


Endocrine Disrupting Chemicals in Polycystic Ovary Syndrome: The Relevant Role of the Theca and Granulosa Cells in the Pathogenesis of the Ovarian Dysfunction


 ,
,  and
and
Abstract
1. Introduction
2. Pathophysiology of Polycystic Ovary Syndrome
2.1. Neuroendocrine Dysfunctions and Reproductive Abnormalities in PCOS
2.2. Metabolic Disorders in PCOS
2.3. Environmental Factors and PCOS Development
3. The Role of Granulosa and Theca Cells in PCOS
3.1. Granulosa and Theca Cells—Two Cell, Two Gonadotropin Theory
3.2. The Role of AMH-Mediated SMAD Signaling Pathway in PCOS
3.3. The Role of the PI3K/AKT/FOXO Signaling Pathway in PCOS
3.4. The Role of the HMGA2/ IGF2BP2 Signaling Pathway in PCOS
3.5. The Role of Theca Cells in PCOS Development
3.6. The Role of Circadian Rhythm in PCOS Development
4. Endocrine Disrupting Chemicals (EDCs)
4.1. Reproductive and Neuroendocrine Dysfunctions Associated with Exposure to EDCs
4.2. Bisphenol A
4.3. Phthalates
4.4. 2,3,7,8-Tetrachlorodibenzo-p-dioxin
4.5. Tributyltin
4.6. Glyphosate
4.7. Other EDCs That Affect the Female Reproductive System
4.8. Endocrine Disrupting Chemicals and Pregnancy
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spritzer, P.M. Polycystic Ovary Syndrome: Reviewing Diagnosis and Management of Metabolic Disturbances. Arq. Bras. Endocrinol. Metabol. 2014, 58, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Azziz, R.; Woods, K.S.; Reyna, R.; Key, T.J.; Knochenhauer, E.S.; Yildiz, B.O. The Prevalence and Features of the Polycystic Ovary Syndrome in an Unselected Population. J. Clin. Endocrinol. Metab. 2004, 89, 2745–2749. [Google Scholar] [CrossRef] [PubMed]
- Deswal, R.; Narwal, V.; Dang, A.; Pundir, C. The Prevalence of Polycystic Ovary Syndrome: A Brief Systematic Review. J. Hum. Reprod. Sci. 2020, 13, 261. [Google Scholar] [CrossRef] [PubMed]
- Engmann, L.; Jin, S.; Sun, F.; Legro, R.S.; Polotsky, A.J.; Hansen, K.R.; Coutifaris, C.; Diamond, M.P.; Eisenberg, E.; Zhang, H.; et al. Racial and Ethnic Differences in the Polycystic Ovary Syndrome Metabolic Phenotype. Am. J. Obstet. Gynecol. 2017, 216, 493.e1–493.e13. [Google Scholar] [CrossRef]
- Zawadski, J.K.; Dunaif, A. Diagnostic Criteria for Polycystic Ovary Syndrome: Towards a Rational Approach. In Polycystic Ovary Syndrome; Dunaif, A., Givens, J.R., Haseltine, F.P., Merriam, G.R., Eds.; Blackwell Scientific: Boston, MA, USA, 1992; pp. 377–384. [Google Scholar]
- Azziz, R. Diagnosis of Polycystic Ovarian Syndrome: The Rotterdam Criteria Are Premature. J. Clin. Endocrinol. Metab. 2006, 91, 781–785. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, F.; Xiong, J.; Shi, X.; Fu, S. Characteristics of Different Phenotypes of Polycystic Ovary Syndrome Based on the Rotterdam Criteria in a Large-Scale Chinese Population. BJOG 2009, 116, 1633–1639. [Google Scholar] [CrossRef]
- Teede, H.J.; Misso, M.L.; Costello, M.F.; Dokras, A.; Laven, J.; Moran, L.; Piltonen, T.; Norman, R.J.; Andersen, M.; Azziz, R.; et al. Recommendations from the International Evidence-Based Guideline for the Assessment and Management of Polycystic Ovary Syndrome. Hum. Reprod. 2018, 33, 1602–1618. [Google Scholar] [CrossRef]
- Yildiz, B.O.; Bolour, S.; Woods, K.; Moore, A.; Azziz, R. Visually Scoring Hirsutism. Hum. Reprod. Update 2010, 16, 51–64. [Google Scholar] [CrossRef]
- DeUgarte, C.M.; Woods, K.S.; Bartolucci, A.A.; Azziz, R. Degree of Facial and Body Terminal Hair Growth in Unselected Black and White Women: Toward a Populational Definition of Hirsutism. J. Clin. Endocrinol. Metab. 2006, 91, 1345–1350. [Google Scholar] [CrossRef]
- Zhao, X.; Ni, R.; Li, L.; Mo, Y.; Huang, J.; Huang, M.; Azziz, R.; Yang, D. Defining Hirsutism in Chinese Women: A Cross-Sectional Study. Fertil. Steril. 2011, 96, 792–796. [Google Scholar] [CrossRef]
- Peña, A.S.; Codner, E.; Witchel, S. Criteria for Diagnosis of Polycystic Ovary Syndrome during Adolescence: Literature Review. Diagnostics 2022, 12, 1931. [Google Scholar] [CrossRef] [PubMed]
- Peña, A.S.; Witchel, S.F.; Hoeger, K.M.; Oberfield, S.E.; Vogiatzi, M.G.; Misso, M.; Garad, R.; Dabadghao, P.; Teede, H. Adolescent Polycystic Ovary Syndrome According to the International Evidence-Based Guideline. BMC Med. 2020, 18, 72. [Google Scholar] [CrossRef] [PubMed]
- Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 Consensus on Diagnostic Criteria and Long-Term Health Risks Related to Polycystic Ovary Syndrome. Fertil. Steril. 2004, 81, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Witchel, S.F.; Oberfield, S.; Rosenfield, R.L.; Codner, E.; Bonny, A.; Ibáñez, L.; Pena, A.; Horikawa, R.; Gomez-Lobo, V.; Joel, D.; et al. The Diagnosis of Polycystic Ovary Syndrome during Adolescence. Horm. Res. Paediatr. 2015, 83, 376–389. [Google Scholar] [CrossRef]
- American Academy of Pediatrics; Committee on Adolescence; American College of Obstetricians and Gynecologists; Committee on Adolescent Health Care. Menstruation in Girls and Adolescents: Using the Menstrual Cycle as a Vital Sign. Pediatrics 2006, 118, 2245–2250. [Google Scholar] [CrossRef]
- Carmina, E.; Lobo, R.A. Comparing Lean and Obese PCOS in Different PCOS Phenotypes: Evidence That the Body Weight Is More Important than the Rotterdam Phenotype in Influencing the Metabolic Status. Diagnostics 2022, 12, 2313. [Google Scholar] [CrossRef] [PubMed]
- Carmina, E. Need to Introduce the Finding of Obesity or Normal Body Weight in the Current Diagnostic Criteria and in the Classification of PCOS. Diagnostics 2022, 12, 2555. [Google Scholar] [CrossRef]
- Papadakis, M.A.; McPhee, S.J.; Bernstein, J. Polycystic Ovary Syndrome (Persistent Anovulation). In Quick Medical Diagnosis & Treatment; Papadakis, M.A., McPhee, S.J., Bernstein, J., Eds.; McGraw Hill: New York, NY, USA, 2020. [Google Scholar]
- Dumesic, D.A.; Padmanabhan, V.; Chazenbalk, G.D.; Abbott, D.H. Polycystic Ovary Syndrome as a Plausible Evolutionary Outcome of Metabolic Adaptation. Reprod. Biol. Endocrinol. 2022, 20, 12. [Google Scholar] [CrossRef]
- Parker, J.; O’Brien, C.; Hawrelak, J.; Gersh, F.L. Polycystic Ovary Syndrome: An Evolutionary Adaptation to Lifestyle and the Environment. Int. J. Environ. Res. Public Health 2022, 19, 1336. [Google Scholar] [CrossRef]
- Abbott, D.; Dumesic, D.; Franks, S. Developmental Origin of Polycystic Ovary Syndrome—A Hypothesis. J. Endocrinol. 2002, 174, 1–5. [Google Scholar] [CrossRef]
- Dumesic, D.A.; Abbott, D.H.; Padmanabhan, V. Polycystic Ovary Syndrome and Its Developmental Origins. Rev. Endocr. Metab. Disord. 2007, 8, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Bourguignon, J.-P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement. Endocr. Rev. 2009, 30, 293–342. [Google Scholar] [CrossRef]
- Cui, L.; Zhao, H.; Zhang, B.; Qu, Z.; Liu, J.; Liang, X.; Zhao, X.; Zhao, J.; Sun, Y.; Wang, P.; et al. Genotype–Phenotype Correlations of PCOS Susceptibility SNPs Identified by GWAS in a Large Cohort of Han Chinese Women. Hum. Reprod. 2013, 28, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-J.; Zhao, H.; He, L.; Shi, Y.; Qin, Y.; Shi, Y.; Li, Z.; You, L.; Zhao, J.; Liu, J.; et al. Genome-Wide Association Study Identifies Susceptibility Loci for Polycystic Ovary Syndrome on Chromosome 2p16.3, 2p21 and 9q33.3. Nature Genet. 2011, 43, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhao, H.; Shi, Y.; Cao, Y.; Yang, D.; Li, Z.; Zhang, B.; Liang, X.; Li, T.; Chen, J.; et al. Genome-Wide Association Study Identifies Eight New Risk Loci for Polycystic Ovary Syndrome. Nature Genet. 2012, 44, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- McAllister, J.M.; Legro, R.S.; Modi, B.P.; Strauss, J.F. Functional Genomics of PCOS: From GWAS to Molecular Mechanisms. Trends. Endocrinol. Metab. 2015, 26, 118–124. [Google Scholar] [CrossRef]
- Day, F.R.; Hinds, D.A.; Tung, J.Y.; Stolk, L.; Styrkarsdottir, U.; Saxena, R.; Bjonnes, A.; Broer, L.; Dunger, D.B.; Halldorsson, B.V.; et al. Causal Mechanisms and Balancing Selection Inferred from Genetic Associations with Polycystic Ovary Syndrome. Nat. Commun. 2015, 6, 8464. [Google Scholar] [CrossRef]
- Hayes, M.G.; Urbanek, M.; Ehrmann, D.A.; Armstrong, L.L.; Lee, J.Y.; Sisk, R.; Karaderi, T.; Barber, T.M.; McCarthy, M.I.; Franks, S.; et al. Genome-Wide Association of Polycystic Ovary Syndrome Implicates Alterations in Gonadotropin Secretion in European Ancestry Populations. Nat. Commun. 2015, 6, 7502. [Google Scholar] [CrossRef]
- Dapas, M.; Lin, F.T.J.; Nadkarni, G.N.; Sisk, R.; Legro, R.S.; Urbanek, M.; Hayes, M.G.; Dunaif, A. Distinct Subtypes of Polycystic Ovary Syndrome with Novel Genetic Associations: An Unsupervised, Phenotypic Clustering Analysis. PLoS Med. 2020, 17, e1003132. [Google Scholar] [CrossRef]
- Goodarzi, M.O.; Dumesic, D.A.; Chazenbalk, G.; Azziz, R. Polycystic Ovary Syndrome: Etiology, Pathogenesis and Diagnosis. Nat. Rev. Endocrinol. 2011, 7, 219–231. [Google Scholar] [CrossRef]
- Blank, S.K.; McCartney, C.R.; Marshall, J.C. The Origins and Sequelae of Abnormal Neuroendocrine Function in Polycystic Ovary Syndrome. Hum. Reprod. Update 2006, 12, 351–361. [Google Scholar] [CrossRef] [PubMed]
- McCartney, C.R.; Eagleson, C.A.; Marshall, J.C. Regulation of Gonadotropin Secretion: Implications for Polycystic Ovary Syndrome. Semin. Reprod. Med. 2002, 20, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Waldstreicher, J.; Santoro, N.F.; Hall, J.E.; Filicori, M.; Crowley, W.F. Hyperfunction of the Hypothalamic-Pituitary Axis in Women with Polycystic Ovarian Disease: Indirect Evidence for Partial Gonadotroph Desensitization. J. Clin. Endocrinol. Metab. 1988, 66, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.; Chávez, M.; Olivar, L.; Rojas, M.; Morillo, J.; Mejías, J.; Calvo, M.; Bermúdez, V. Polycystic Ovary Syndrome, Insulin Resistance, and Obesity: Navigating the Pathophysiologic Labyrinth. Int. J. Reprod. Med. 2014, 2014, 719050. [Google Scholar] [CrossRef]
- Eagleson, C.A.; Gingrich, M.B.; Pastor, C.L.; Arora, T.K.; Burt, C.M.; Evans, W.S.; Marshall, J.C. Polycystic Ovarian Syndrome: Evidence That Flutamide Restores Sensitivity of the Gonadotropin-Releasing Hormone Pulse Generator to Inhibition by Estradiol and Progesterone. J. Clin. Endocrinol. Metab. 2000, 85, 4047–4052. [Google Scholar] [CrossRef][Green Version]
- Walters, K.A.; Allan, C.M.; Handelsman, D.J. Androgen Actions and the Ovary. Biol. Reprod. 2008, 78, 380–389. [Google Scholar] [CrossRef]
- Vendola, K.A.; Zhou, J.; Adesanya, O.O.; Weil, S.J.; Bondy, C.A. Androgens Stimulate Early Stages of Follicular Growth in the Primate Ovary. J. Clin. Investig. 1998, 101, 2622–2629. [Google Scholar] [CrossRef]
- Wijeyaratne, C.N.; Balen, A.H.; Barth, J.H.; Belchetz, P.E. Clinical Manifestations and Insulin Resistance (IR) in Polycystic Ovary Syndrome (PCOS) among South Asians and Caucasians: Is There a Difference? Clin. Endocrinol. 2002, 57, 343–350. [Google Scholar] [CrossRef]
- Sadrzadeh, S.; Klip, W.A.; Broekmans, F.J.; Schats, R.; Willemsen, W.N.; Burger, C.W.; van Leeuwen, F.E.; Lambalk, C.B. OMEGA Project group Birth Weight and Age at Menarche in Patients with Polycystic Ovary Syndrome or Diminished Ovarian Reserve, in a Retrospective Cohort. Human. Reprod. 2003, 18, 2225–2230. [Google Scholar] [CrossRef]
- Abbott, D.H.; Nicol, L.E.; Levine, J.E.; Xu, N.; Goodarzi, M.O.; Dumesic, D.A. Nonhuman Primate Models of Polycystic Ovary Syndrome. Mol. Cell. Endocrinol. 2013, 373, 21–28. [Google Scholar] [CrossRef]
- Ehrmann, D.A.; Barnes, R.B.; Rosenfield, R.L. Polycystic Ovary Syndrome as a Form of Functional Ovarian Hyperandrogenism Due to Dysregulation of Androgen Secretion. Endocr. Rev. 1995, 16, 322–353. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Brennan, K.; Azziz, R. Prevalence of Hyperandrogenemia in the Polycystic Ovary Syndrome Diagnosed by the National Institutes of Health 1990 Criteria. Fertil. Steril. 2010, 93, 1938–1941. [Google Scholar] [CrossRef] [PubMed]
- Azziz, R.; Carmina, E.; Dewailly, D.; Diamanti-Kandarakis, E.; Escobar-Morreale, H.F.; Futterweit, W.; Janssen, O.E.; Legro, R.S.; Norman, R.J.; Taylor, A.E.; et al. The Androgen Excess and PCOS Society Criteria for the Polycystic Ovary Syndrome: The Complete Task Force Report. Fertil. Steril. 2009, 91, 456–488. [Google Scholar] [CrossRef] [PubMed]
- Azziz, R.; Sanchez, L.A.; Knochenhauer, E.S.; Moran, C.; Lazenby, J.; Stephens, K.C.; Taylor, K.; Boots, L.R. Androgen Excess in Women: Experience with Over 1000 Consecutive Patients. J. Clin. Endocrinol. Metab. 2004, 89, 453–462. [Google Scholar] [CrossRef]
