
The Past and Present Lives of the Intraocular Transmembrane Protein CD36


Abstract
1. Introduction
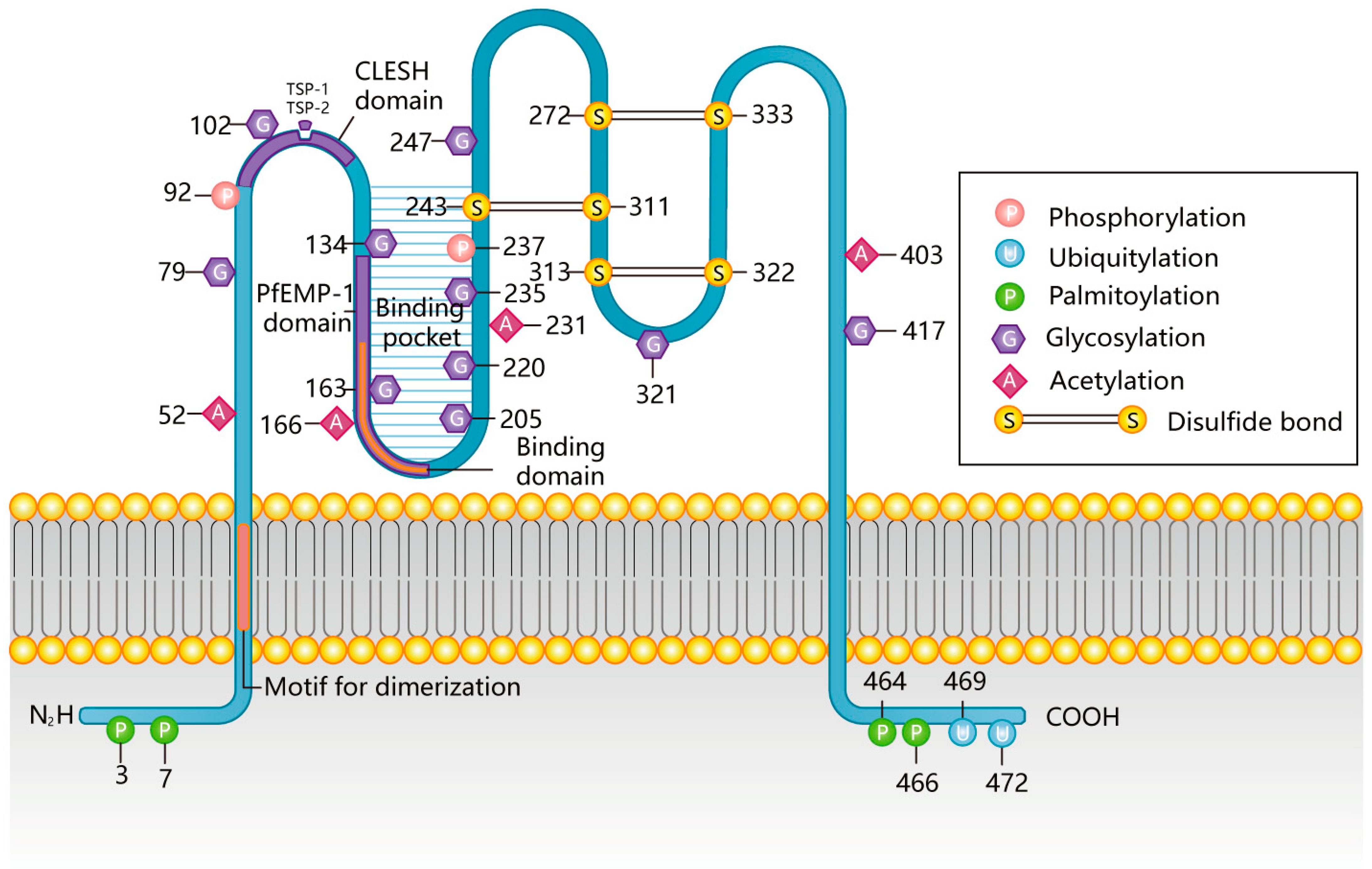
2. Molecular Characteristics of CD36
2.1. Structural Features of CD36
2.2. Posttranslational Modifications of CD36
2.2.1. CD36 Palmitoylation
2.2.2. CD36 Ubiquitination
2.2.3. CD36 Glycosylation
2.2.4. CD36 Phosphorylation
2.2.5. CD36 Acetylation
2.3. CD36 Distribution
2.4. CD36 Ligands
- CD36, limP-2, Emp sequence homology (CLESH) binding sites (93–120 and 155–183): CLESH is a 30-residue, long, negatively charged domain in CD36 that interacts with thrombospondin structural homology repeat with a positively charged surface ridge with high affinity. After binding, macrophages initiate the binding and entrainment of apoptotic neutrophils to produce IL-10 [78,86,87,88].
