et al. Developing in Vitro Expanded CD45RA+ Regulatory T Cells as an Adoptive Cell Therapy for Crohn’s Disease. Gut 2016, 65, 584–594. [Google Scholar] [CrossRef]
- Goldberg, R.; Scotta, C.; Cooper, D.; Nissim-Eliraz, E.; Nir, E.; Tasker, S.; Irving, P.M.; Sanderson, J.; Lavender, P.; Ibrahim, F.; et al. Correction of Defective T-Regulatory Cells From Patients With Crohn’s Disease by Ex Vivo Ligation of Retinoic Acid Receptor-α. Gastroenterology 2019, 156, 1775–1787. [Google Scholar] [CrossRef]
- Marek-Trzonkowska, N.; Myśliwiec, M.; Dobyszuk, A.; Grabowska, M.; Derkowska, I.; Juścińska, J.; Owczuk, R.; Szadkowska, A.; Witkowski, P.; Młynarski, W.; et al. Therapy of Type 1 Diabetes with CD4(+)CD25(High)CD127-Regulatory T Cells Prolongs Survival of Pancreatic Islets—Results of One Year Follow-Up. Clin. Immunol. 2014, 153, 23–30. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 Diabetes Immunotherapy Using Polyclonal Regulatory T Cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef]
- Dong, S.; Hiam-Galvez, K.J.; Mowery, C.T.; Herold, K.C.; Gitelman, S.E.; Esensten, J.H.; Liu, W.; Lares, A.P.; Leinbach, A.S.; Lee, M.; et al. The Effect of Low-Dose IL-2 and Treg Adoptive Cell Therapy in Patients with Type 1 Diabetes. JCI Insight 2021, 6, e147474. [Google Scholar] [CrossRef] [PubMed]
- Dall’Era, M.; Pauli, M.L.; Remedios, K.; Taravati, K.; Sandova, P.M.; Putnam, A.L.; Lares, A.; Haemel, A.; Tang, Q.; Hellerstein, M.; et al. Adoptive Treg Cell Therapy in a Patient With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Hou, Y.; Gong, W.; Liu, S.; Li, J.; Yuan, Y.; Zhang, D.; Chen, Q.; Yan, X. Adoptive Induced Antigen-Specific Treg Cells Reverse Inflammation in Collagen-Induced Arthritis Mouse Model. Inflammation 2018, 41, 485–495. [Google Scholar] [CrossRef]
- Lifshitz, G.V.; Zhdanov, D.D.; Lokhonina, A.V.; Eliseeva, D.D.; Lyssuck, E.Y.; Zavalishin, I.A.; Bykovskaia, S.N. Ex Vivo Expanded Regulatory T Cells CD4+CD25+FoxP3+CD127Low Develop Strong Immunosuppressive Activity in Patients with Remitting-Relapsing Multiple Sclerosis. Autoimmunity 2016, 49, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Goswami, T.K.; Singh, M.; Dhawan, M.; Mitra, S.; Emran, T.B.; Rabaan, A.A.; Al Mutair, A.; Al Alawi, Z.; Alhumaid, S.; Dhama, K. Regulatory T Cells (Tregs) and Their Therapeutic Potential against Autoimmune Disorders—Advances and Challenges. Hum. Vaccines Immunother. 2022, 18, e2035117. [Google Scholar] [CrossRef] [PubMed]
- Retamozo, S.; Flores-Chavez, A.; Consuegra-Fernández, M.; Lozano, F.; Ramos-Casals, M.; Brito-Zerón, P. Cytokines as Therapeutic Targets in Primary Sjögren Syndrome. Pharmacol. Ther. 2018, 184, 81–97. [Google Scholar] [CrossRef] [PubMed]