- Rosenfield, R.L.; Ehrmann, D.A. The Pathogenesis of Polycystic Ovary Syndrome (PCOS): The Hypothesis of PCOS as Functional Ovarian Hyperandrogenism Revisited. Endocr. Rev. 2016, 37, 467–520. [Google Scholar] [CrossRef]
- Dunaif, A. Insulin Resistance and the Polycystic Ovary Syndrome: Mechanism and Implications for Pathogenesis. Endocr. Rev. 1997, 18, 774–800. [Google Scholar] [CrossRef]
- Ciaraldi, T.P.; Aroda, V.; Mudaliar, S.; Chang, R.J.; Henry, R.R. Polycystic Ovary Syndrome Is Associated with Tissue-Specific Differences in Insulin Resistance. J. Clin. Endocrinol. Metab. 2009, 94, 157–163. [Google Scholar] [CrossRef]
- Poretsky, L.; Seto-Young, D.; Shrestha, A.; Dhillon, S.; Mirjany, M.; Liu, H.-C.; Yih, M.C.; Rosenwaks, Z. Phosphatidyl-Inositol-3 Kinase-Independent Insulin Action Pathway(s) in the Human Ovary. J. Clin. Endocrinol. Metab. 2001, 86, 3115–3119. [Google Scholar] [CrossRef]
- Ciaraldi, T.P.; el-Roeiy, A.; Madar, Z.; Reichart, D.; Olefsky, J.M.; Yen, S.S. Cellular Mechanisms of Insulin Resistance in Polycystic Ovarian Syndrome. J. Clin. Endocrinol. Metab. 1992, 75, 577–583. [Google Scholar] [CrossRef]
- Dunaif, A.; Wu, X.; Lee, A.; Diamanti-Kandarakis, E. Defects in Insulin Receptor Signaling in Vivo in the Polycystic Ovary Syndrome (PCOS). Am. J. Physiol. Endocrinol. Metab. 2001, 281, E392–E399. [Google Scholar] [CrossRef]
- Rosenbaum, D.; Haber, R.S.; Dunaif, A. Insulin Resistance in Polycystic Ovary Syndrome: Decreased Expression of GLUT-4 Glucose Transporters in Adipocytes. Am. J. Physiol. Endocrinol. Metab. 1993, 264, E197–E202. [Google Scholar] [CrossRef] [PubMed]
- Ciaraldi, T.P.; Morales, A.J.; Hickman, M.G.; Odom-Ford, R.; Olefsky, J.M.; Yen, S.S.C. Cellular Insulin Resistance in Adipocytes from Obese Polycystic Ovary Syndrome Subjects Involves Adenosine Modulation of Insulin Sensitivity. J. Clin. Endocrinol. Metab. 1997, 82, 1421–1425. [Google Scholar] [CrossRef] [PubMed]
- Yildiz, B.O.; Knochenhauer, E.S.; Azziz, R. Impact of Obesity on the Risk for Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2008, 93, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Diamanti-Kandarakis, E.; Dunaif, A. Insulin Resistance and the Polycystic Ovary Syndrome Revisited: An Update on Mechanisms and Implications. Endocr. Rev. 2012, 33, 981–1030. [Google Scholar] [CrossRef]
- Zhang, L.H.; Rodriguez, H.; Ohno, S.; Miller, W.L. Serine Phosphorylation of Human P450c17 Increases 17,20-Lyase Activity: Implications for Adrenarche and the Polycystic Ovary Syndrome. Proc. Natl. Acad. Sci. USA 1995, 92, 10619–10623. [Google Scholar] [CrossRef]
- Glintborg, D.; Højlund, K.; Andersen, N.R.; Hansen, B.F.; Beck-Nielsen, H.; Wojtaszewski, J.F.P. Impaired Insulin Activation and Dephosphorylation of Glycogen Synthase in Skeletal Muscle of Women with Polycystic Ovary Syndrome Is Reversed by Pioglitazone Treatment. J. Clin. Endocrinol. Metab. 2008, 93, 3618–3626. [Google Scholar] [CrossRef]
- Højlund, K.; Glintborg, D.; Andersen, N.R.; Birk, J.B.; Treebak, J.T.; Frøsig, C.; Beck-Nielsen, H.; Wojtaszewski, J.F.P. Impaired Insulin-Stimulated Phosphorylation of Akt and AS160 in Skeletal Muscle of Women With Polycystic Ovary Syndrome Is Reversed by Pioglitazone Treatment. Diabetes 2008, 57, 357–366. [Google Scholar] [CrossRef]
- Zhao, X.; Zhong, J.; Mo, Y.; Chen, X.; Chen, Y.; Yang, D. Association of Biochemical Hyperandrogenism with Type 2 Diabetes and Obesity in Chinese Women with Polycystic Ovary Syndrome. Int. J. Gynaecol. Obstet. 2010, 108, 148–151. [Google Scholar] [CrossRef]
- Dokras, A.; Clifton, S.; Futterweit, W.; Wild, R. Increased Risk for Abnormal Depression Scores in Women With Polycystic Ovary Syndrome. Obstet. Gynecol. 2011, 117, 145–152. [Google Scholar] [CrossRef]
- Pasquali, R.; Stener-Victorin, E.; Yildiz, B.O.; Duleba, A.J.; Hoeger, K.; Mason, H.; Homburg, R.; Hickey, T.; Franks, S.; Tapanainen, J.S.; et al. PCOS Forum: Research in Polycystic Ovary Syndrome Today and Tomorrow. Clin. Endocrinol. 2011, 74, 424–433. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Piperi, C. Genetics of Polycystic Ovary Syndrome: Searching for the Way out of the Labyrinth. Hum. Reprod. Update 2005, 11, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Merkin, S.S.; Azziz, R.; Seeman, T.; Calderon-Margalit, R.; Daviglus, M.; Kiefe, C.; Matthews, K.; Sternfeld, B.; Siscovick, D. Socioeconomic Status and Polycystic Ovary Syndrome. J. Womens Health 2011, 20, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.H.; Habtewold, T.D.; Birhanu, M.M.; Sissay, T.A.; Tegegne, B.S.; Abuzerr, S.; Esmaillzadeh, A. Neighbourhood Socioeconomic Status and Overweight/Obesity: A Systematic Review and Meta-Analysis of Epidemiological Studies. BMJ Open 2019, 9, e028238. [Google Scholar] [CrossRef] [PubMed]
- Newton, S.; Braithwaite, D.; Akinyemiju, T.F. Socio-Economic Status over the Life Course and Obesity: Systematic Review and Meta-Analysis. PLoS ONE 2017, 12, e0177151. [Google Scholar] [CrossRef]
- Carmina, E.; Bucchieri, S.; Mansueto, P.; Rini, G.; Ferin, M.; Lobo, R.A. Circulating Levels of Adipose Products and Differences in Fat Distribution in the Ovulatory and Anovulatory Phenotypes of Polycystic Ovary Syndrome. Fertil. Steril. 2009, 91, 1332–1335. [Google Scholar] [CrossRef] [PubMed]
- Dewailly, D.; Catteau-Jonard, S.; Reyss, A.-C.; Leroy, M.; Pigny, P. Oligoanovulation with Polycystic Ovaries But Not Overt Hyperandrogenism. J. Clin. Endocrinol. Metab. 2006, 91, 3922–3927. [Google Scholar] [CrossRef] [PubMed]
- Moran, L.; Teede, H. Metabolic Features of the Reproductive Phenotypes of Polycystic Ovary Syndrome. Hum. Reprod. Update 2009, 15, 477–488. [Google Scholar] [CrossRef]
- Rizzo, M.; Berneis, K.; Hersberger, M.; Pepe, I.; di Fede, G.; Rini, G.B.; Spinas, G.A.; Carmina, E. Milder Forms of Atherogenic Dyslipidemia in Ovulatory versus Anovulatory Polycystic Ovary Syndrome Phenotype. Hum. Reprod. 2009, 24, 2286–2292. [Google Scholar] [CrossRef]
- Welt, C.K.; Gudmundsson, J.A.; Arason, G.; Adams, J.; Palsdottir, H.; Gudlaugsdottir, G.; Ingadottir, G.; Crowley, W.F. Characterizing Discrete Subsets of Polycystic Ovary Syndrome as Defined by the Rotterdam Criteria: The Impact of Weight on Phenotype and Metabolic Features. J. Clin. Endocrinol. Metab. 2006, 91, 4842–4848. [Google Scholar] [CrossRef]
- Carmina, E.; Legro, R.S.; Stamets, K.; Lowell, J.; Lobo, R.A. Difference in Body Weight between American and Italian Women with Polycystic Ovary Syndrome: Influence of the Diet. Hum. Reprod. 2003, 18, 2289–2293. [Google Scholar] [CrossRef]
- Clark, A.M.; Thornley, B.; Tomlinson, L.; Galletley, C.; Norman, R.J. Weight Loss in Obese Infertile Women Results in Improvement in Reproductive Outcome for All Forms of Fertility Treatment. Hum. Reprod. 1998, 13, 1502–1505. [Google Scholar] [CrossRef] [PubMed]
- Guzick, D.S.; Wing, R.; Smith, D.; Berga, S.L.; Winters, S.J. Endocrine Consequences of Weight Loss in Obese, Hyperandrogenic, Anovulatory Women. Fertil. Steril. 1994, 61, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Pasquali, R.; Antenucci, D.; Casimirri, F.; Venturoli, S.; Paradisi, R.; Fabbri, R.; Balestra, V.; Melchionda, N.; Barbara, L. Clinical and Hormonal Characteristics of Obese Amenorrheic Hyperandrogenic Women Before and After Weight Loss. J. Clin. Endocrinol. Metab. 1989, 68, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Moran, L.J.; Pasquali, R.; Teede, H.J.; Hoeger, K.M.; Norman, R.J. Treatment of Obesity in Polycystic Ovary Syndrome: A Position Statement of the Androgen Excess and Polycystic Ovary Syndrome Society. Fertil. Steril. 2009, 92, 1966–1982. [Google Scholar] [CrossRef]
- Huber-Buchholz, M.-M.; Carey, D.G.P.; Norman, R.J. Restoration of Reproductive Potential by Lifestyle Modification in Obese Polycystic Ovary Syndrome: Role of Insulin Sensitivity and Luteinizing Hormone1. J. Clin. Endocrinol. Metab. 1999, 84, 1470–1474. [Google Scholar] [CrossRef]
- Bruner, B.; Chad, K.; Chizen, D. Effects of Exercise and Nutritional Counseling in Women with Polycystic Ovary Syndrome. Appl. Physiol. Nutr. Metab. 2006, 31, 384–391. [Google Scholar] [CrossRef]
- Palioura, E.; Diamanti-Kandarakis, E. Polycystic Ovary Syndrome (PCOS) and Endocrine Disrupting Chemicals (EDCs). Rev. Endocr. Metab. Disord. 2015, 16, 365–371. [Google Scholar] [CrossRef]
- Kechagias, K.S.; Semertzidou, A.; Athanasiou, A.; Paraskevaidi, M.; Kyrgiou, M. Bisphenol-A and Polycystic Ovary Syndrome: A Review of the Literature. Rev. Environ. Health 2020, 35, 323–331. [Google Scholar] [CrossRef]
- Vandenberg, L.N.; Hauser, R.; Marcus, M.; Olea, N.; Welshons, W.V. Human Exposure to Bisphenol A (BPA). Reprod. Toxicol. 2007, 24, 139–177. [Google Scholar] [CrossRef]
- Zhou, W.; Liu, J.; Liao, L.; Han, S.; Liu, J. Effect of Bisphenol A on Steroid Hormone Production in Rat Ovarian Theca-Interstitial and Granulosa Cells. Mol. Cell. Endocrinol. 2008, 283, 12–18. [Google Scholar] [CrossRef]
- Alonso-Magdalena, P.; Morimoto, S.; Ripoll, C.; Fuentes, E.; Nadal, A. The Estrogenic Effect of Bisphenol A Disrupts Pancreatic β-Cell Function In Vivo and Induces Insulin Resistance. Environ. Health Perspect. 2006, 114, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Tsutsumi, O.; Ikezuki, Y.; Takai, Y.; Taketani, Y. Positive Relationship between Androgen and the Endocrine Disruptor, Bisphenol A, in Normal Women and Women with Ovarian Dysfunction. Endocr. J. 2004, 51, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Tsutsumi, O.; Nakamura, N.; Ikezuki, Y.; Takai, Y.; Yano, T.; Taketani, Y. Gender Difference in Serum Bisphenol A Levels May Be Caused by Liver UDP-Glucuronosyltransferase Activity in Rats. Biochem. Biophys. Res. Commun. 2004, 325, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Fabozzi, G.; Rebuzzini, P.; Cimadomo, D.; Allori, M.; Franzago, M.; Stuppia, L.; Garagna, S.; Ubaldi, F.M.; Zuccotti, M.; Rienzi, L. Endocrine-Disrupting Chemicals, Gut Microbiota, and Human (In)Fertility—It Is Time to Consider the Triad. Cells 2022, 11, 3335. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.; Gasbarrini, A.; Mele, M. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Hooper, L.V.; Gordon, J.I. Commensal Host-Bacterial Relationships in the Gut. Science (1979) 2001, 292, 1115–1118. [Google Scholar] [CrossRef]
- Claus, S.P.; Guillou, H.; Ellero-Simatos, S. The Gut Microbiota: A Major Player in the Toxicity of Environmental Pollutants? NPJ Biofilms Microbiomes 2016, 2, 16003. [Google Scholar] [CrossRef]
- Li, N.; Li, J.; Zhang, Q.; Gao, S.; Quan, X.; Liu, P.; Xu, C. Effects of Endocrine Disrupting Chemicals in Host Health: Three-Way Interactions between Environmental Exposure, Host Phenotypic Responses, and Gut Microbiota. Environ. Pollut. 2021, 271, 116387. [Google Scholar] [CrossRef]
- He, S.; Li, H.; Yu, Z.; Zhang, F.; Liang, S.; Liu, H.; Chen, H.; Lü, M. The Gut Microbiome and Sex Hormone-Related Diseases. Front. Microbiol. 2021, 12, 2699. [Google Scholar] [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut Microbiota, Metabolites and Host Immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- García-Peñarrubia, P.; Ruiz-Alcaraz, A.J.; Martínez-Esparza, M.; Marín, P.; Machado-Linde, F. Hypothetical Roadmap towards Endometriosis: Prenatal Endocrine-Disrupting Chemical Pollutant Exposure, Anogenital Distance, Gut-Genital Microbiota and Subclinical Infections. Hum. Reprod. Update 2020, 26, 214–246. [Google Scholar] [CrossRef] [PubMed]
- Giampaolino, P.; Foreste, V.; di Filippo, C.; Gallo, A.; Mercorio, A.; Serafino, P.; Improda, F.P.; Verrazzo, P.; Zara, G.; Buonfantino, C.; et al. Microbiome and PCOS: State-of-Art and Future Aspects. Int. J. Mol. Sci. 2021, 22, 2048. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Shao, J.; Yang, Y.; Niu, X.; Liao, J.; Zhao, Q.; Wang, D.; Li, S.; Hu, J. Gut Microbiota in Patients with Polycystic Ovary Syndrome: A Systematic Review. Reprod. Sci. 2022, 29, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Yurtdaş, G.; Akdevelioğlu, Y. A New Approach to Polycystic Ovary Syndrome: The Gut Microbiota. J. Am. Coll. Nutr. 2020, 39, 371–382. [Google Scholar] [CrossRef]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; van Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 2546. [Google Scholar] [CrossRef]
- Metcalfe, C.D.; Bayen, S.; Desrosiers, M.; Muñoz, G.; Sauvé, S.; Yargeau, V. An Introduction to the Sources, Fate, Occurrence and Effects of Endocrine Disrupting Chemicals Released into the Environment. Environ. Res. 2022, 207, 112658. [Google Scholar] [CrossRef]