- P. falciparum erythrocyte membrane protein-1 (PfEMP-1)-binding site (139–184, 146-164AA, or 145-171AA, to be exact): This region binds PfEMP-1, a membrane protein specifically expressed by erythrocytes infected with Plasmodium falciparum [89].
- Lipid and protein binding site (155–183): This region contains a positive groove formed by a lysine cluster and hydrophobic amino acids that can recognize bound lipids such as ox-LDL (155–183) [90], oxidized phospholipids (157–171) [85], and long-chain fatty acids (127–279) [91]. In addition, this region can bind proteins such as advanced glycosylation end products [92] and members of the synthetic-growth-hormone-releasing peptide families such as hexarelin and EP 80317 [93]. Other possible ox-LDL binding sites on CD36 are 28–93 and 120–155 [94].
3. The Function of CD36
3.1. Mediation of Lipid Recognition and Intake
3.2. Involved in Inflammation
3.3. Regulation of Apoptosis and Angiogenesis
4. CD36 with Eye Diseases and Pathological Changes
4.1. Fundus Diseases and Pathological Changes
4.1.1. CD36 and Age-Related Macular Degeneration (AMD)
4.1.2. CD36 and Diabetic Retinopathy (DR)
4.1.3. CD36 and Glaucoma
4.1.4. CD36 and Retinal Neovascularization
4.1.5. CD36 and Subretinal Inflammation
4.2. Ocular Surface Diseases and Pathological Changes
4.2.1. CD36 and Corneal Neovascularization (CNV)
4.2.2. CD36 and Keratitis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Goldstein, J.L.; Ho, Y.K.; Basu, S.K.; Brown, M.S. Binding site on macrophages that mediates uptake and degradation of acetylated low density lipoprotein, producing massive cholesterol deposition. Proc. Natl. Acad. Sci. USA 1979, 76, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Lipoprotein metabolism in the macrophage: Implications for cholesterol deposition in atherosclerosis. Annu. Rev. Biochem. 1983, 52, 223–261. [Google Scholar] [CrossRef] [PubMed]
- PrabhuDas, M.R.; Baldwin, C.L.; Bollyky, P.L.; Bowdish, D.M.E.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; et al. A Consensus Definitive Classification of Scavenger Receptors and Their Roles in Health and Disease. J. Immunol. 2017, 198, 3775–3789. [Google Scholar] [CrossRef]
- Prabhudas, M.; Bowdish, D.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; Means, T.K.; Moestrup, S.K.; et al. Standardizing scavenger receptor nomenclature. J. Immunol. 2014, 192, 1997–2006. [Google Scholar] [CrossRef]
- Hsieh, F.L.; Turner, L.; Bolla, J.R.; Robinson, C.V.; Lavstsen, T.; Higgins, M.K. The structural basis for CD36 binding by the malaria parasite. Nat. Commun. 2016, 7, 12837. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef]
- Atzeni, F.; Rodríguez-Carrio, J.; Popa, C.D.; Nurmohamed, M.T.; Szűcs, G.; Szekanecz, Z. Cardiovascular effects of approved drugs for rheumatoid arthritis. Nat. Rev. Rheumatol. 2021, 17, 270–290. [Google Scholar] [CrossRef]
- Li, J.; Yu, C.; Wang, R.; Xu, J.; Chi, Y.; Qin, J.; Liu, Q. The ω-carboxyl group of 7-ketocholesteryl-9-carboxynonanoate mediates the binding of oxLDL to CD36 receptor and enhances caveolin-1 expression in macrophages. Int. J. Biochem. Cell Biol. 2017, 90, 121–135. [Google Scholar] [CrossRef]
- Moon, J.S.; Karunakaran, U.; Suma, E.; Chung, S.M.; Won, K.C. The Role of CD36 in Type 2 Diabetes Mellitus: β-Cell Dysfunction and Beyond. Diabetes Metab. J. 2020, 44, 222–233. [Google Scholar] [CrossRef]
- Rada, P.; González-Rodríguez, Á.; García-Monzón, C.; Valverde, Á.M. Understanding lipotoxicity in NAFLD pathogenesis: Is CD36 a key driver? Cell Death Dis. 2020, 11, 802. [Google Scholar] [CrossRef]