- Mariette, X.; Ravaud, P.; Steinfeld, S.; Baron, G.; Goetz, J.; Hachulla, E.; Combe, B.; Puéchal, X.; Pennec, Y.; Sauvezie, B.; et al. Inefficacy of Infliximab in Primary Sjögren’s Syndrome: Results of the Randomized, Controlled Trial of Remicade in Primary Sjögren’s Syndrome (TRIPSS). Arthritis Rheum. 2004, 50, 1270–1276. [Google Scholar] [CrossRef]
- Sankar, V.; Brennan, M.T.; Kok, M.R.; Leakan, R.A.; Smith, J.A.; Manny, J.; Baum, B.J.; Pillemer, S.R. Etanercept in Sjögren’s Syndrome: A Twelve-Week Randomized, Double-Blind, Placebo-Controlled Pilot Clinical Trial. Arthritis Rheum. 2004, 50, 2240–2245. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
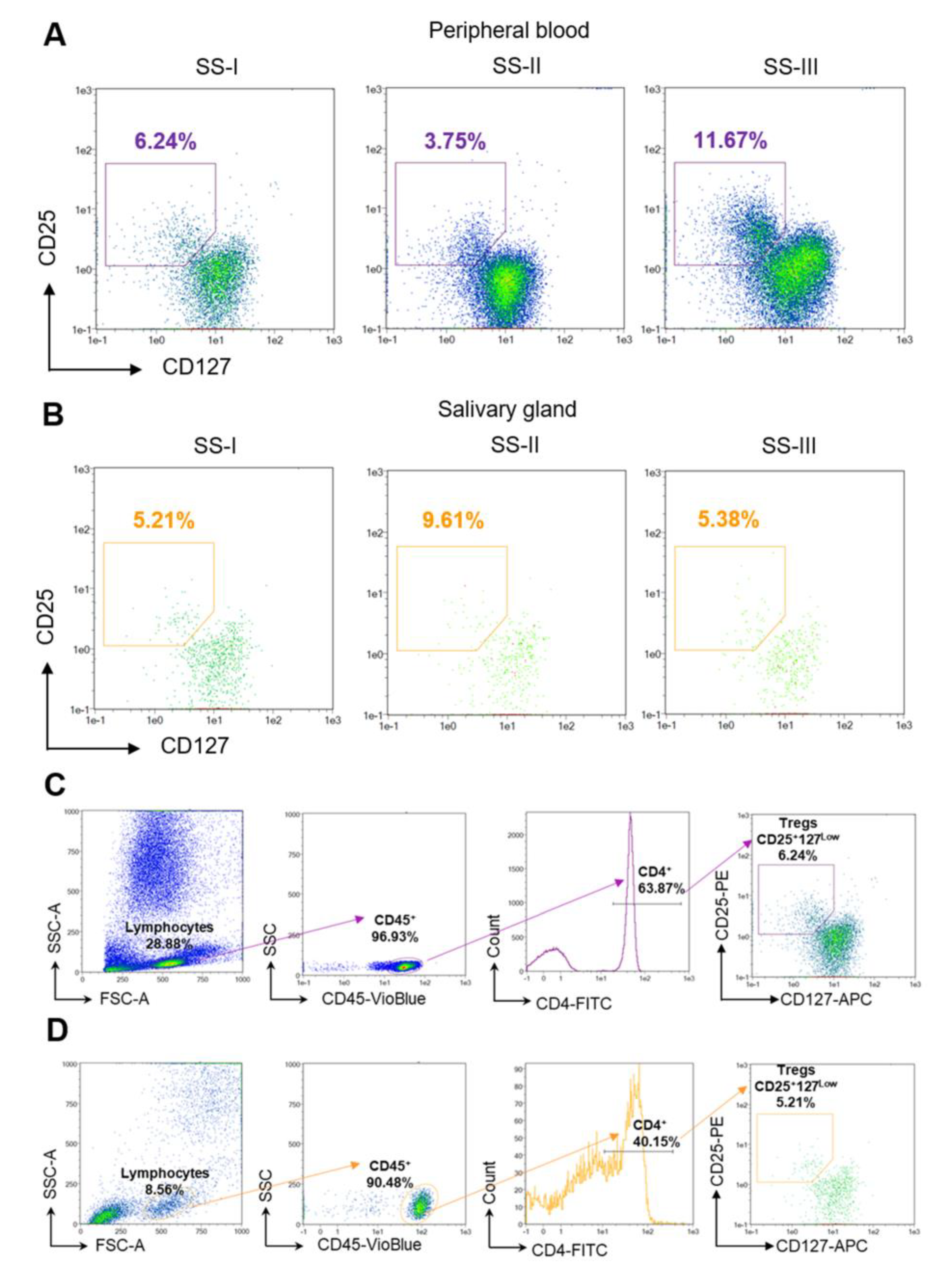{kind=link}
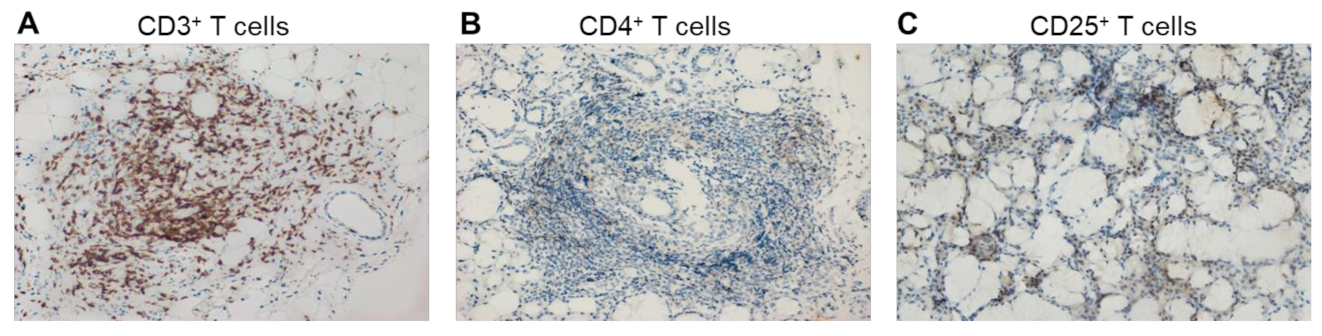{kind=link}
| Treg Population in Peripheral Blood | Number According to Disease State | Suppressive Activity | Reference |
|---|---|---|---|
| CD4+CD25+FoxP3+ | Decreased | Decreased | [86] |
| Decreased | Unchanged | [87] | |
| Decreased | Not studied | [88] | |
| Decreased | Not studied | [89] | |
| Decreased; no correlation with disease activity | Not studied | [90] | |
| Increased | Not studied | [91] | |
| Increased | Unchanged | [92] | |
| CD4+CD25lowGITR+ | Increased in patients with inactive disease | Unchanged | [90] |
| Helios+FoxP3+ | Increased | Increased | [93,94] |
| Tr1 cells | Decreased | Not studied | [95] |
| Drug | Target | Mechanism | Efficacy | Reference |
|---|---|---|---|---|
| Rituximab | CD20 on B-cell surface | Chimeric anti-CD20 antibody. Causes antibody-dependent cellular cytotoxicity, complement-mediated cytotoxicity, and apoptosis-mediated transient depletion of B cells in peripheral blood, salivary glands, and other target tissues. | Depends on the dose and duration of therapy. With a well-chosen course, stimulation of salivation and improvement in the function of lacrimal glands, a decrease in the activity of the disease according to the ESSDAI, and a reduction in infiltrates and GCs. | [108,109,110,111,112,113,114,115] |
| Epratuzumab | CD22 on B-cell surface | Humanized anti-CD22 antibody. Modulates B-cell activity (CD22 regulates B-cell function via CD19 and B-cell antigen receptor (BCR) signaling and induces BCR-induced cell death. CD22 also regulates TLR signaling and controls B-cell survival in peripheral organs). | Significant improvement in the function of the lacrimal glands, unstimulated salivation, and elimination of symptoms of fatigue. | [116,117] |
| Belimumab | BAFF | Human monoclonal antibody. Inhibits BAFF, thereby preventing their activation and proliferation. | Decrease in parotid edema and levels of B-cell activation biomarkers. No change in unstimulated salivation or Schirmer test. | [118] |