- Kranc, W.; Brązert, M.; Celichowski, P.; Bryja, A.; Nawrocki, M.; Ożegowska, K.; Jankowski, M.; Jeseta, M.; Pawelczyk, L.; Bręborowicz, A.; et al. ‘Heart Development and Morphogenesis’ Is a Novel Pathway for Human Ovarian Granulosa Cell Differentiation during Long-term in Vitro Cultivation-a Microarray Approach. Mol. Med. Rep. 2019, 19, 1705–1715. [Google Scholar] [CrossRef]
- Kossowska-Tomaszczuk, K.; de Geyter, C. Cells with Stem Cell Characteristics in Somatic Compartments of the Ovary. Biomed. Res. Int. 2013, 2013, 310859. [Google Scholar] [CrossRef]
- Richards, J.S. Maturation of Ovarian Follicles: Actions and Interactions of Pituitary and Ovarian Hormones on Follicular Cell Differentiation. Physiol. Rev. 1980, 60, 51–89. [Google Scholar] [CrossRef]
- Clark, B.J.; Stocco, D.M. The Steroidogenic Acute Regulatory Protein (StAR). In Cholesterol Transporters of the START Domain Protein Family in Health and Disease: START Proteins-Structure and Function; Springer: New York, NY, USA, 2014; Volume 9781493911127, pp. 15–47. ISBN 9781493911127. [Google Scholar]
- Hanukoglu, I. Steroidogenic Enzymes: Structure, Function, and Role in Regulation of Steroid Hormone Biosynthesis. J. Steroid. Biochem. Mol. Biol. 1992, 43, 779–804. [Google Scholar] [CrossRef]
- Penning, T.M. Molecular Endocrinology of Hydroxysteroid Dehydrogenases. Endocr. Rev. 1997, 18, 281–305. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.L.; Spink, D.C.; Spink, B.C.; Cao, J.Q.; Walker, N.J.; Sutter, T.R. 17 Beta-Estradiol Hydroxylation Catalyzed by Human Cytochrome P450 1B1. Proc. Natl. Acad. Sci. USA 1996, 93, 9776–9781. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Nakajima, M.; Yokoi, T. Cytochrome P450-Mediated Metabolism of Estrogens and Its Regulation in Human. Cancer Lett. 2005, 227, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Guarnotta, V.; Amodei, R.; Frasca, F.; Aversa, A.; Giordano, C. Impact of Chemical Endocrine Disruptors and Hormone Modulators on the Endocrine System. Int. J. Mol. Sci. 2022, 23, 5710. [Google Scholar] [CrossRef]
- Moolhuijsen, L.M.E.; Visser, J.A. Anti-Müllerian Hormone and Ovarian Reserve: Update on Assessing Ovarian Function. J. Clin. Endocrinol. Metab. 2020, 105, 3361–3373. [Google Scholar] [CrossRef]
- Parco, S. Serum Anti-Müllerian Hormone as a Predictive Marker of Polycystic Ovarian Syndrome. Int. J. Gen. Med. 2011, 4, 759. [Google Scholar] [CrossRef]
- Pellatt, L.; Rice, S.; Mason, H.D. Anti-Müllerian Hormone and Polycystic Ovary Syndrome: A Mountain Too High? Reproduction 2010, 139, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Catteau-Jonard, S.; Pigny, P.; Reyss, A.-C.; Decanter, C.; Poncelet, E.; Dewailly, D. Changes in Serum Anti-Müllerian Hormone Level during Low-Dose Recombinant Follicular-Stimulating Hormone Therapy for Anovulation in Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2007, 92, 4138–4143. [Google Scholar] [CrossRef]
- Pigny, P.; Merlen, E.; Robert, Y.; Cortet-Rudelli, C.; Decanter, C.; Jonard, S.; Dewailly, D. Elevated Serum Level of Anti-Mullerian Hormone in Patients with Polycystic Ovary Syndrome: Relationship to the Ovarian Follicle Excess and to the Follicular Arrest. J. Clin. Endocrinol. Metab. 2003, 88, 5957–5962. [Google Scholar] [CrossRef]
- Laven, J.S.E.; Mulders, A.G.M.G.J.; Visser, J.A.; Themmen, A.P.; de Jong, F.H.; Fauser, B.C.J.M. Anti-Müllerian Hormone Serum Concentrations in Normoovulatory and Anovulatory Women of Reproductive Age. J. Clin. Endocrinol. Metab. 2004, 89, 318–323. [Google Scholar] [CrossRef]
- Park, A.S.; Lawson, M.A.; Chuan, S.S.; Oberfield, S.E.; Hoeger, K.M.; Witchel, S.F.; Chang, R.J. Serum Anti-Müllerian Hormone Concentrations Are Elevated in Oligomenorrheic Girls without Evidence of Hyperandrogenism. J. Clin. Endocrinol. Metab. 2010, 95, 1786–1792. [Google Scholar] [CrossRef] [PubMed]
- Pellatt, L.; Rice, S.; Dilaver, N.; Heshri, A.; Galea, R.; Brincat, M.; Brown, K.; Simpson, E.R.; Mason, H.D. Anti-Müllerian Hormone Reduces Follicle Sensitivity to Follicle-Stimulating Hormone in Human Granulosa Cells. Fertil. Steril. 2011, 96, 1246–1251.e1. [Google Scholar] [CrossRef] [PubMed]
- Sacchi, S.; D’Ippolito, G.; Sena, P.; Marsella, T.; Tagliasacchi, D.; Maggi, E.; Argento, C.; Tirelli, A.; Giulini, S.; la Marca, A. The Anti-Müllerian Hormone (AMH) Acts as a Gatekeeper of Ovarian Steroidogenesis Inhibiting the Granulosa Cell Response to Both FSH and LH. J. Assist. Reprod. Genet. 2016, 33, 95–100. [Google Scholar] [CrossRef]
- Roy, S.; Gandra, D.; Seger, C.; Biswas, A.; Kushnir, V.A.; Gleicher, N.; Kumar, T.R.; Sen, A. Oocyte-Derived Factors (GDF9 and BMP15) and FSH Regulate AMH Expression Via Modulation of H3K27AC in Granulosa Cells. Endocrinology 2018, 159, 3433–3445. [Google Scholar] [CrossRef]
- Pellatt, L.; Hanna, L.; Brincat, M.; Galea, R.; Brain, H.; Whitehead, S.; Mason, H. Granulosa Cell Production of Anti-Müllerian Hormone Is Increased in Polycystic Ovaries. J. Clin. Endocrinol. Metab. 2007, 92, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Pierre, A.; Peigne, M.; Grynberg, M.; Arouche, N.; Taieb, J.; Hesters, L.; Gonzales, J.; Picard, J.-Y.; Dewailly, D.; Fanchin, R.; et al. Loss of LH-Induced down-Regulation of Anti-Mullerian Hormone Receptor Expression May Contribute to Anovulation in Women with Polycystic Ovary Syndrome. Hum. Reprod. 2013, 28, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Dilaver, N.; Pellatt, L.; Jameson, E.; Ogunjimi, M.; Bano, G.; Homburg, R.; D Mason, H.; Rice, S. The Regulation and Signalling of Anti-Müllerian Hormone in Human Granulosa Cells: Relevance to Polycystic Ovary Syndrome. Hum. Reprod. 2019, 34, 2467–2479. [Google Scholar] [CrossRef]
- Josso, N.; di Clemente, N.; Gouédard, L. Anti-Müllerian Hormone and Its Receptors. Mol. Cell. Endocrinol. 2001, 179, 25–32. [Google Scholar] [CrossRef]
- Attisano, L.; Wrana, J.L. Signal Transduction by the TGF-β Superfamily. Science 2002, 296, 1646–1647. [Google Scholar] [CrossRef]
- Ni, X.-R.; Sun, Z.-J.; Hu, G.-H.; Wang, R.-H. High Concentration of Insulin Promotes Apoptosis of Primary Cultured Rat Ovarian Granulosa Cells Via Its Increase in Extracellular HMGB1. Reprod. Sci. 2015, 22, 271–277. [Google Scholar] [CrossRef]
- Wang, M.; Sun, J.; Xu, B.; Chrusciel, M.; Gao, J.; Bazert, M.; Stelmaszewska, J.; Xu, Y.; Zhang, H.; Pawelczyk, L.; et al. Functional Characterization of MicroRNA-27a-3p Expression in Human Polycystic Ovary Syndrome. Endocrinology 2018, 159, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Slack, C.; Werz, C.; Wieser, D.; Alic, N.; Foley, A.; Stocker, H.; Withers, D.J.; Thornton, J.M.; Hafen, E.; Partridge, L. Regulation of Lifespan, Metabolism, and Stress Responses by the Drosophila SH2B Protein, Lnk. PLoS Genet. 2010, 6, e1000881. [Google Scholar] [CrossRef] [PubMed]
- Devallière, J.; Charreau, B. The Adaptor Lnk (SH2B3): An Emerging Regulator in Vascular Cells and a Link between Immune and Inflammatory Signaling. Biochem. Pharmacol. 2011, 82, 1391–1402. [Google Scholar] [CrossRef]
- Fox, E.R.; Young, J.H.; Li, Y.; Dreisbach, A.W.; Keating, B.J.; Musani, S.K.; Liu, K.; Morrison, A.C.; Ganesh, S.; Kutlar, A.; et al. Association of Genetic Variation with Systolic and Diastolic Blood Pressure among African Americans: The Candidate Gene Association Resource Study. Hum. Mol. Genet. 2011, 20, 2273–2284. [Google Scholar] [CrossRef]
- Jiang, J.; Balcerek, J.; Rozenova, K.; Cheng, Y.; Bersenev, A.; Wu, C.; Song, Y.; Tong, W. 14-3-3 Regulates the LNK/JAK2 Pathway in Mouse Hematopoietic Stem and Progenitor Cells. J. Clin. Investig. 2012, 122, 2079–2091. [Google Scholar] [CrossRef] [PubMed]
- Bersenev, A.; Wu, C.; Balcerek, J.; Jing, J.; Kundu, M.; Blobel, G.A.; Chikwava, K.R.; Tong, W. Lnk Constrains Myeloproliferative Diseases in Mice. J. Clin. Investig. 2010, 120, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- Hao, M.; Yuan, F.; Jin, C.; Zhou, Z.; Cao, Q.; Xu, L.; Wang, G.; Huang, H.; Yang, D.; Xie, M.; et al. Overexpression of Lnk in the Ovaries Is Involved in Insulin Resistance in Women With Polycystic Ovary Syndrome. Endocrinology 2016, 157, 3709–3718. [Google Scholar] [CrossRef]
- Tan, M.; Cheng, Y.; Zhong, X.; Yang, D.; Jiang, S.; Ye, Y.; Ding, M.; Guan, G.; Yang, D.; Zhao, X. LNK Promotes Granulosa Cell Apoptosis in PCOS via Negatively Regulating Insulin-Stimulated AKT-FOXO3 Pathway. Aging (Albany N.Y.) 2021, 13, 4617–4633. [Google Scholar] [CrossRef]
- Hu, C.-L.; Cowan, R.G.; Harman, R.M.; Quirk, S.M. Cell Cycle Progression and Activation of Akt Kinase Are Required for Insulin-Like Growth Factor I-Mediated Suppression of Apoptosis in Granulosa Cells. Mol. Endocrinol. 2004, 18, 326–338. [Google Scholar] [CrossRef]
- John, G.B.; Shidler, M.J.; Besmer, P.; Castrillon, D.H. Kit Signaling via PI3K Promotes Ovarian Follicle Maturation but Is Dispensable for Primordial Follicle Activation. Dev. Biol. 2009, 331, 292–299. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. FOXO Transcription Factors at the Interface between Longevity and Tumor Suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and Regulation of Apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Nagaraju, G.; Liu, Z.; Liu, K. Functional Roles of the Phosphatidylinositol 3-Kinases (PI3Ks) Signaling in the Mammalian Ovary. Mol. Cell. Endocrinol. 2012, 356, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Mo, H.; Chen, W.; Li, L.; Xiao, Y.; Zhang, J.; Li, X.; Lu, Y. Role of the PI3K-Akt Signaling Pathway in the Pathogenesis of Polycystic Ovary Syndrome. Reprod. Sci. 2017, 24, 646–655. [Google Scholar] [CrossRef]
- Gong, Y.; Luo, S.; Fan, P.; Zhu, H.; Li, Y.; Huang, W. Growth Hormone Activates PI3K/Akt Signaling and Inhibits ROS Accumulation and Apoptosis in Granulosa Cells of Patients with Polycystic Ovary Syndrome. Reprod. Biol. Endocrinol. 2020, 18, 121. [Google Scholar] [CrossRef]
- Nekoonam, S.; Naji, M.; Nashtaei, M.S.; Mortezaee, K.; Koruji, M.; Safdarian, L.; Amidi, F. Expression of AKT1 along with AKT2 in Granulosa-Lutein Cells of Hyperandrogenic PCOS Patients. Arch. Gynecol. Obstet. 2017, 295, 1041–1050. [Google Scholar] [CrossRef]
- Villavicencio, A.; Goyeneche, A.; Telleria, C.; Bacallao, K.; Gabler, F.; Fuentes, A.; Vega, M. Involvement of Akt, Ras and Cell Cycle Regulators in the Potential Development of Endometrial Hyperplasia in Women with Polycystic Ovarian Syndrome. Gynecol. Oncol. 2009, 115, 102–107. [Google Scholar] [CrossRef]
- Brants, J.R.; Ayoubi, T.A.Y.; Chada, K.; Marchal, K.; van de Ven, W.J.M.; Petit, M.M.R. Differential Regulation of the Insulin-like Growth Factor II MRNA-Binding Protein Genes by Architectural Transcription Factor HMGA2. FEBS Lett. 2004, 569, 277–283. [Google Scholar] [CrossRef]
- Cleynen, I.; Brants, J.R.; Peeters, K.; Deckers, R.; Debiec-Rychter, M.; Sciot, R.; van de Ven, W.J.M.; Petit, M.M.R. HMGA2 Regulates Transcription of the Imp2 Gene via an Intronic Regulatory Element in Cooperation with Nuclear Factor-ΚB. Mol. Cancer. Res. 2007, 5, 363–372. [Google Scholar] [CrossRef]
- Reeves, R.; Nissen, M.S. The A.T-DNA-Binding Domain of Mammalian High Mobility Group I Chromosomal Proteins. A Novel Peptide Motif for Recognizing DNA Structure. J. Biol. Chem. 1990, 265, 8573–8582. [Google Scholar] [CrossRef]
- Fedele, M.; Visone, R.; de Martino, I.; Troncone, G.; Palmieri, D.; Battista, S.; Ciarmiello, A.; Pallante, P.; Arra, C.; Melillo, R.M.; et al. HMGA2 Induces Pituitary Tumorigenesis by Enhancing E2F1 Activity. Cancer Cell 2006, 9, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.W.; Chen, H.; Roix, J.J.; Kathiresan, S.; Hirschhorn, J.N.; Daly, M.J.; et al. Genome-Wide Association Analysis Identifies Loci for Type 2 Diabetes and Triglyceride Levels. Science 2007, 316, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, H.; Zhao, S.-G.; Wei, D.-M.; Zhao, Y.-R.; Huang, T.; Muhammad, T.; Yan, L.; Gao, F.; Li, L.; et al. The HMGA2-IMP2 Pathway Promotes Granulosa Cell Proliferation in Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 1049–1059. [Google Scholar] [CrossRef]
- Jakubowicz, D.J.; Nestler, J.E. 17α-Hydroxyprogesterone Responses to Leuprolide and Serum Androgens in Obese Women with and without Polycystic Ovary Syndrome after Dietary Weight Loss. J. Clin. Endocrinol. Metab. 1997, 82, 556–560. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gilling-Smith, C.; Story, H.; Rogers, V.; Franks, S. Evidence for a Primary Abnormality of Thecal Cell Steroidogenesis in the Polycystic Ovary Syndrome. Clin. Endocrinol. 1997, 47, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Nestler, J.E.; Jakubowicz, D.J.; Falcon de Vargas, A.; Brik, C.; Quintero, N.; Medina, F. Insulin Stimulates Testosterone Biosynthesis by Human Thecal Cells from Women with Polycystic Ovary Syndrome by Activating Its Own Receptor and Using Inositolglycan Mediators as the Signal Transduction System. J. Clin. Endocrinol. Metab. 1998, 83, 2001–2005. [Google Scholar] [CrossRef] [PubMed]