- Dorion, M.F.; Mulumba, M.; Kasai, S.; Itoh, K.; Lubell, W.D.; Ong, H. The CD36 Ligand-Promoted Autophagy Protects Retinal Pigment Epithelial Cells from Oxidative Stress. Oxidative Med. Cell. Longev. 2021, 2021, 6691402. [Google Scholar] [CrossRef] [PubMed]
- Roggia, M.F.; Ueta, T. αvβ5 Integrin/FAK/PGC-1α Pathway Confers Protective Effects on Retinal Pigment Epithelium. PLoS ONE 2015, 10, e0134870. [Google Scholar] [CrossRef] [PubMed]
- Mwaikambo, B.R.; Sennlaub, F.; Ong, H.; Chemtob, S.; Hardy, P. Genetic ablation of CD36 induces age-related corneal neovascularization. Cornea 2008, 27, 1037–1041. [Google Scholar] [CrossRef]
- Lu, Z.; Li, Y.; Ru, J.H.; Lopes-Virella, M.F.; Lyons, T.J.; Huang, Y. Interaction of palmitate and LPS regulates cytokine expression and apoptosis through sphingolipids in human retinal microvascular endothelial cells. Exp. Eye Res. 2019, 178, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef]
- Kindzelskii, A.L.; Elner, V.M.; Elner, S.G.; Yang, D.; Hughes, B.A.; Petty, H.R. Toll-like receptor 4 (TLR4) of retinal pigment epithelial cells participates in transmembrane signaling in response to photoreceptor outer segments. J. Gen. Physiol. 2004, 124, 139–149. [Google Scholar] [CrossRef]
- Mast, N.; Bederman, I.R.; Pikuleva, I.A. Retinal Cholesterol Content Is Reduced in Simvastatin-Treated Mice Due to Inhibited Local Biosynthesis Albeit Increased Uptake of Serum Cholesterol. Drug Metab. Dispos. Biol. Fate Chem. 2018, 46, 1528–1537. [Google Scholar] [CrossRef]
- Collot-Teixeira, S.; Martin, J.; McDermott-Roe, C.; Poston, R.; McGregor, J.L. CD36 and macrophages in atherosclerosis. Cardiovasc. Res. 2007, 75, 468–477. [Google Scholar] [CrossRef]
- Armesilla, A.L.; Vega, M.A. Structural organization of the gene for human CD36 glycoprotein. J. Biol. Chem. 1994, 269, 18985–18991. [Google Scholar] [CrossRef]
- Qiao, L.; Zou, C.; Shao, P.; Schaack, J.; Johnson, P.F.; Shao, J. Transcriptional regulation of fatty acid translocase/CD36 expression by CCAAT/enhancer-binding protein alpha. J. Biol. Chem. 2008, 283, 8788–8795. [Google Scholar] [CrossRef]
- Wang, J.; Qin, X.; Sun, H.; He, M.; Lv, Q.; Gao, C.; He, X.; Liao, H. Nogo receptor impairs the clearance of fibril amyloid-β by microglia and accelerates Alzheimer’s-like disease progression. Aging Cell 2021, 20, e13515. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Yu, T.; Pietronigro, E.C.; Yuan, J.; Arioli, J.; Pei, Y.; Luo, X.; Ye, J.; Constantin, G.; Mao, C.; et al. Peli1 impairs microglial Aβ phagocytosis through promoting C/EBPβ degradation. PLoS Biol. 2020, 18, e3000837. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Mun, B.R.; Lee, S.J.; Joh, Y.; Lee, H.Y.; Ji, K.Y.; Choi, H.R.; Lee, E.H.; Kim, E.M.; Jang, J.H.; et al. TREM2 promotes Aβ phagocytosis by upregulating C/EBPα-dependent CD36 expression in microglia. Sci. Rep. 2017, 7, 11118. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Su, W.; Ding, Z.; Wang, X.; Mercanti, F.; Chen, M.; Raina, S.; Mehta, J.L. Regulation of MSR-1 and CD36 in macrophages by LOX-1 mediated through PPAR-γ. Biochem. Biophys. Res. Commun. 2013, 431, 496–500. [Google Scholar] [CrossRef]