| Ianalumab (VAY736) | BAFF | Inhibits BAFF, leading to blockade of BAFF-mediated signaling and deletion of B cells. Direct lysis of B cells via antibody-dependent cellular cytotoxicity. | Dose-dependent reduction in disease activity according to the ESSDAI. | [119] |
| Remibrutinib | BTK | Inhibits BTK on B cells, leading to impaired BCR signaling that regulates B-cell proliferation and survival. | Improvement in the ESSDAI, salivary flow, and pathologically elevated immunoglobulins as signatures of activity. | [120,121] |
| Baminercept | Lymphotoxin-β receptor (LTβR) on B-cell surface | Recombinant lymphotoxin-β receptor fusion protein. Blockade of LTβR-mediated signaling inhibits lymphocytic infiltration and formation of ectopic GCs. | There was no significant decrease in disease activity according to the ESSDAI, no significant improvement in the secretion of the salivary and lacrimal glands, and extraglandular manifestations. Significant changes in the number of circulating T and B lymphocytes. | [122] |
| Drug | Target | Mechanism | Efficacy | Reference |
|---|---|---|---|---|
| Abatacept | CD80/86 on APC surface | Blocks the interaction between CD80/86 on APC surface with CD28 of T-cell surface, which is important for proliferation of T lymphocytes and production of cytokines. | Reduction in inflammation in salivary glands, improved salivation, reduction in the number of cTfh and Tregs, no changes in the foci of lymphoplasmacytic infiltration, and reduction in GCs. | [123,124,125,126] |
| Alefacept | CD2 on T-cell surface | Binds to CD2, inhibiting the interaction between LFA-3 and CD2, preventing the activation of T lymphocytes | Dose-dependent depletion of CD4+ and CD8+ cells in psoriasis | [127] |
| CFZ533 (iscalimab) | CD40 on APC and B-cell surfaces | Binds to CD40, blocking the interaction of APCs and B cells with CD40L of T lymphocytes | In phase 2 of clinical trials | [107] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blinova, V.G.; Vasilyev, V.I.; Rodionova, E.B.; Zhdanov, D.D. The Role of Regulatory T Cells in the Onset and Progression of Primary Sjögren’s Syndrome. Cells 2023, 12, 1359. https://doi.org/10.3390/cells12101359
Blinova VG, Vasilyev VI, Rodionova EB, Zhdanov DD. The Role of Regulatory T Cells in the Onset and Progression of Primary Sjögren’s Syndrome. Cells. 2023; 12(10):1359. https://doi.org/10.3390/cells12101359
Chicago/Turabian StyleBlinova, Varvara G., Vladimir I. Vasilyev, Ekaterina B. Rodionova, and Dmitry D. Zhdanov. 2023. "The Role of Regulatory T Cells in the Onset and Progression of Primary Sjögren’s Syndrome" Cells 12, no. 10: 1359. https://doi.org/10.3390/cells12101359
APA StyleBlinova, V. G., Vasilyev, V. I., Rodionova, E. B., & Zhdanov, D. D. (2023). The Role of Regulatory T Cells in the Onset and Progression of Primary Sjögren’s Syndrome. Cells, 12(10), 1359. https://doi.org/10.3390/cells12101359