- Gilling-Smith, C.; Willis, D.S.; Beard, R.W.; Franks, S. Hypersecretion of Androstenedione by Isolated Thecal Cells from Polycystic Ovaries. J. Clin. Endocrinol. Metab. 1994, 79, 1158–1165. [Google Scholar] [CrossRef]
- Nelson, V.L.; Legro, R.S.; Strauss, J.F.; McAllister, J.M. Augmented Androgen Production Is a Stable Steroidogenic Phenotype of Propagated Theca Cells from Polycystic Ovaries. Mol. Endocrinol. 1999, 13, 946–957. [Google Scholar] [CrossRef] [PubMed]
- McAllister, J.M.; Modi, B.; Miller, B.A.; Biegler, J.; Bruggeman, R.; Legro, R.S.; Strauss, J.F. Overexpression of a DENND1A Isoform Produces a Polycystic Ovary Syndrome Theca Phenotype. Proc. Natl. Acad. Sci. USA 2014, 111, E1519–E1527. [Google Scholar] [CrossRef]
- Nelson, V.L.; Qin, K.; Rosenfield, R.L.; Wood, J.R.; Penning, T.M.; Legro, R.S.; Strauss, J.F.; McAllister, J.M. The Biochemical Basis for Increased Testosterone Production in Theca Cells Propagated from Patients with Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2001, 86, 5925–5933. [Google Scholar] [CrossRef]
- Wickenheisser, J.K.; Biegler, J.M.; Nelson-DeGrave, V.L.; Legro, R.S.; Strauss, J.F.; McAllister, J.M. Cholesterol Side-Chain Cleavage Gene Expression in Theca Cells: Augmented Transcriptional Regulation and MRNA Stability in Polycystic Ovary Syndrome. PLoS ONE 2012, 7, e48963. [Google Scholar] [CrossRef] [PubMed]
- Wickenheisser, J.K.; Quinn, P.G.; Nelson, V.L.; Legro, R.S.; Strauss, J.F.; McAllister, J.M. Differential Activity of the Cytochrome P450 17α-Hydroxylase and Steroidogenic Acute Regulatory Protein Gene Promoters in Normal and Polycystic Ovary Syndrome Theca Cells. J. Clin. Endocrinol. Metab. 2000, 85, 2304–2311. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, M.O.; Jones, M.R.; Li, X.; Chua, A.K.; Garcia, O.A.; Chen, Y.-D.I.; Krauss, R.M.; Rotter, J.I.; Ankener, W.; Legro, R.S.; et al. Replication of Association of DENND1A and THADA Variants with Polycystic Ovary Syndrome in European Cohorts. J. Med. Genet. 2012, 49, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Welt, C.K.; Styrkarsdottir, U.; Ehrmann, D.A.; Thorleifsson, G.; Arason, G.; Gudmundsson, J.A.; Ober, C.; Rosenfield, R.L.; Saxena, R.; Thorsteinsdottir, U.; et al. Variants in DENND1A Are Associated with Polycystic Ovary Syndrome in Women of European Ancestry. J. Clin. Endocrinol. Metab. 2012, 97, E1342–E1347. [Google Scholar] [CrossRef]
- Lerchbaum, E.; Trummer, O.; Giuliani, A.; Gruber, H.-J.; Pieber, T.; Obermayer-Pietsch, B. Susceptibility Loci for Polycystic Ovary Syndrome on Chromosome 2p16.3, 2p21, and 9q33.3 in a Cohort of Caucasian Women. Horm. Metab. Res. 2011, 43, 743–747. [Google Scholar] [CrossRef]
- Eriksen, M.B.; Brusgaard, K.; Andersen, M.; Tan, Q.; Altinok, M.L.; Gaster, M.; Glintborg, D. Association of Polycystic Ovary Syndrome Susceptibility Single Nucleotide Polymorphism Rs2479106 and PCOS in Caucasian Patients with PCOS or Hirsutism as Referral Diagnosis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2012, 163, 39–42. [Google Scholar] [CrossRef]
- Kosova, G.; Urbanek, M. Genetics of the Polycystic Ovary Syndrome. Mol. Cell. Endocrinol. 2013, 373, 29–38. [Google Scholar] [CrossRef]
- Tian, Y.; Li, J.; Su, S.; Cao, Y.; Wang, Z.; Zhao, S.; Zhao, H. PCOS-GWAS Susceptibility Variants in THADA, INSR, TOX3, and DENND1A Are Associated With Metabolic Syndrome or Insulin Resistance in Women With PCOS. Front. Endocrinol. 2020, 11, 274. [Google Scholar] [CrossRef]
- Khan, S.; Duan, P.; Yao, L.; Hou, H. Shiftwork-Mediated Disruptions of Circadian Rhythms and Sleep Homeostasis Cause Serious Health Problems. Int. J. Genom. 2018, 2018, 8576890. [Google Scholar] [CrossRef]
- Wang, F.; Xie, N.; Wu, Y.; Zhang, Q.; Zhu, Y.; Dai, M.; Zhou, J.; Pan, J.; Tang, M.; Cheng, Q.; et al. Association between Circadian Rhythm Disruption and Polycystic Ovary Syndrome. Fertil. Steril. 2021, 115, 771–781. [Google Scholar] [CrossRef]
- Johnson, B.S.; Krishna, M.B.; Padmanabhan, R.A.; Pillai, S.M.; Jayakrishnan, K.; Laloraya, M. Derailed Peripheral Circadian Genes in Polycystic Ovary Syndrome Patients Alters Peripheral Conversion of Androgens Synthesis. Hum. Reprod. 2022, 37, 1835–1855. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.; Li, S.; Geng, X.; Wang, D.; Zhai, J.; Lu, G.; Chan, W.-Y.; Chen, Z.-J.; Du, Y. Long-Term Environmental Exposure of Darkness Induces Hyperandrogenism in PCOS via Melatonin Receptor 1A and Aromatase Reduction. Front. Cell Dev. Biol. 2022, 10, 954186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hirano, A.; Hsu, P.-K.; Jones, C.R.; Sakai, N.; Okuro, M.; McMahon, T.; Yamazaki, M.; Xu, Y.; Saigoh, N.; et al. A PERIOD3 Variant Causes a Circadian Phenotype and Is Associated with a Seasonal Mood Trait. Proc. Natl. Acad. Sci. USA 2016, 113, E1536–E1544. [Google Scholar] [CrossRef] [PubMed]
- Solt, L.A.; Kojetin, D.J.; Burris, T.P. The REV-ERBs and RORs: Molecular Links between Circadian Rhythms and Lipid Homeostasis. Future Med. Chem. 2011, 3, 623–638. [Google Scholar] [CrossRef]
- Guillaumond, F.; Dardente, H.; Giguère, V.; Cermakian, N. Differential Control of Bmal1 Circadian Transcription by REV-ERB and ROR Nuclear Receptors. J. Biol. Rhythm. 2005, 20, 391–403. [Google Scholar] [CrossRef]
- Sun, L.; Tian, H.; Xue, S.; Ye, H.; Xue, X.; Wang, R.; Liu, Y.; Zhang, C.; Chen, Q.; Gao, S. Circadian Clock Genes REV-ERBs Inhibits Granulosa Cells Apoptosis by Regulating Mitochondrial Biogenesis and Autophagy in Polycystic Ovary Syndrome. Front. Cell Dev. Biol. 2021, 9, 2079. [Google Scholar] [CrossRef]
- Preitner, N.; Damiola, F.; Luis-Lopez-Molina; Zakany, J.; Duboule, D.; Albrecht, U.; Schibler, U. The Orphan Nuclear Receptor REV-ERBα Controls Circadian Transcription within the Positive Limb of the Mammalian Circadian Oscillator. Cell 2002, 110, 251–260. [Google Scholar] [CrossRef]
- Kohsaka, A.; Bass, J. A Sense of Time: How Molecular Clocks Organize Metabolism. Trends Endocrinol. Metab. 2007, 18, 4–11. [Google Scholar] [CrossRef]
- Li, C.; Shi, Y.; You, L.; Wang, L.; Chen, Z.-J. Melatonin Receptor 1A Gene Polymorphism Associated with Polycystic Ovary Syndrome. Gynecol. Obstet. 2011, 72, 130–134. [Google Scholar] [CrossRef]
- Li, C.; Shi, Y.; You, L.; Wang, L.; Chen, Z.-J. Association of Rs10830963 and Rs10830962 SNPs in the Melatonin Receptor (MTNR1B) Gene among Han Chinese Women with Polycystic Ovary Syndrome. Mol. Hum. Reprod. 2011, 17, 193–198. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Zhang, X.; Shi, J.; Wang, M.; Wei, Z.; Zhao, A.; Li, B.; Zhao, X.; Xing, Q.; et al. Common Genetic Variation in MTNR1B Is Associated with Serum Testosterone, Glucose Tolerance, and Insulin Secretion in Polycystic Ovary Syndrome Patients. Fertil. Steril. 2010, 94, 2486–2489.e2. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.Y.; Lenn, N.J. A Retinohypothalamic Projection in the Rat. J. Comp. Neurol. 1972, 146, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Debono, M. Review: Replication of Cortisol Circadian Rhythm: New Advances in Hydrocortisone Replacement Therapy. Ther. Adv. Endocrinol. Metab. 2010, 1, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Asghari, M.H.; Moloudizargari, M.; Bahadar, H.; Abdollahi, M. A Review of the Protective Effect of Melatonin in Pesticide-Induced Toxicity. Expert. Opin. Drug Metab. Toxicol. 2017, 13, 545–554. [Google Scholar] [CrossRef]
- Asghari, M.H.; Moloudizargari, M.; Ghobadi, E.; Fallah, M.; Abdollahi, M. Melatonin as a Multifunctional Anti-Cancer Molecule: Implications in Gastric Cancer. Life Sci. 2017, 185, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Asghari, M.H.; Moloudizargari, M.; Baeeri, M.; Baghaei, A.; Rahimifard, M.; Solgi, R.; Jafari, A.; Aminjan, H.H.; Hassani, S.; Moghadamnia, A.A.; et al. On the Mechanisms of Melatonin in Protection of Aluminum Phosphide Cardiotoxicity. Arch. Toxicol. 2017, 91, 3109–3120. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.-X.; Tamura, H.; Cruz, M.H.C.; Fuentes-Broto, L. Clinical Relevance of Melatonin in Ovarian and Placental Physiology: A Review. Gynecol. Endocrinol. 2014, 30, 83–89. [Google Scholar] [CrossRef]
- Sack, R.L.; Blood, M.L.; Lewy, A.J. Melatonin Rhythms in Night Shift Workers. Sleep 1992, 15, 434–441. [Google Scholar] [CrossRef]
- Fernandez, R.; Moore, V.; van Ryswyk, E.; Varcoe, T.; Rodgers, R.; March, W.; Moran, L.; Avery, J.; McEvoy, D.; Davies, M. Sleep Disturbances in Women with Polycystic Ovary Syndrome: Prevalence, Pathophysiology, Impact and Management Strategies. Nat. Sci. Sleep 2018, 10, 45–64. [Google Scholar] [CrossRef]
- Terzieva, D.D.; Orbetzova, M.M.; Mitkov, M.D.; Mateva, N.G. Serum Melatonin in Women with Polycystic Ovary Syndrome. Folia Med. 2013, 55, 10–15. [Google Scholar] [CrossRef]
- Jain, M.; Jain, S.; Singh, T.; Haldar, C.; Jain, P. Melatonin and Its Correlation with Testosterone in Polycystic Ovarian Syndrome. J. Hum. Reprod. Sci. 2013, 6, 253. [Google Scholar] [CrossRef] [PubMed]
- Shreeve, N.; Cagampang, F.; Sadek, K.; Tolhurst, M.; Houldey, A.; Hill, C.M.; Brook, N.; Macklon, N.; Cheong, Y. Poor Sleep in PCOS; Is Melatonin the Culprit? Hum. Reprod. 2013, 28, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Luboshitzky, R.; Qupti, G.; Ishay, A.; Shen-Orr, Z.; Futerman, B.; Linn, S. Increased 6-Sulfatoxymelatonin Excretion in Women with Polycystic Ovary Syndrome. Fertil. Steril. 2001, 76, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Nakamura, Y.; Korkmaz, A.; Manchester, L.C.; Tan, D.-X.; Sugino, N.; Reiter, R.J. Melatonin and the Ovary: Physiological and Pathophysiological Implications. Fertil. Steril. 2009, 92, 328–343. [Google Scholar] [CrossRef]
- Kim, M.K.; Park, E.A.; Kim, H.J.; Choi, W.Y.; Cho, J.H.; Lee, W.S.; Cha, K.Y.; Kim, Y.S.; Lee, D.R.; Yoon, T.K. Does Supplementation of In-Vitro Culture Medium with Melatonin Improve IVF Outcome in PCOS? Reprod. Biomed. Online 2013, 26, 22–29. [Google Scholar] [CrossRef]
- Polson, D.W.; Wadsworth, J.; Adams, J.; Franks, S. Polycystic ovaries—A common finding in normal women. Lancet 1988, 331, 870–872. [Google Scholar] [CrossRef]
- Mojaverrostami, S.; Asghari, N.; Khamisabadi, M.; Heidari Khoei, H. The Role of Melatonin in Polycystic Ovary Syndrome: A Review. Int. J. Reprod. Biomed. 2019, 17, 865–882. [Google Scholar] [CrossRef]
- Uzumcu, M.; Zama, A.; Oruc, E. Epigenetic Mechanisms in the Actions of Endocrine-Disrupting Chemicals: Gonadal Effects and Role in Female Reproduction. Reprod. Domest. Anim. 2012, 47, 338–347. [Google Scholar] [CrossRef]
- Craig, Z.R.; Wang, W.; Flaws, J.A. Endocrine-Disrupting Chemicals in Ovarian Function: Effects on Steroidogenesis, Metabolism and Nuclear Receptor Signaling. Reproduction 2011, 142, 633–646. [Google Scholar] [CrossRef]
- Piazza, M.J.; Urbanetz, A.A. Environmental Toxins and the Impact of Other Endocrine Disrupting Chemicals in Women’s Reproductive Health. JBRA Assist. Reprod. 2019, 23, 154–164. [Google Scholar] [CrossRef]
- Uzumcu, M.; Zachow, R. Developmental Exposure to Environmental Endocrine Disruptors: Consequences within the Ovary and on Female Reproductive Function. Reprod. Toxicol. 2007, 23, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Fowler, P.A.; Bellingham, M.; Sinclair, K.D.; Evans, N.P.; Pocar, P.; Fischer, B.; Schaedlich, K.; Schmidt, J.-S.; Amezaga, M.R.; Bhattacharya, S.; et al. Impact of Endocrine-Disrupting Compounds (EDCs) on Female Reproductive Health. Mol. Cell. Endocrinol. 2012, 355, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Bolli, A.; Bulzomi, P.; Galluzzo, P.; Acconcia, F.; Marino, M. Bisphenol A Impairs Estradiol-Induced Protective Effects against DLD-1 Colon Cancer Cell Growth. IUBMB Life 2010, 62, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Bolli, A.; Galluzzo, P.; Ascenzi, P.; del Pozzo, G.; Manco, I.; Vietri, M.T.; Mita, L.; Altucci, L.; Mita, D.G.; Marino, M. Laccase Treatment Impairs Bisphenol A-Induced Cancer Cell Proliferation Affecting Estrogen Receptor α-Dependent Rapid Signals. IUBMB Life 2008, 60, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Pallottini, V.; Marino, M. Molecular Mechanisms of Action of BPA. Dose-Response 2015, 13, 155932581561058. [Google Scholar] [CrossRef] [PubMed]