- Goto, K.; Iso, T.; Hanaoka, H.; Yamaguchi, A.; Suga, T.; Hattori, A.; Irie, Y.; Shinagawa, Y.; Matsui, H.; Syamsunarno, M.R.; et al. Peroxisome proliferator-activated receptor-γ in capillary endothelia promotes fatty acid uptake by heart during long-term fasting. J. Am. Heart Assoc. 2013, 2, e004861. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Gu, C.; Li, Y.; Huang, J. High Glucose Promotes CD36 Expression by Upregulating Peroxisome Proliferator-Activated Receptor γ Levels to Exacerbate Lipid Deposition in Renal Tubular Cells. BioMed Res. Int. 2017, 2017, 1414070. [Google Scholar] [CrossRef]
- Luiken, J.J.; Chanda, D.; Nabben, M.; Neumann, D.; Glatz, J.F. Post-translational modifications of CD36 (SR-B2): Implications for regulation of myocellular fatty acid uptake. Biochim. Et Biophys. Acta 2016, 1862, 2253–2258. [Google Scholar] [CrossRef]
- Abumrad, N.A.; el-Maghrabi, M.R.; Amri, E.Z.; Lopez, E.; Grimaldi, P.A. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J. Biol. Chem. 1993, 268, 17665–17668. [Google Scholar] [CrossRef]
- Cao, D.; Luo, J.; Chen, D.; Xu, H.; Shi, H.; Jing, X.; Zang, W. CD36 regulates lipopolysaccharide-induced signaling pathways and mediates the internalization of Escherichia coli in cooperation with TLR4 in goat mammary gland epithelial cells. Sci Rep. 2016, 6, 23132. [Google Scholar] [CrossRef]
- Xing, Q.; Feng, Y.; Sun, H.; Yang, S.; Sun, T.; Guo, X.; Ji, F.; Wu, B.; Zhou, D. Scavenger receptor MARCO contributes to macrophage phagocytosis and clearance of tumor cells. Exp. Cell Res. 2021, 408, 112862. [Google Scholar] [CrossRef]
- Thorne, R.F.; Ralston, K.J.; de Bock, C.E.; Mhaidat, N.M.; Zhang, X.D.; Boyd, A.W.; Burns, G.F. Palmitoylation of CD36/FAT regulates the rate of its post-transcriptional processing in the endoplasmic reticulum. Biochim. Et Biophys. Acta 2010, 1803, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.W.; Wang, J.; Guo, H.; Zhao, Y.Y.; Sun, H.H.; Li, Y.F.; Lai, X.Y.; Zhao, N.; Wang, X.; Xie, C.; et al. CD36 facilitates fatty acid uptake by dynamic palmitoylation-regulated endocytosis. Nat. Commun. 2020, 11, 4765. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Wu, F.; Chen, M.; Li, Y.; You, M.; Zhang, Y.; Yang, P.; Wei, L.; Ruan, X.Z.; Zhao, L.; et al. Inhibition of Fatty Acid Translocase (FAT/CD36) Palmitoylation Enhances Hepatic Fatty Acid β-Oxidation by Increasing Its Localization to Mitochondria and Interaction with Long-Chain Acyl-CoA Synthetase 1. Antioxid. Redox Signal. 2022, 36, 1081–1100. [Google Scholar] [CrossRef] [PubMed]
- Aicart-Ramos, C.; Valero, R.A.; Rodriguez-Crespo, I. Protein palmitoylation and subcellular trafficking. Biochim. Et Biophys. Acta 2011, 1808, 2981–2994. [Google Scholar] [CrossRef]
- Meiler, S.; Baumer, Y.; Huang, Z.; Hoffmann, F.W.; Fredericks, G.J.; Rose, A.H.; Norton, R.L.; Hoffmann, P.R.; Boisvert, W.A. Selenoprotein K is required for palmitoylation of CD36 in macrophages: Implications in foam cell formation and atherogenesis. J. Leukoc. Biol. 2013, 93, 771–780. [Google Scholar] [CrossRef]
- van Oort, M.M.; Drost, R.; Janβen, L.; Van Doorn, J.M.; Kerver, J.; Van der Horst, D.J.; Luiken, J.J.; Rodenburg, K.C. Each of the four intracellular cysteines of CD36 is essential for insulin- or AMP-activated protein kinase-induced CD36 translocation. Arch. Physiol. Biochem. 2014, 120, 40–49. [Google Scholar] [CrossRef]
- Zhang, Y.; Dong, D.; Xu, X.; He, H.; Zhu, Y.; Lei, T.; Ou, H. Oxidized high-density lipoprotein promotes CD36 palmitoylation and increases lipid uptake in macrophages. J. Biol. Chem. 2022, 298, 102000. [Google Scholar] [CrossRef]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Smith, J.; Su, X.; El-Maghrabi, R.; Stahl, P.D.; Abumrad, N.A. Opposite regulation of CD36 ubiquitination by fatty acids and insulin: Effects on fatty acid uptake. J. Biol. Chem. 2008, 283, 13578–13585. [Google Scholar] [CrossRef]