- Koch, H.M.; Calafat, A.M. Human Body Burdens of Chemicals Used in Plastic Manufacture. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 2063–2078. [Google Scholar] [CrossRef] [PubMed]
- Ikezuki, Y.; Tsutsumi, O.; Takai, Y.; Kamei, Y.; Taketani, Y. Determination of Bisphenol A Concentrations in Human Biological Fluids Reveals Significant Early Prenatal Exposure. Hum. Reprod. 2002, 17, 2839–2841. [Google Scholar] [CrossRef]
- Zhou, W.; Fang, F.; Zhu, W.; Chen, Z.-J.; Du, Y.; Zhang, J. Bisphenol A and Ovarian Reserve among Infertile Women with Polycystic Ovarian Syndrome. Int. J. Environ. Res. Public Health 2016, 14, 18. [Google Scholar] [CrossRef]
- Fernández, M.; Bourguignon, N.; Lux-Lantos, V.; Libertun, C. Neonatal Exposure to Bisphenol A and Reproductive and Endocrine Alterations Resembling the Polycystic Ovarian Syndrome in Adult Rats. Environ. Health Perspect. 2010, 118, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.A.; Birnbaum, L.S.; Farabollini, F.; Newbold, R.R.; Rubin, B.S.; Talsness, C.E.; Vandenbergh, J.G.; Walser-Kuntz, D.R.; vom Saal, F.S. In Vivo Effects of Bisphenol A in Laboratory Rodent Studies. Reprod. Toxicol. 2007, 24, 199–224. [Google Scholar] [CrossRef]
- Kato, H.; Ota, T.; Furuhashi, T.; Ohta, Y.; Iguchi, T. Changes in Reproductive Organs of Female Rats Treated with Bisphenol A during the Neonatal Period. Reprod. Toxicol. 2003, 17, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Adewale, H.B.; Jefferson, W.N.; Newbold, R.R.; Patisaul, H.B. Neonatal Bisphenol-A Exposure Alters Rat Reproductive Development and Ovarian Morphology Without Impairing Activation of Gonadotropin-Releasing Hormone Neurons1. Biol. Reprod. 2009, 81, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, W.; Liu, J.; Wang, W.; Li, H.; Zhu, J.; Weng, S.; Xiao, S.; Wu, T. Prepubertal Bisphenol A Exposure Interferes with Ovarian Follicle Development and Its Relevant Gene Expression. Reprod. Toxicol. 2014, 44, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Peretz, J.; Gupta, R.K.; Singh, J.; Hernández-Ochoa, I.; Flaws, J.A. Bisphenol A Impairs Follicle Growth, Inhibits Steroidogenesis, and Downregulates Rate-Limiting Enzymes in the Estradiol Biosynthesis Pathway. Toxicol. Sci. 2011, 119, 209–217. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, Q.; Dang, X.; He, Y.; Li, X.; Sun, Y. Local Effect of Bisphenol A on the Estradiol Synthesis of Ovarian Granulosa Cells from PCOS. Gynecol. Endocrinol. 2017, 33, 21–25. [Google Scholar] [CrossRef]
- Mlynarčíková, A.; Kolena, J.; Ficková, M.; Scsuková, S. Alterations in Steroid Hormone Production by Porcine Ovarian Granulosa Cells Caused by Bisphenol A and Bisphenol A Dimethacrylate. Mol. Cell. Endocrinol. 2005, 244, 57–62. [Google Scholar] [CrossRef]
- Déchaud, H.; Ravard, C.; Claustrat, F.; de la Perrière, A.B.; Pugeat, M. Xenoestrogen Interaction with Human Sex Hormone-Binding Globulin (HSHBG). Steroids 1999, 64, 328–334. [Google Scholar] [CrossRef]
- Rochester, J.R. Bisphenol A and Human Health: A Review of the Literature. Reprod. Toxicol. 2013, 42, 132–155. [Google Scholar] [CrossRef]
- Kandaraki, E.; Chatzigeorgiou, A.; Livadas, S.; Palioura, E.; Economou, F.; Koutsilieris, M.; Palimeri, S.; Panidis, D.; Diamanti-Kandarakis, E. Endocrine Disruptors and Polycystic Ovary Syndrome (PCOS): Elevated Serum Levels of Bisphenol A in Women with PCOS. J. Clin. Endocrinol. Metab. 2011, 96, E480–E484. [Google Scholar] [CrossRef]
- Akın, L.; Kendirci, M.; Narin, F.; Kurtoglu, S.; Saraymen, R.; Kondolot, M.; Koçak, S.; Elmalı, F. The Endocrine Disruptor Bisphenol A May Play a Role in the Aetiopathogenesis of Polycystic Ovary Syndrome in Adolescent Girls. Acta Paediatr. 2015, 104, e171–e177. [Google Scholar] [CrossRef]
- Konieczna, A.; Rachoń, D.; Owczarek, K.; Kubica, P.; Kowalewska, A.; Kudłak, B.; Wasik, A.; Namieśnik, J. Serum Bisphenol A Concentrations Correlate with Serum Testosterone Levels in Women with Polycystic Ovary Syndrome. Reprod. Toxicol. 2018, 82, 32–37. [Google Scholar] [CrossRef]
- Hossein Rashidi, B.; Amanlou, M.; Behrouzi Lak, T.; Ghazizadeh, M.; Haghollahi, F.; Bagheri, M.; Eslami, B. The Association Between Bisphenol A and Polycystic Ovarian Syndrome: A Case-Control Study. Acta Med. Iran. 2017, 55, 759–764. [Google Scholar] [PubMed]
- Tsutsumi, O. Assessment of Human Contamination of Estrogenic Endocrine-Disrupting Chemicals and Their Risk for Human Reproduction. J. Steroid. Biochem. Mol. Biol. 2005, 93, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Tsutsumi, O.; Ikezuki, Y.; Kamei, Y.; Osuga, Y.; Fujiwara, T.; Takai, Y.; Momoeda, M.; Yano, T.; Taketani, Y. Elevated Serum Bisphenol A Levels under Hyperandrogenic Conditions May Be Caused by Decreased UDP-Glucuronosyltransferase Activity. Endocr. J. 2006, 53, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Yokota, H.; Iwano, H.; Endo, M.; Kobayashi, T.; Inoue, H.; Ikushiro, S.; Yuasa, A. Glucuronidation of the Environmental Oestrogen Bisphenol A by an Isoform of UDP-Glucuronosyltransferase, UGT2B1, in the Rat Liver. Biochem. J. 1999, 340 Pt 2, 405–409. [Google Scholar] [CrossRef]
- North, E.J.; Halden, R.U. Plastics and Environmental Health: The Road Ahead. Rev. Environ. Health 2013, 28, 1–8. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Chou, Y.-Y.; Wu, Y.-M.; Lin, C.-C.; Lin, S.-J.; Lee, C.-C. Phthalates May Promote Female Puberty by Increasing Kisspeptin Activity. Hum. Reprod. 2013, 28, 2765–2773. [Google Scholar] [CrossRef]
- Kim, S.H.; Park, M.J. Phthalate Exposure and Childhood Obesity. Ann. Pediatr. Endocrinol. Metab. 2014, 19, 69. [Google Scholar] [CrossRef]
- Huang, L.-P.; Lee, C.-C.; Hsu, P.-C.; Shih, T.-S. The Association between Semen Quality in Workers and the Concentration of Di(2-Ethylhexyl) Phthalate in Polyvinyl Chloride Pellet Plant Air. Fertil. Steril. 2011, 96, 90–94. [Google Scholar] [CrossRef]
- Marsee, K.; Woodruff, T.J.; Axelrad, D.A.; Calafat, A.M.; Swan, S.H. Estimated Daily Phthalate Exposures in a Population of Mothers of Male Infants Exhibiting Reduced Anogenital Distance. Environ. Health Perspect. 2006, 114, 805–809. [Google Scholar] [CrossRef]
- Xu, C.; Chen, J.-A.; Qiu, Z.; Zhao, Q.; Luo, J.; Yang, L.; Zeng, H.; Huang, Y.; Zhang, L.; Cao, J.; et al. Ovotoxicity and PPAR-Mediated Aromatase Downregulation in Female Sprague–Dawley Rats Following Combined Oral Exposure to Benzo[a]Pyrene and Di-(2-Ethylhexyl) Phthalate. Toxicol. Lett. 2010, 199, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.J.; Maronpot, R.R.; Heindel, J.J. Di-(2-Ethylhexyl) Phthalate Suppresses Estradiol and Ovulation in Cycling Rats. Toxicol. Appl. Pharmacol. 1994, 128, 216–223. [Google Scholar] [CrossRef]
- Svechnikova, I.; Svechnikov, K.; Söder, O. The Influence of Di-(2-Ethylhexyl) Phthalate on Steroidogenesis by the Ovarian Granulosa Cells of Immature Female Rats. J. Endocrinol. 2007, 194, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Lovekamp-Swan, T.; Jetten, A.M.; Davis, B.J. Dual Activation of PPARα and PPARγ by Mono-(2-Ethylhexyl) Phthalate in Rat Ovarian Granulosa Cells. Mol. Cell. Endocrinol. 2003, 201, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Reinsberg, J.; Wegener-Toper, P.; van der Ven, K.; van der Ven, H.; Klingmueller, D. Effect of Mono-(2-Ethylhexyl) Phthalate on Steroid Production of Human Granulosa Cells. Toxicol. Appl. Pharmacol. 2009, 239, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhang, Q.; Pan, J.-X.; Wang, F.-F.; Qu, F. The Effects of Di(2-Ethylhexyl) Phthalate Exposure In Women with Polycystic Ovary Syndrome Undergoing in Vitro Fertilization. J. Int. Med. Res. 2019, 47, 6278–6293. [Google Scholar] [CrossRef] [PubMed]
- Vagi, S.J.; Azziz-Baumgartner, E.; Sjödin, A.; Calafat, A.M.; Dumesic, D.; Gonzalez, L.; Kato, K.; Silva, M.J.; Ye, X.; Azziz, R. Exploring the Potential Association between Brominated Diphenyl Ethers, Polychlorinated Biphenyls, Organochlorine Pesticides, Perfluorinated Compounds, Phthalates, and Bisphenol a in Polycystic Ovary Syndrome: A Case–Control Study. BMC Endocr. Disord. 2014, 14, 86. [Google Scholar] [CrossRef] [PubMed]
- Parks, L.G. The Plasticizer Diethylhexyl Phthalate Induces Malformations by Decreasing Fetal Testosterone Synthesis during Sexual Differentiation in the Male Rat. Toxicol. Sci. 2000, 58, 339–349. [Google Scholar] [CrossRef]
- Joensen, U.N.; Frederiksen, H.; Jensen, M.B.; Lauritsen, M.P.; Olesen, I.A.; Lassen, T.H.; Andersson, A.-M.; Jørgensen, N. Phthalate Excretion Pattern and Testicular Function: A Study of 881 Healthy Danish Men. Environ. Health Perspect. 2012, 120, 1397–1403. [Google Scholar] [CrossRef]
- Hannon, P.R.; Flaws, J.A. The Effects of Phthalates on the Ovary. Front. Endocrinol. 2015, 6, 8. [Google Scholar] [CrossRef]
- Kay, V.R.; Chambers, C.; Foster, W.G. Reproductive and Developmental Effects of Phthalate Diesters in Females. Crit. Rev. Toxicol. 2013, 43, 200–219. [Google Scholar] [CrossRef] [PubMed]
- Orlowska, K.; Swigonska, S.; Sadowska, A.; Ruszkowska, M.; Nynca, A.; Molcan, T.; Zmijewska, A.; Ciereszko, R.E. Proteomic Changes of Aryl Hydrocarbon Receptor (AhR)-Silenced Porcine Granulosa Cells Exposed to 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD). PLoS ONE 2019, 14, e0223420. [Google Scholar] [CrossRef] [PubMed]
- Schecter, A.; Birnbaum, L.; Ryan, J.J.; Constable, J.D. Dioxins: An Overview. Environ. Res. 2006, 101, 419–428. [Google Scholar] [CrossRef]
- ben Maamar, M.; Nilsson, E.; Thorson, J.L.M.; Beck, D.; Skinner, M.K. Transgenerational Disease Specific Epigenetic Sperm Biomarkers after Ancestral Exposure to Dioxin. Environ. Res. 2021, 192, 110279. [Google Scholar] [CrossRef] [PubMed]
- Michalek, J.E.; Pirkle, J.L.; Needham, L.L.; Patterson JR, D.G.; Caudill, S.P.; Tripathi, R.C.; Mocarelli, P. Pharmacokinetics of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin in Seveso Adults and Veterans of Operation Ranch Hand. J. Expo. Anal. Environ. Epidemiol. 2002, 12, 44–53. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gaspari, L.; Paris, F.; Kalfa, N.; Soyer-Gobillard, M.-O.; Sultan, C.; Hamamah, S. Experimental Evidence of 2,3,7,8-Tetrachlordibenzo-p-Dioxin (TCDD) Transgenerational Effects on Reproductive Health. Int. J. Mol. Sci. 2021, 22, 9091. [Google Scholar] [CrossRef]
- Patel, S.; Zhou, C.; Rattan, S.; Flaws, J.A. Effects of Endocrine-Disrupting Chemicals on the Ovary. Biol. Reprod. 2015, 93, 20. [Google Scholar] [CrossRef]
- Grochowalski, A.; Piekło, R.; Gasińska, A.; Chrzaszcz, R.; Gregoraszczuk, E.L. Accumulation of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin (TCDD) in Porcine Preovulatory Follicles after in Vitro Exposure to TCDD: Effects on Steroid Secretion and Cell Proliferation. Cytobios 2000, 102, 21–31. [Google Scholar]
- Heimler, I.; Trewin, A.L.; Chaffin, C.L.; Rawlins, R.G.; Hutz, R.J. Modulation of Ovarian Follicle Maturation and Effects on Apoptotic Cell Death in Holtzman Rats Exposed to 2,3,7,8-Tetrachlorodibenzo-p-Dioxin(TCDD) in Utero and Lactationally. Reprod. Toxicol. 1998, 12, 69–73. [Google Scholar] [CrossRef]
- Karman, B.N.; Basavarajappa, M.S.; Craig, Z.R.; Flaws, J.A. 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Activates the Aryl Hydrocarbon Receptor and Alters Sex Steroid Hormone Secretion without Affecting Growth of Mouse Antral Follicles in Vitro. Toxicol. Appl. Pharmacol. 2012, 261, 88–96. [Google Scholar] [CrossRef]
- Valdez, K.E.; Shi, Z.; Ting, A.Y.; Petroff, B.K. Effect of Chronic Exposure to the Aryl Hydrocarbon Receptor Agonist 2,3,7,8-Tetrachlorodibenzo-p-Dioxin in Female Rats on Ovarian Gene Expression. Reprod. Toxicol. 2009, 28, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Petroff, B.K.; Rozman, K.K.; Terranova, P.F. Gonadotropin-Releasing Hormone (GnRH) Partially Reverses the Inhibitory Effect of 2,3,7,8-Tetrachlorodibenzo-p-Dioxin on Ovulation in the Immature Gonadotropin-Treated Rat. Toxicology 2000, 147, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Jung, N.-K.; Park, J.-Y.; Park, J.-H.; Kim, S.-Y.; Park, J.-K.; Chang, W.-K.; Lee, H.-C.; Kim, S.-W.; Chun, S.-Y. Attenuation of Cell Cycle Progression by 2,3,7,8-Tetrachlorodibenzo-p-Dioxin Eliciting Ovulatory Blockade in Gonadotropin-Primed Immature Rats. Endocr. J. 2010, 57, 863–871. [Google Scholar] [CrossRef]
- Karman, B.N.; Basavarajappa, M.S.; Hannon, P.; Flaws, J.A. Dioxin Exposure Reduces the Steroidogenic Capacity of Mouse Antral Follicles Mainly at the Level of HSD17B1 without Altering Atresia. Toxicol. Appl. Pharmacol. 2012, 264, 1–12. [Google Scholar] [CrossRef]
- Garavaglia, E.; Sala, C.; Taccagni, G.; Traglia, M.; Barbieri, C.; Ferrari, S.; Candiani, M.; Panina-Bordignon, P.; Toniolo, D. Fertility Preservation in Endometriosis Patients: Anti-Müllerian Hormone Is a Reliable Marker of the Ovarian Follicle Density. Front. Surg. 2017, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.-L.; Chen, L.-R.; Tsao, H.-M.; Chen, K.-H. Polycystic Ovarian Syndrome and the Risk of Subsequent Primary Ovarian Insufficiency: A Nationwide Population-Based Study. Menopause 2017, 24, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, E.; Larsen, G.; Manikkam, M.; Guerrero-Bosagna, C.; Savenkova, M.I.; Skinner, M.K. Environmentally Induced Epigenetic Transgenerational Inheritance of Ovarian Disease. PLoS ONE 2012, 7, e36129. [Google Scholar] [CrossRef] [PubMed]
- Merlo, E.; Podratz, P.L.; Sena, G.C.; de Araújo, J.F.P.; Lima, L.C.F.; Alves, I.S.S.; Gama-de-Souza, L.N.; Pelição, R.; Rodrigues, L.C.M.; Brandão, P.A.A.; et al. The Environmental Pollutant Tributyltin Chloride Disrupts the Hypothalamic-Pituitary-Adrenal Axis at Different Levels in Female Rats. Endocrinology 2016, 157, 2978–2995. [Google Scholar] [CrossRef]
- Chen, C.; Chen, L.; Li, Y.; Fu, W.; Shi, X.; Duan, J.; Zhang, W. Impacts of Microplastics on Organotins’ Photodegradation in Aquatic Environments. Environ. Pollut. 2020, 267, 115686. [Google Scholar] [CrossRef]
- Santillo David, J.P.L.W.J. TBT Antifoulants: A Tale of Ships, Snails and Imposex. In The Precautionary Principle in the 20th Century; Harremoes, P., Gee, D., MacGarvin, M., Stirling, A., Keys, J., Wynne, B., Vaz, S.G., Eds.; Routledge: London, UK, 2002; pp. 135–148. ISBN 9781134207787. [Google Scholar]
- Ferraz da Silva, I.; Merlo, E.; Costa, C.S.; Graceli, J.B.; Rodrigues, L.C.M. Tributyltin Exposure Is Associated With Recognition Memory Impairments, Alterations in Estrogen Receptor α Protein Levels, and Oxidative Stress in the Brain of Female Mice. Front. Toxicol. 2021, 3, 654077. [Google Scholar] [CrossRef]
- Batista-Andrade, J.A.; Caldas, S.S.; Batista, R.M.; Castro, I.B.; Fillmann, G.; Primel, E.G. From TBT to Booster Biocides: Levels and Impacts of Antifouling along Coastal Areas of Panama. Environ. Pollut. 2018, 234, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Filipkowska, A.; Kowalewska, G. Butyltins in Sediments from the Southern Baltic Coastal Zone: Is It Still a Matter of Concern, 10 Years after Implementation of the Total Ban? Mar. Pollut. Bull. 2019, 146, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Filipkowska, A.; Złoch, I.; Wawrzyniak-Wydrowska, B.; Kowalewska, G. Organotins in Fish Muscle and Liver from the Polish Coast of the Baltic Sea: Is the Total Ban Successful? Mar. Pollut. Bull. 2016, 111, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Whalen, M.M.; Loganathan, B.G.; Kannan, K. Immunotoxicity of Environmentally Relevant Concentrations of Butyltins on Human Natural Killer Cells In Vitro. Environ. Res. 1999, 81, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Podratz, P.L.; Filho, V.S.D.; Lopes, P.F.I.; Sena, G.C.; Matsumoto, S.T.; Samoto, V.Y.; Takiya, C.M.; Miguel, E.D.C.; Silva, I.V.; Graceli, J.B. Tributyltin Impairs the Reproductive Cycle in Female Rats. J. Toxicol. Environ. Health A 2012, 75, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Sena, G.C.; Freitas-Lima, L.C.; Merlo, E.; Podratz, P.L.; de Araújo, J.F.P.; Brandão, P.A.A.; Carneiro, M.T.W.D.; Zicker, M.C.; Ferreira, A.V.M.; Takiya, C.M.; et al. Environmental Obesogen Tributyltin Chloride Leads to Abnormal Hypothalamic-Pituitary-Gonadal Axis Function by Disruption in Kisspeptin/Leptin Signaling in Female Rats. Toxicol. Appl. Pharmacol. 2017, 319, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Cui, Y.; Ma, J.; Ge, Z.; Shen, W.; Yin, S. Tributyltin Oxide Exposure Impairs Mouse Oocyte Maturation and Its Possible Mechanisms. J. Cell. Biochem. 2019, 120, 715–726. [Google Scholar] [CrossRef]
- Ema, M.; Miyawaki, E.; Harazono, A. Effects of Triphenyltin Chloride on Implantation and Pregnancy in Rats. Reprod. Toxicol. 1997, 11, 201–206. [Google Scholar] [CrossRef]
- Harazono, A.; Ema, M.; Ogawa, Y. Evaluation of Early Embryonic Loss Induced by Tributyltin Chloride in Rats: Phase- and Dose-Dependent Antifertility Effects. Arch. Environ. Contam. Toxicol. 1998, 34, 94–99. [Google Scholar] [CrossRef]
- de Araújo, J.F.P.; Podratz, P.L.; Sena, G.C.; Merlo, E.; Freitas-Lima, L.C.; Ayub, J.G.M.; Pereira, A.F.Z.; Santos-Silva, A.P.; Miranda-Alves, L.; Silva, I.V.; et al. The Obesogen Tributyltin Induces Abnormal Ovarian Adipogenesis in Adult Female Rats. Toxicol. Lett. 2018, 295, 99–114. [Google Scholar] [CrossRef]
- Janesick, A.; Blumberg, B. Minireview: PPARγ as the Target of Obesogens. J. Steroid. Biochem. Mol. Biol. 2011, 127, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Grün, F.; Blumberg, B. Environmental Obesogens: Organotins and Endocrine Disruption via Nuclear Receptor Signaling. Endocrinology 2006, 147, s50–s55. [Google Scholar] [CrossRef] [PubMed]
- Madsen, M.S.; Siersbæk, R.; Boergesen, M.; Nielsen, R.; Mandrup, S. Peroxisome Proliferator-Activated Receptor γ and C/EBPα Synergistically Activate Key Metabolic Adipocyte Genes by Assisted Loading. Mol. Cell. Biol. 2014, 34, 939–954. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.D.; Walkey, C.J.; Puigserver, P.; Spiegelman, B.M. Transcriptional Regulation of Adipogenesis. Genes Dev. 2000, 14, 1293–1307. [Google Scholar] [CrossRef]
- Orio, F.; Matarese, G.; di Biase, S.; Palomba, S.; Labella, D.; Sanna, V.; Savastano, S.; Zullo, F.; Colao, A.; Lombardi, G. Exon 6 and 2 Peroxisome Proliferator-Activated Receptor-γ Polymorphisms in Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2003, 88, 5887–5892. [Google Scholar] [CrossRef][Green Version]
- Bertuloso, B.D.; Podratz, P.L.; Merlo, E.; de Araújo, J.F.P.; Lima, L.C.F.; de Miguel, E.C.; de Souza, L.N.; Gava, A.L.; de Oliveira, M.; Miranda-Alves, L.; et al. Tributyltin Chloride Leads to Adiposity and Impairs Metabolic Functions in the Rat Liver and Pancreas. Toxicol. Lett. 2015, 235, 45–59. [Google Scholar] [CrossRef]
- Caldwell, A.S.L.; Middleton, L.J.; Jimenez, M.; Desai, R.; McMahon, A.C.; Allan, C.M.; Handelsman, D.J.; Walters, K.A. Characterization of Reproductive, Metabolic, and Endocrine Features of Polycystic Ovary Syndrome in Female Hyperandrogenic Mouse Models. Endocrinology 2014, 155, 3146–3159. [Google Scholar] [CrossRef]
- Rantakokko, P.; Main, K.M.; Wohlfart-Veje, C.; Kiviranta, H.; Airaksinen, R.; Vartiainen, T.; Skakkebæk, N.E.; Toppari, J.; Virtanen, H.E. Association of Placenta Organotin Concentrations with Growth and Ponderal Index in 110 Newborn Boys from Finland during the First 18 Months of Life: A Cohort Study. Environ. Health 2014, 13, 45. [Google Scholar] [CrossRef]
- Pu, Y.; Pearl, S.; Gingrich, J.; Jing, J.; Martin, D.; Murga-Zamalloa, C.A.; Veiga-Lopez, A. Multispecies Study: Low-Dose Tributyltin Impairs Ovarian Theca Cell Cholesterol Homeostasis through the RXR Pathway in Five Mammalian Species Including Humans. Arch. Toxicol. 2019, 93, 1665–1677. [Google Scholar] [CrossRef]
- Singh, S.; Kumar, V.; Gill, J.P.K.; Datta, S.; Singh, S.; Dhaka, V.; Kapoor, D.; Wani, A.B.; Dhanjal, D.S.; Kumar, M.; et al. Herbicide Glyphosate: Toxicity and Microbial Degradation. Int. J. Environ. Res. Public Health 2020, 17, 7519. [Google Scholar] [CrossRef]
- Milesi, M.M.; Lorenz, V.; Durando, M.; Rossetti, M.F.; Varayoud, J. Glyphosate Herbicide: Reproductive Outcomes and Multigenerational Effects. Front. Endocrinol. 2021, 12, 672532. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority (EFSA) Peer Review of the Pesticide Risk Assessment of the Potential Endocrine Disrupting Properties of Glyphosate. EFSA J. 2017, 15, e04979. [CrossRef]
- Environmental Protection Agency (EPA). Chemical: Glyphosate. EDSP: Weight of Evidence Analysis of Potential Interaction With the Estrogen, Androgen or Thyroid Pathways. Available online: https://www.regulations.gov/document/EPA-HQ-OPP-2009-0361-0047 (accessed on 17 December 2022).
- Kiyama, R.; Wada-Kiyama, Y. Estrogenic Endocrine Disruptors: Molecular Mechanisms of Action. Environ. Int. 2015, 83, 11–40. [Google Scholar] [CrossRef] [PubMed]
- Richard, S.; Moslemi, S.; Sipahutar, H.; Benachour, N.; Seralini, G.-E. Differential Effects of Glyphosate and Roundup on Human Placental Cells and Aromatase. Environ. Health. Perspect. 2005, 113, 716–720. [Google Scholar] [CrossRef] [PubMed]
- Benachour, N.; Sipahutar, H.; Moslemi, S.; Gasnier, C.; Travert, C.; Séralini, G.E. Time- and Dose-Dependent Effects of Roundup on Human Embryonic and Placental Cells. Arch. Environ. Contam. Toxicol. 2007, 53, 126–133. [Google Scholar] [CrossRef]
- Perego, M.C.; Schutz, L.F.; Caloni, F.; Cortinovis, C.; Albonico, M.; Spicer, L.J. Evidence for Direct Effects of Glyphosate on Ovarian Function: Glyphosate Influences Steroidogenesis and Proliferation of Bovine Granulosa but Not Theca Cells In Vitro. J. Appl. Toxicol. 2017, 37, 692–698. [Google Scholar] [CrossRef]
- Perego, M.C.; Caloni, F.; Cortinovis, C.; Schutz, L.F.; Albonico, M.; Tsuzukibashi, D.; Spicer, L.J. Influence of a Roundup Formulation on Glyphosate Effects on Steroidogenesis and Proliferation of Bovine Granulosa Cells In Vitro. Chemosphere 2017, 188, 274–279. [Google Scholar] [CrossRef]
- Gigante, P.; Berni, M.; Bussolati, S.; Grasselli, F.; Grolli, S.; Ramoni, R.; Basini, G. Glyphosate Affects Swine Ovarian and Adipose Stromal Cell Functions. Anim. Reprod. Sci. 2018, 195, 185–196. [Google Scholar] [CrossRef]
- Thongprakaisang, S.; Thiantanawat, A.; Rangkadilok, N.; Suriyo, T.; Satayavivad, J. Glyphosate Induces Human Breast Cancer Cells Growth via Estrogen Receptors. Food. Chem. Toxicol. 2013, 59, 129–136. [Google Scholar] [CrossRef]
- Mesnage, R.; Phedonos, A.; Biserni, M.; Arno, M.; Balu, S.; Corton, J.C.; Ugarte, R.; Antoniou, M.N. Evaluation of Estrogen Receptor Alpha Activation by Glyphosate-Based Herbicide Constituents. Food Chem. Toxicol. 2017, 108, 30–42. [Google Scholar] [CrossRef]
- Guerrero Schimpf, M.; Milesi, M.M.; Luque, E.H.; Varayoud, J. Glyphosate-Based Herbicide Enhances the Uterine Sensitivity to Estradiol in Rats. J. Endocrinol. 2018, 239, 197–213. [Google Scholar] [CrossRef]
- Lorenz, V.; Pacini, G.; Luque, E.H.; Varayoud, J.; Milesi, M.M. Perinatal Exposure to Glyphosate or a Glyphosate-Based Formulation Disrupts Hormonal and Uterine Milieu during the Receptive State in Rats. Food Chem. Toxicol. 2020, 143, 111560. [Google Scholar] [CrossRef]
- Guerrero Schimpf, M.; Milesi, M.M.; Ingaramo, P.I.; Luque, E.H.; Varayoud, J. Neonatal Exposure to a Glyphosate Based Herbicide Alters the Development of the Rat Uterus. Toxicology 2017, 376, 2–14. [Google Scholar] [CrossRef]
- Ren, X.; Li, R.; Liu, J.; Huang, K.; Wu, S.; Li, Y.; Li, C. Effects of Glyphosate on the Ovarian Function of Pregnant Mice, the Secretion of Hormones and the Sex Ratio of Their Fetuses. Environ. Pollut. 2018, 243, 833–841. [Google Scholar] [CrossRef]
- Alarcón, R.; Rivera, O.E.; Ingaramo, P.I.; Tschopp, M.V.; Dioguardi, G.H.; Milesi, M.M.; Muñoz-de-Toro, M.; Luque, E.H. Neonatal Exposure to a Glyphosate-Based Herbicide Alters the Uterine Differentiation of Prepubertal Ewe Lambs. Environ. Pollut. 2020, 265, 114874. [Google Scholar] [CrossRef]
- Gomez, A.L.; Altamirano, G.A.; Leturia, J.; Bosquiazzo, V.L.; Muñoz-de-Toro, M.; Kass, L. Male Mammary Gland Development and Methylation Status of Estrogen Receptor Alpha in Wistar Rats Are Modified by the Developmental Exposure to a Glyphosate-Based Herbicide. Mol. Cell. Endocrinol. 2019, 481, 14–25. [Google Scholar] [CrossRef]
- Hamdaoui, L.; Oudadesse, H.; Lefeuvre, B.; Mahmoud, A.; Naifer, M.; Badraoui, R.; Ayadi, F.; Rebai, T. Sub-Chronic Exposure to Kalach 360 SL, Glyphosate-Based Herbicide, Induced Bone Rarefaction in Female Wistar Rats. Toxicology 2020, 436, 152412. [Google Scholar] [CrossRef]
- Alarcón, R.; Ingaramo, P.I.; Rivera, O.E.; Dioguardi, G.H.; Repetti, M.R.; Demonte, L.D.; Milesi, M.M.; Varayoud, J.; Muñoz-de-Toro, M.; Luque, E.H. Neonatal Exposure to a Glyphosate-Based Herbicide Alters the Histofunctional Differentiation of the Ovaries and Uterus in Lambs. Mol. Cell. Endocrinol. 2019, 482, 45–56. [Google Scholar] [CrossRef]
- Parker J A New Hypothesis for the Mechanism of Glyphosate Induced Intestinal Permeability in the Pathogenesis of Polycystic Ovary Syndrome. J. Australas. Coll. Nutr. Environ. Med. 2015, 34, 3–7.