- Wu, W.; Wang, S.; Liu, Q.; Shan, T.; Wang, X.; Feng, J.; Wang, Y. AMPK facilitates intestinal long-chain fatty acid uptake by manipulating CD36 expression and translocation. FASEB J. 2020, 34, 4852–4869. [Google Scholar] [CrossRef]
- Kim, K.Y.; Stevens, M.V.; Akter, M.H.; Rusk, S.E.; Huang, R.J.; Cohen, A.; Noguchi, A.; Springer, D.; Bocharov, A.V.; Eggerman, T.L.; et al. Parkin is a lipid-responsive regulator of fat uptake in mice and mutant human cells. J. Clin. Investig. 2011, 121, 3701–3712. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Hu, T.; He, J.; Xu, Q.; Yu, C.; Liu, X.; Shao, Z.; Liao, Y.; Huang, H.; Liu, N. USP10 deletion inhibits macrophage-derived foam cell formation and cellular-oxidized low density lipoprotein uptake by promoting the degradation of CD36. Aging 2020, 12, 22892–22905. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Xia, X.; Chai, R.; Xu, R.; Xu, Q.; Liu, M.; Chen, X.; Liu, B.; Liu, S.; Liu, N. Inhibition of USP14 suppresses the formation of foam cell by promoting CD36 degradation. J. Cell. Mol. Med. 2020, 24, 3292–3302. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Xu, Q.; Liu, M.; Chen, X.; Liu, X.; He, J.; Hu, T.; Yu, C.; Huang, H.; Liu, S.; et al. Deubiquitination of CD36 by UCHL1 promotes foam cell formation. Cell Death Dis. 2020, 11, 636. [Google Scholar] [CrossRef]
- Moremen, K.W.; Tiemeyer, M.; Nairn, A.V. Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. [Google Scholar] [CrossRef]
- Hoosdally, S.J.; Andress, E.J.; Wooding, C.; Martin, C.A.; Linton, K.J. The Human Scavenger Receptor CD36: Glycosylation status and its role in trafficking and function. J. Biol. Chem. 2009, 284, 16277–16288. [Google Scholar] [CrossRef] [PubMed]
- Lauzier, B.; Merlen, C.; Vaillant, F.; McDuff, J.; Bouchard, B.; Beguin, P.C.; Dolinsky, V.W.; Foisy, S.; Villeneuve, L.R.; Labarthe, F.; et al. Post-translational modifications, a key process in CD36 function: Lessons from the spontaneously hypertensive rat heart. J. Mol. Cell. Cardiol. 2011, 51, 99–108. [Google Scholar] [CrossRef]
- Jiang, M.; Wu, N.; Xu, B.; Chu, Y.; Li, X.; Su, S.; Chen, D.; Li, W.; Shi, Y.; Gao, X.; et al. Fatty acid-induced CD36 expression via O-GlcNAcylation drives gastric cancer metastasis. Theranostics 2019, 9, 5359–5373. [Google Scholar] [CrossRef]
- Viñals, M.; Xu, S.; Vasile, E.; Krieger, M. Identification of the N-linked glycosylation sites on the high density lipoprotein (HDL) receptor SR-BI and assessment of their effects on HDL binding and selective lipid uptake. J. Biol. Chem. 2003, 278, 5325–5332. [Google Scholar] [CrossRef]
- Laczy, B.; Fülöp, N.; Onay-Besikci, A.; Des Rosiers, C.; Chatham, J.C. Acute regulation of cardiac metabolism by the hexosamine biosynthesis pathway and protein O-GlcNAcylation. PLoS ONE 2011, 6, e18417. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Hiasa, Y.; Murakami, H.; Ikeda, Y.; Yamanishi, H.; Abe, M.; Matsuura, B.; Onji, M. Rapid alternative absorption of dietary long-chain fatty acids with upregulation of intestinal glycosylated CD36 in liver cirrhosis. Am. J. Clin. Nutr. 2012, 96, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.Y.; Silverstein, R.L. CD36 ectodomain phosphorylation blocks thrombospondin-1 binding: Structure-function relationships and regulation by protein kinase C. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Hatmi, M.; Gavaret, J.M.; Elalamy, I.; Vargaftig, B.B.; Jacquemin, C. Evidence for cAMP-dependent platelet ectoprotein kinase activity that phosphorylates platelet glycoprotein IV (CD36). J. Biol. Chem. 1996, 271, 24776–24780. [Google Scholar] [CrossRef] [PubMed]
- Lynes, M.; Narisawa, S.; Millán, J.L.; Widmaier, E.P. Interactions between CD36 and global intestinal alkaline phosphatase in mouse small intestine and effects of high-fat diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1738–R1747. [Google Scholar] [CrossRef]
- Asch, A.S.; Liu, I.; Briccetti, F.M.; Barnwell, J.W.; Kwakye-Berko, F.; Dokun, A.; Goldberger, J.; Pernambuco, M. Analysis of CD36 binding domains: Ligand specificity controlled by dephosphorylation of an ectodomain. Science 1993, 262, 1436–1440. [Google Scholar] [CrossRef]
- Guthmann, F.; Maehl, P.; Preiss, J.; Kolleck, I.; Rüstow, B. Ectoprotein kinase-mediated phosphorylation of FAT/CD36 regulates palmitate uptake by human platelets. Cell. Mol. Life Sci. 2002, 59, 1999–2003. [Google Scholar] [CrossRef]
- Pfleger, J.; Gross, P.; Johnson, J.; Carter, R.L.; Gao, E.; Tilley, D.G.; Houser, S.R.; Koch, W.J. G protein-coupled receptor kinase 2 contributes to impaired fatty acid metabolism in the failing heart. J. Mol. Cell. Cardiol. 2018, 123, 108–117. [Google Scholar] [CrossRef]
- Lundby, A.; Lage, K.; Weinert, B.T.; Bekker-Jensen, D.B.; Secher, A.; Skovgaard, T.; Kelstrup, C.D.; Dmytriyev, A.; Choudhary, C.; Lundby, C.; et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012, 2, 419–431. [Google Scholar] [CrossRef]
- Kuda, O.; Pietka, T.A.; Demianova, Z.; Kudova, E.; Cvacka, J.; Kopecky, J.; Abumrad, N.A. Sulfo-N-succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 lysine 164: SSO also inhibits oxidized low density lipoprotein uptake by macrophages. J. Biol. Chem. 2013, 288, 15547–15555. [Google Scholar] [CrossRef]
- Khan, S.; Kowluru, A. CD36 mediates lipid accumulation in pancreatic beta cells under the duress of glucolipotoxic conditions: Novel roles of lysine deacetylases. Biochem. Biophys. Res. Commun. 2018, 495, 2221–2226. [Google Scholar] [CrossRef]
- Du, X.; Jiang, S.; Zeng, X.; Zhang, J.; Pan, K.; Zhou, J.; Xie, Y.; Kan, H.; Song, W.; Sun, Q.; et al. Air pollution is associated with the development of atherosclerosis via the cooperation of CD36 and NLRP3 inflammasome in ApoE(-/-) mice. Toxicol Lett 2018, 290, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, U.; Elumalai, S.; Moon, J.S.; Won, K.C. CD36 Signal Transduction in Metabolic Diseases: Novel Insights and Therapeutic Targeting. Cells 2021, 10, 1833. [Google Scholar] [CrossRef] [PubMed]
- Bonen, A.; Luiken, J.J.; Arumugam, Y.; Glatz, J.F.; Tandon, N.N. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J. Biol. Chem. 2000, 275, 14501–14508. [Google Scholar] [CrossRef]
- Chabowski, A.; Coort, S.L.; Calles-Escandon, J.; Tandon, N.N.; Glatz, J.F.; Luiken, J.J.; Bonen, A. Insulin stimulates fatty acid transport by regulating expression of FAT/CD36 but not FABPpm. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E781–E789. [Google Scholar] [CrossRef]
- Jeppesen, J.; Albers, P.H.; Rose, A.J.; Birk, J.B.; Schjerling, P.; Dzamko, N.; Steinberg, G.R.; Kiens, B. Contraction-induced skeletal muscle FAT/CD36 trafficking and FA uptake is AMPK independent. J. Lipid Res. 2011, 52, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Houssier, M.; Raoul, W.; Lavalette, S.; Keller, N.; Guillonneau, X.; Baragatti, B.; Jonet, L.; Jeanny, J.C.; Behar-Cohen, F.; Coceani, F.; et al. CD36 deficiency leads to choroidal involution via COX2 down-regulation in rodents. PLoS Med. 2008, 5, e39. [Google Scholar] [CrossRef] [PubMed]