- Kubsad, D.; Nilsson, E.E.; King, S.E.; Sadler-Riggleman, I.; Beck, D.; Skinner, M.K. Assessment of Glyphosate Induced Epigenetic Transgenerational Inheritance of Pathologies and Sperm Epimutations: Generational Toxicology. Sci. Rep. 2019, 9, 6372. [Google Scholar] [CrossRef]
- ben Maamar, M.; Beck, D.; Nilsson, E.E.; Kubsad, D.; Skinner, M.K. Epigenome-Wide Association Study for Glyphosate Induced Transgenerational Sperm DNA Methylation and Histone Retention Epigenetic Biomarkers for Disease. Epigenetics 2021, 16, 1150–1167. [Google Scholar] [CrossRef]
- Mose, T.; Kjaerstad, M.B.; Mathiesen, L.; Nielsen, J.B.; Edelfors, S.; Knudsen, L.E. Placental Passage of Benzoic Acid, Caffeine, and Glyphosate in an Ex Vivo Human Perfusion System. J. Toxicol. Environ. Health A 2008, 71, 984–991. [Google Scholar] [CrossRef]
- Valle, A.L.; Mello, F.C.C.; Alves-Balvedi, R.P.; Rodrigues, L.P.; Goulart, L.R. Glyphosate Detection: Methods, Needs and Challenges. Environ. Chem. Lett. 2019, 17, 291–317. [Google Scholar] [CrossRef]
- Kongtip, P.; Nankongnab, N.; Phupancharoensuk, R.; Palarach, C.; Sujirarat, D.; Sangprasert, S.; Sermsuk, M.; Sawattrakool, N.; Woskie, S.R. Glyphosate and Paraquat in Maternal and Fetal Serums in Thai Women. J. Agromed. 2017, 22, 282–289. [Google Scholar] [CrossRef]
- Arbuckle, T.E.; Lin, Z.; Mery, L.S. An Exploratory Analysis of the Effect of Pesticide Exposure on the Risk of Spontaneous Abortion in an Ontario Farm Population. Environ. Health Perspect. 2001, 109, 851–857. [Google Scholar] [CrossRef]
- Parvez, S.; Gerona, R.R.; Proctor, C.; Friesen, M.; Ashby, J.L.; Reiter, J.L.; Lui, Z.; Winchester, P.D. Glyphosate Exposure in Pregnancy and Shortened Gestational Length: A Prospective Indiana Birth Cohort Study. Environ. Health 2018, 17, 23. [Google Scholar] [CrossRef]
- Rochester, J.R.; Bolden, A.L.; Pelch, K.E.; Kwiatkowski, C.F. Potential Developmental and Reproductive Impacts of Triclocarban: A Scoping Review. J. Toxicol. 2017, 2017, 9679738. [Google Scholar] [CrossRef]
- Huang, H.; Du, G.; Zhang, W.; Hu, J.; Wu, D.; Song, L.; Xia, Y.; Wang, X. The in Vitro Estrogenic Activities of Triclosan and Triclocarban. J. Appl. Toxicol. 2014, 34, 1060–1067. [Google Scholar] [CrossRef]
- Geer, L.A.; Pycke, B.F.G.; Waxenbaum, J.; Sherer, D.M.; Abulafia, O.; Halden, R.U. Association of Birth Outcomes with Fetal Exposure to Parabens, Triclosan and Triclocarban in an Immigrant Population in Brooklyn, New York. J. Hazard. Mater. 2017, 323, 177–183. [Google Scholar] [CrossRef]
- Wei, L.; Qiao, P.; Shi, Y.; Ruan, Y.; Yin, J.; Wu, Q.; Shao, B. Triclosan/Triclocarban Levels in Maternal and Umbilical Blood Samples and Their Association with Fetal Malformation. Clin. Chim. Acta 2017, 466, 133–137. [Google Scholar] [CrossRef]
- Dann, A.B.; Hontela, A. Triclosan: Environmental Exposure, Toxicity and Mechanisms of Action. J. Appl. Toxicol. 2011, 31, 285–311. [Google Scholar] [CrossRef]
- Moss, T.; Howes, D.; Williams, F.M. Percutaneous Penetration and Dermal Metabolism of Triclosan (2,4,4′-Trichloro-2′-Hydroxydiphenyl Ether). Food Chem. Toxicol. 2000, 38, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Sandborgh-Englund, G.; Adolfsson-Erici, M.; Odham, G.; Ekstrand, J. Pharmacokinetics of Triclosan Following Oral Ingestion in Humans. J. Toxicol. Environ. Health A 2006, 69, 1861–1873. [Google Scholar] [CrossRef] [PubMed]
- Geens, T.; Neels, H.; Covaci, A. Distribution of Bisphenol-A, Triclosan and n-Nonylphenol in Human Adipose Tissue, Liver and Brain. Chemosphere 2012, 87, 796–802. [Google Scholar] [CrossRef]
- Hovander, T.M.M.A.L. Identification of Hydroxylated PCB Metabolites and Other Phenolic Halogenated Pollutants in Human Blood Plasma. Arch. Environ. Contam. Toxicol. 2002, 42, 105–117. [Google Scholar] [CrossRef]
- Allmyr, M.; Adolfsson-Erici, M.; McLachlan, M.S.; Sandborgh-Englund, G. Triclosan in Plasma and Milk from Swedish Nursing Mothers and Their Exposure via Personal Care Products. Sci. Total Environ. 2006, 372, 87–93. [Google Scholar] [CrossRef]
- Dayan, A.D. Risk Assessment of Triclosan [Irgasan®] in Human Breast Milk. Food Chem. Toxicol. 2007, 45, 125–129. [Google Scholar] [CrossRef]
- Adolfsson-Erici, M.; Pettersson, M.; Parkkonen, J.; Sturve, J. Triclosan, a Commonly Used Bactericide Found in Human Milk and in the Aquatic Environment in Sweden. Chemosphere 2002, 46, 1485–1489. [Google Scholar] [CrossRef]
- Calafat, A.M.; Ye, X.; Wong, L.-Y.; Reidy, J.A.; Needham, L.L. Urinary Concentrations of Triclosan in the U.S. Population: 2003–2004. Environ. Health Perspect. 2008, 116, 303–307. [Google Scholar] [CrossRef]
- Maksymowicz, M.; Ręka, G.; Machowiec, P.; Piecewicz-Szczęsna, H. Impact of Triclosan on Female and Male Reproductive System and Its Consequences on Fertility; A Literature Review. J. Fam. Reprod. Health 2022, 16, 33–42. [Google Scholar] [CrossRef]
- Hipwell, A.E.; Kahn, L.G.; Factor-Litvak, P.; Porucznik, C.A.; Siegel, E.L.; Fichorova, R.N.; Hamman, R.F.; Klein-Fedyshin, M.; Harley, K.G. Exposure to Non-Persistent Chemicals in Consumer Products and Fecundability: A Systematic Review. Hum. Reprod. Update 2019, 25, 51–71. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-J.; Song, B.-S.; Kim, J.-W.; Yang, S.-G.; Kim, S.-U.; Koo, D.-B. Exposure of Triclosan in Porcine Oocyte Leads to Superoxide Production and Mitochondrial-Mediated Apoptosis during In Vitro Maturation. Int. J. Mol. Sci. 2020, 21, 3050. [Google Scholar] [CrossRef]
- Ha, M.; Zhang, P.; Li, L.; Liu, C. Triclosan Suppresses Testicular Steroidogenesis via the MiR-6321/JNK/ Nur77 Cascade. Cell. Physiol. Biochem. 2018, 50, 2029–2045. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, G.; Kayani, J.; Taylor, H.S.; Pal, L. Implications of Triclosan for Female Fertility: Results from the National Health and Nutrition Examination Survey, 2013–2016. F&S Rep. 2022, 3, 204–210. [Google Scholar] [CrossRef]
- Ye, J.; Zhu, W.; Liu, H.; Mao, Y.; Jin, F.; Zhang, J. Environmental Exposure to Triclosan and Polycystic Ovary Syndrome: A Cross-Sectional Study in China. BMJ Open 2018, 8, e019707. [Google Scholar] [CrossRef]
- Zhu, W.; Zhou, W.; Huo, X.; Zhao, S.; Gan, Y.; Wang, B.; Cheng, W.; Ouyang, F.; Wang, W.; Tian, Y.; et al. Triclosan and Female Reproductive Health. Epidemiology 2019, 30, S24–S31. [Google Scholar] [CrossRef]
- Gu, J.; Yuan, T.; Ni, N.; Ma, Y.; Shen, Z.; Yu, X.; Shi, R.; Tian, Y.; Zhou, W.; Zhang, J. Urinary Concentration of Personal Care Products and Polycystic Ovary Syndrome: A Case-Control Study. Environ. Res. 2019, 168, 48–53. [Google Scholar] [CrossRef]
- Kennedy, R.C.M.; Menn, F.-M.; Healy, L.; Fecteau, K.A.; Hu, P.; Bae, J.; Gee, N.A.; Lasley, B.L.; Zhao, L.; Chen, J. Early Life Triclocarban Exposure During Lactation Affects Neonate Rat Survival. Reprod. Sci. 2015, 22, 75–89. [Google Scholar] [CrossRef]
- Costa, N.O.; Borges, L.I.; Cavalcanti, L.F.; Montagnini, B.G.; Anselmo Franci, J.A.; Kiss, A.C.I.; Gerardin, D.C.C. In Utero and Lactational Exposure to Triclocarban: Reproductive Effects on Female Rat Offspring. J. Appl. Toxicol. 2020, 40, 504–514. [Google Scholar] [CrossRef]
- Yueh, M.-F.; Li, T.; Evans, R.M.; Hammock, B.; Tukey, R.H. Triclocarban Mediates Induction of Xenobiotic Metabolism through Activation of the Constitutive Androstane Receptor and the Estrogen Receptor Alpha. PLoS ONE 2012, 7, e37705. [Google Scholar] [CrossRef]
- Enright, H.; Lao, V.; Falso, M.; Walsworth, V.; Buchholz, B.; Malfatti, M.; Bench, G.; Turteltaub, K. Transfer of Triclocarban from Mother to Offspring during Periods of Development; Lawrence Livermore National Lab. (LLNL): Livermore, CA, USA, 2014. [Google Scholar]
- Chung, E.; Genco, M.C.; Megrelis, L.; Ruderman, J.V. Effects of Bisphenol A and Triclocarban on Brain-Specific Expression of Aromatase in Early Zebrafish Embryos. Proc. Natl. Acad. Sci. USA 2011, 108, 17732–17737. [Google Scholar] [CrossRef]
- Villeneuve, D.L.; Jensen, K.M.; Cavallin, J.E.; Durhan, E.J.; Garcia-Reyero, N.; Kahl, M.D.; Leino, R.L.; Makynen, E.A.; Wehmas, L.C.; Perkins, E.J.; et al. Effects of the Antimicrobial Contaminant Triclocarban, and Co-Exposure with the Androgen 17β-Trenbolone, on Reproductive Function and Ovarian Transcriptome of the Fathead Minnow (Pimephales promelas). Environ. Toxicol. Chem. 2017, 36, 231–242. [Google Scholar] [CrossRef]
- Mandour, D.A.; Aidaros, A.A.-M.; Mohamed, S. Potential Long-Term Developmental Toxicity of in Utero and Lactational Exposure to Triclocarban (TCC) in Hampering Ovarian Folliculogenesis in Rat Offspring. Acta Histochem. 2021, 123, 151772. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zheng, F.; Liu, F. Effects of Triclosan on Antioxidant- and Apoptosis-Related Genes Expression in the Gill and Ovary of Zebrafish. Exp. Anim. 2020, 69, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Priyanka; Trivedi, A.; Maske, P.; Mote, C.; Dighe, V. Gestational and Lactational Exposure to Triclosan Causes Impaired Fertility of F1 Male Offspring and Developmental Defects in F2 Generation. Environ. Pollut. 2020, 257, 113617. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.-Y.; Hua, X.; Xiong, J.-W.; Zhu, W.-T.; Zhang, J.; Chen, L. Impact of Triclosan on Female Reproduction through Reducing Thyroid Hormones to Suppress Hypothalamic Kisspeptin Neurons in Mice. Front. Mol. Neurosci. 2018, 11, 6. [Google Scholar] [CrossRef]
- Rouillon, S.; el Ouazzani, H.; Hardouin, J.-B.; Enjalbert, L.; Rabouan, S.; Migeot, V.; Albouy-Llaty, M. How to Educate Pregnant Women about Endocrine Disruptors? Int. J. Environ. Res. Public Health 2020, 17, 2156. [Google Scholar] [CrossRef]
- Weinberger, B.; Vetrano, A.M.; Archer, F.E.; Marcella, S.W.; Buckley, B.; Wartenberg, D.; Robson, M.G.; Klim, J.; Azhar, S.; Cavin, S.; et al. Effects of Maternal Exposure to Phthalates and Bisphenol A during Pregnancy on Gestational Age. J. Matern. Fetal. Neonatal. Med. 2014, 27, 323–327. [Google Scholar] [CrossRef]
- Veiga-Lopez, A.; Kannan, K.; Liao, C.; Ye, W.; Domino, S.E.; Padmanabhan, V. Gender-Specific Effects on Gestational Length and Birth Weight by Early Pregnancy BPA Exposure. J. Clin. Endocrinol. Metab. 2015, 100, E1394–E1403. [Google Scholar] [CrossRef]
- di Renzo, G.C.; Conry, J.A.; Blake, J.; DeFrancesco, M.S.; DeNicola, N.; Martin, J.N.; McCue, K.A.; Richmond, D.; Shah, A.; Sutton, P.; et al. International Federation of Gynecology and Obstetrics Opinion on Reproductive Health Impacts of Exposure to Toxic Environmental Chemicals. Int. J. Gynecol. Obstet. 2015, 131, 219–225. [Google Scholar] [CrossRef]
- Street, M.E.; Bernasconi, S. Endocrine-Disrupting Chemicals in Human Fetal Growth. Int. J. Mol. Sci. 2020, 21, 1430. [Google Scholar] [CrossRef] [PubMed]
- An Endocrine Society Endocrine-Disrupting Chemicals. An Endocrine Society Position Statement. Available online: https://www.endocrine.org/advocacy/position-statements/endocrine-disrupting-chemicals (accessed on 17 December 2022).




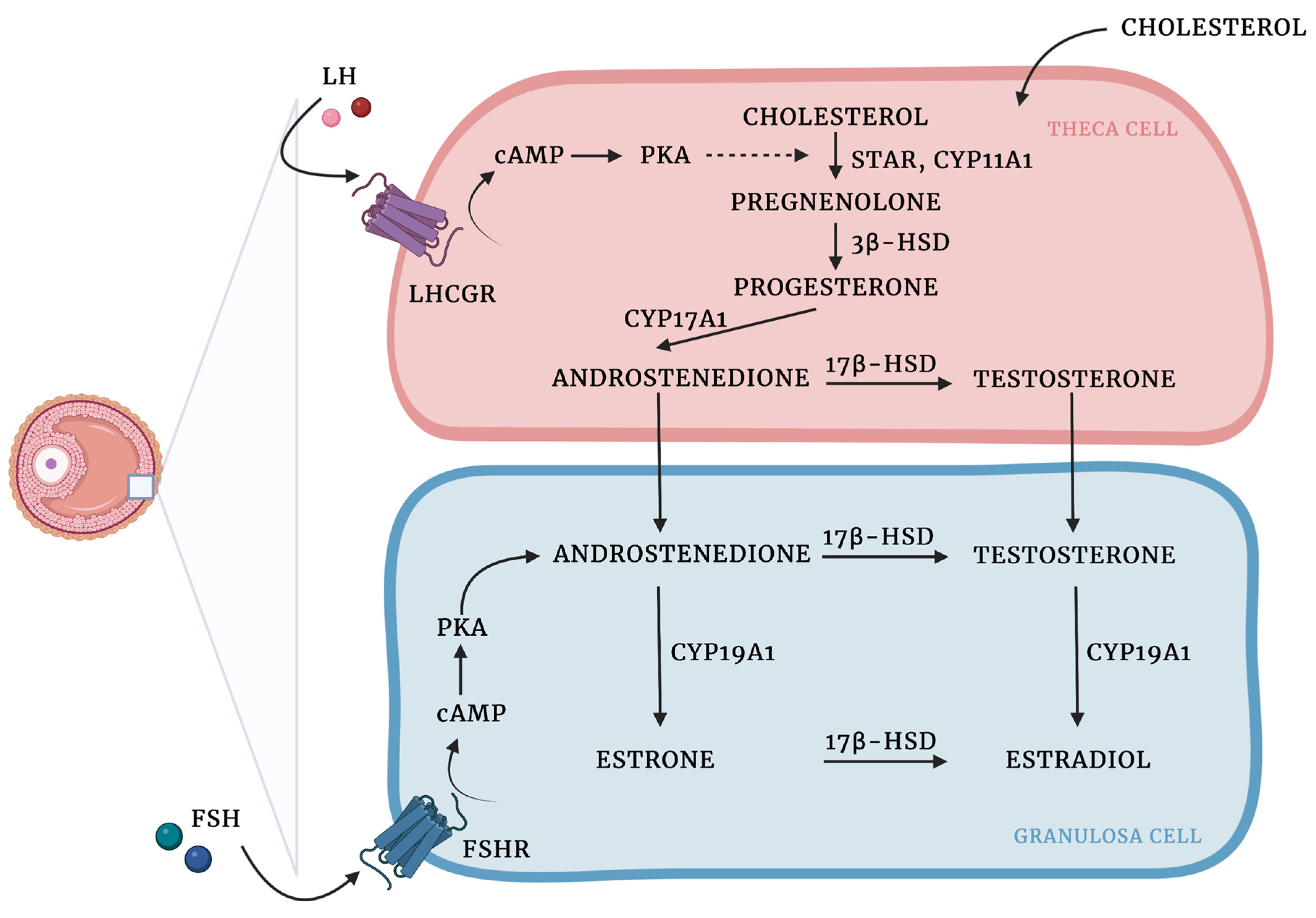{kind=link}
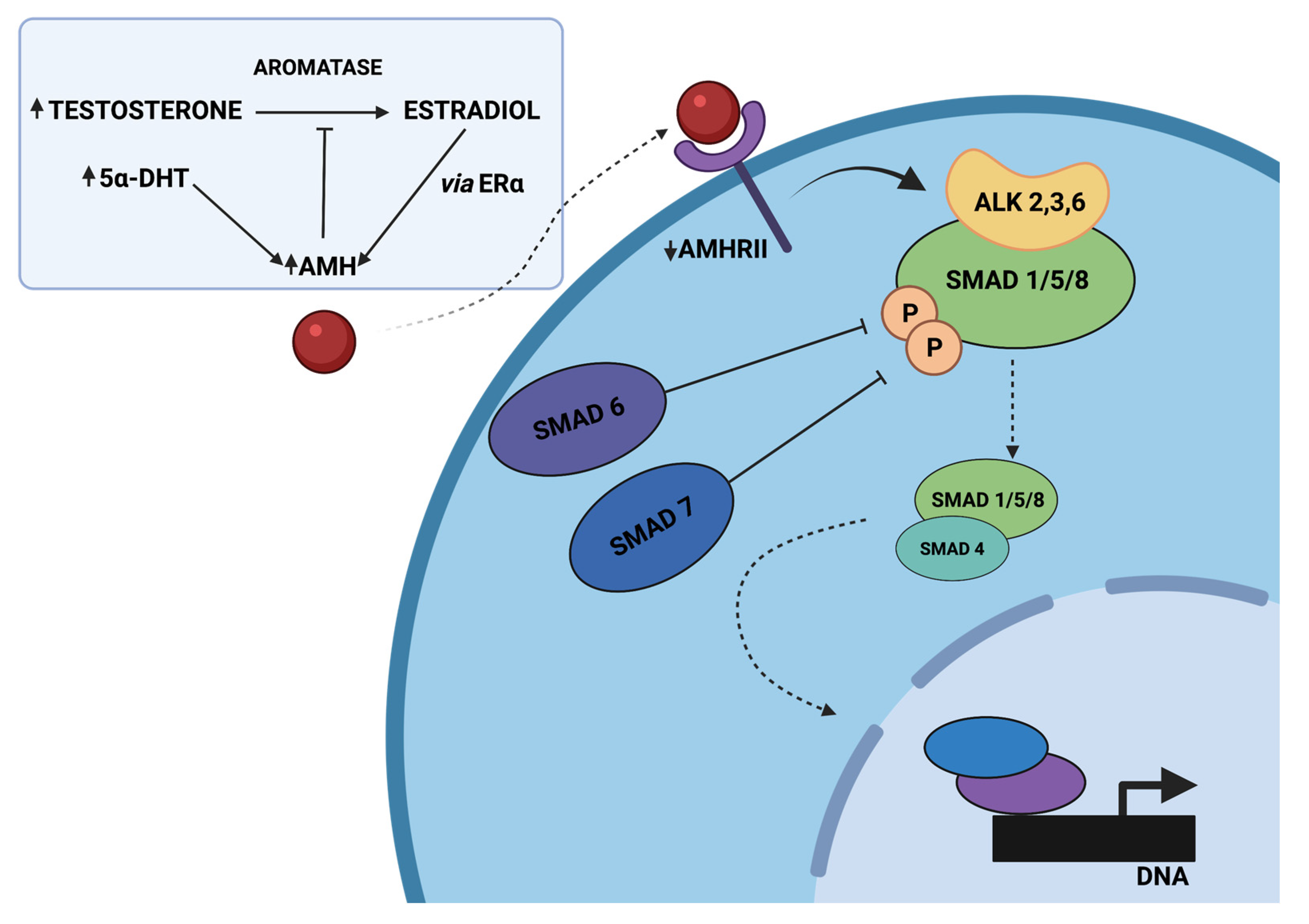{kind=link}
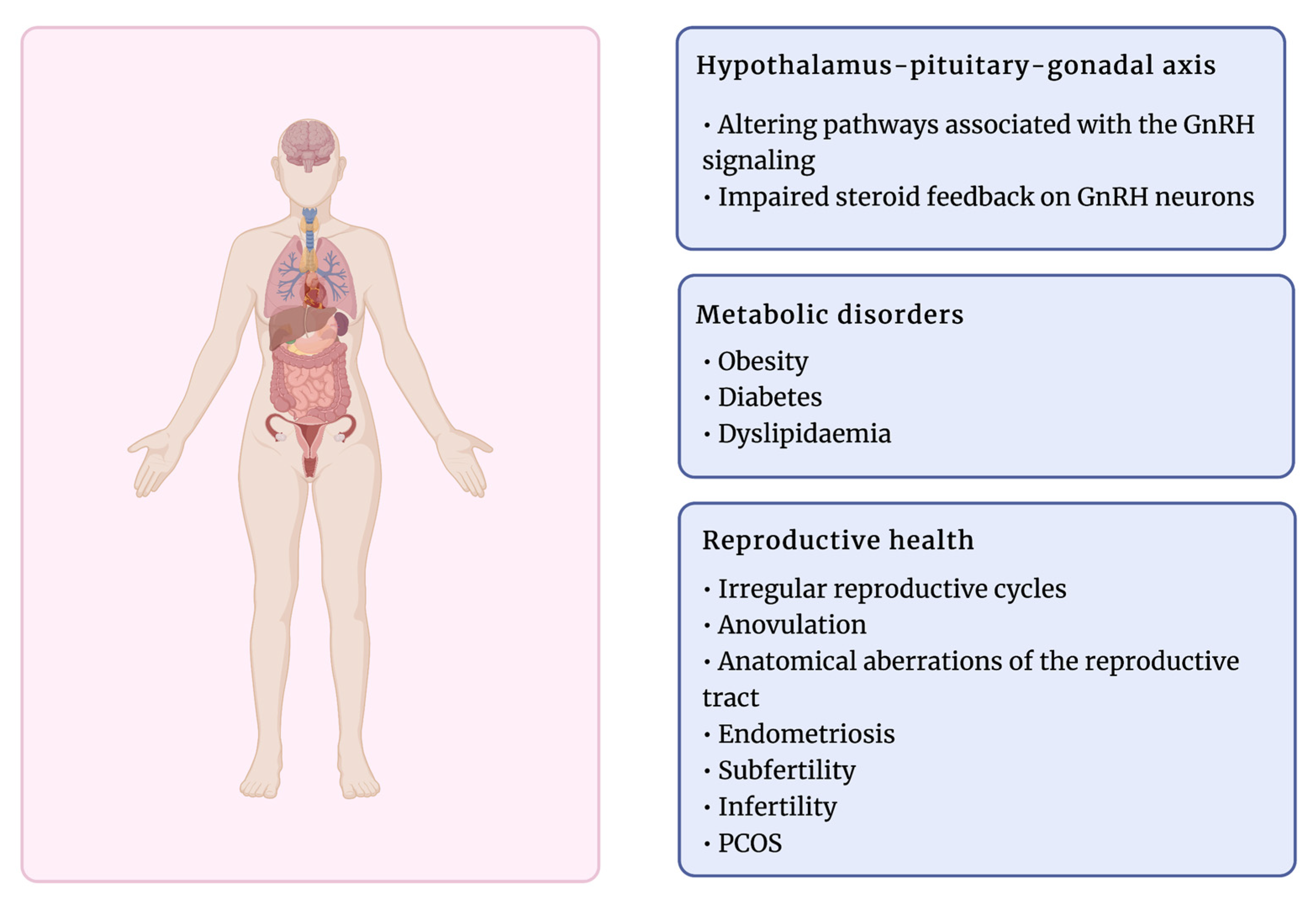{kind=link}
| EDC | Model | Strain | Exposure Duration | Age at Exposure | Route of Exposure | Dosage | End Points | Source |
|---|---|---|---|---|---|---|---|---|
| TCC | Rat | Sprague Dawley | 35d | Embryonic, adult | Oral (food) | 0.2% w/w, 0.5% w/w | ↓ body weight and survival in pups | [323] |
| Rat | Wistar | 21d | Embryonic, adult | Gavage | 0.3 mg/kg, 1.5 mg/kg, 3 mg/kg | ↓ estradiol levels in the TCC 0.3 and TCC 3.0 groups of female pups ↓ progesterone levels ↑preimplantation loss in the TCC 3.0 in adulthood | [324] | |
| Mouse | hUGT1*28 and CAR-null | 2d | Adult | Intra-peritoneal | 16 mg/kg, 20 mg/kg | ↑ hUGT and CYP gene expression via the CAR | [325] | |
| Mouse | CD-1 | GD1-18; PND0-10 | Embryonic, neonate, adult | Oral (water) | 100 nM | ↑ body weight of pups ↓ uterine weight in female offspring ↓ leptin, adiponectin and PPARα gene expression in adipose and liver tissues | [326] | |
| Fish | Zebrafish (Danio rerio) | 24 h | Embryonic | Submersion | 0.25 µM | ↑ E2-induced AroB expression ↓ BPA-induced AroB expression | [327] | |
| Fish | Fathead minnow (Pimephales promelas) | 22d | Adult | Submersion | 1.5 µg/L | No changes in adult body weight. | [328] | |
| Rat | Sprague Dawley | GD5-PND21 | Embryonic, lactational | Oral (water) | 0.5 mg/L | ↓follicle count, proliferation and gonadosomatic index of GCs. Delayed puberty onset. ↓ transition of the primordial follicles to more developed ↑ atresia, apoptosis, AR expression in GCs | [329] | |
| TCS | Fish | Zebrafish (Danio rerio) | 42d | Adult | TCS solution in exposure tank | 0, 17, 34, or 68 µg/L | ↓ expression of SOD, GPx1a, CAT, sMT-B in the ovary of 68 µg/L group ↑ oxidative damage in ovaries ↑ ROS-dependent ovary apoptosis | [330] |
| Rat | Holtzman | GD6-PD21 | Gestational, lactational | subcutaneous injection | 0.1, 4, 40 and 150 mg/kg b. wt./day | ↓ reproductive functions and fertility of F1 male rats ↓ testosterone, sperm count and motility ↓ AR, ERα and ERβ, SAR, aromatase expression ↑ pre- and post-implantation loss | [331] | |
| Mouse | ICR mice | 50d | Adult | Oral | 1, 10 or 100 mg/kg/day | ↓ LH, FSH, progesterone serum levels ↓ GnRH mRNA expression ↑ PRL ↓ kisspeptin immunoreactivity | [332] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jozkowiak, M.; Piotrowska-Kempisty, H.; Kobylarek, D.; Gorska, N.; Mozdziak, P.; Kempisty, B.; Rachon, D.; Spaczynski, R.Z. Endocrine Disrupting Chemicals in Polycystic Ovary Syndrome: The Relevant Role of the Theca and Granulosa Cells in the Pathogenesis of the Ovarian Dysfunction. Cells 2023, 12, 174. https://doi.org/10.3390/cells12010174
Jozkowiak M, Piotrowska-Kempisty H, Kobylarek D, Gorska N, Mozdziak P, Kempisty B, Rachon D, Spaczynski RZ. Endocrine Disrupting Chemicals in Polycystic Ovary Syndrome: The Relevant Role of the Theca and Granulosa Cells in the Pathogenesis of the Ovarian Dysfunction. Cells. 2023; 12(1):174. https://doi.org/10.3390/cells12010174
Chicago/Turabian StyleJozkowiak, Malgorzata, Hanna Piotrowska-Kempisty, Dominik Kobylarek, Natalia Gorska, Paul Mozdziak, Bartosz Kempisty, Dominik Rachon, and Robert Z. Spaczynski. 2023. "Endocrine Disrupting Chemicals in Polycystic Ovary Syndrome: The Relevant Role of the Theca and Granulosa Cells in the Pathogenesis of the Ovarian Dysfunction" Cells 12, no. 1: 174. https://doi.org/10.3390/cells12010174
APA StyleJozkowiak, M., Piotrowska-Kempisty, H., Kobylarek, D., Gorska, N., Mozdziak, P., Kempisty, B., Rachon, D., & Spaczynski, R. Z. (2023). Endocrine Disrupting Chemicals in Polycystic Ovary Syndrome: The Relevant Role of the Theca and Granulosa Cells in the Pathogenesis of the Ovarian Dysfunction. Cells, 12(1), 174. https://doi.org/10.3390/cells12010174