- Tserentsoodol, N.; Gordiyenko, N.V.; Pascual, I.; Lee, J.W.; Fliesler, S.J.; Rodriguez, I.R. Intraretinal lipid transport is dependent on high density lipoprotein-like particles and class B scavenger receptors. Mol. Vis. 2006, 12, 1319–1333. [Google Scholar] [PubMed]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Yoon, H.; Choi, S.I.; Kim, E.K. Uptake of cell debris and enhanced expression of inflammatory factors in response to dead cells in corneal fibroblast cells. Exp. Eye Res. 2020, 194, 108017. [Google Scholar] [CrossRef]
- Soriano-Romaní, L.; Contreras-Ruiz, L.; García-Posadas, L.; López-García, A.; Masli, S.; Diebold, Y. Inflammatory Cytokine-Mediated Regulation of Thrombospondin-1 and CD36 in Conjunctival Cells. J. Ocul. Pharmacol. Ther. Off. J. Assoc. Ocul. Pharmacol. Ther. 2015, 31, 419–428. [Google Scholar] [CrossRef]
- Tanaka, T.; Nakata, T.; Oka, T.; Ogawa, T.; Okamoto, F.; Kusaka, Y.; Sohmiya, K.; Shimamoto, K.; Itakura, K. Defect in human myocardial long-chain fatty acid uptake is caused by FAT/CD36 mutations. J. Lipid Res. 2001, 42, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Wintergerst, E.S.; Jelk, J.; Rahner, C.; Asmis, R. Apoptosis induced by oxidized low density lipoprotein in human monocyte-derived macrophages involves CD36 and activation of caspase-3. Eur J. Biochem 2000, 267, 6050–6059. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yin, Y.; Zhou, Z.; He, M.; Dai, Y. OxLDL-induced IL-1 beta secretion promoting foam cells formation was mainly via CD36 mediated ROS production leading to NLRP3 inflammasome activation. Inflamm. Res. 2014, 63, 33–43. [Google Scholar] [CrossRef]
- Seimon, T.A.; Nadolski, M.J.; Liao, X.; Magallon, J.; Nguyen, M.; Feric, N.T.; Koschinsky, M.L.; Harkewicz, R.; Witztum, J.L.; Tsimikas, S.; et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab 2010, 12, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Iwao, Y.; Nakajou, K.; Nagai, R.; Kitamura, K.; Anraku, M.; Maruyama, T.; Otagiri, M. CD36 is one of important receptors promoting renal tubular injury by advanced oxidation protein products. Am. J. Physiol Ren. Physiol 2008, 295, F1871–F1880. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Li, W.; Silverstein, R.L. Advanced glycation end products induce a prothrombotic phenotype in mice via interaction with platelet CD36. Blood 2012, 119, 6136–6144. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, M.J.; de Witt, S.; Nergiz-Unal, R.; van Kruchten, R.; Korporaal, S.J.; Verhamme, P.; Febbraio, M.; Tjwa, M.; Voshol, P.J.; Hoylaerts, M.F.; et al. Supporting roles of platelet thrombospondin-1 and CD36 in thrombus formation on collagen. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1187–1192. [Google Scholar] [CrossRef]
- Simantov, R.; Febbraio, M.; Silverstein, R.L. The antiangiogenic effect of thrombospondin-2 is mediated by CD36 and modulated by histidine-rich glycoprotein. Matrix Biol. 2005, 24, 27–34. [Google Scholar] [CrossRef]
- Kerkhoff, C.; Sorg, C.; Tandon, N.N.; Nacken, W. Interaction of S100A8/S100A9-arachidonic acid complexes with the scavenger receptor CD36 may facilitate fatty acid uptake by endothelial cells. Biochemistry 2001, 40, 241–248. [Google Scholar] [CrossRef]
- Tondera, C.; Laube, M.; Pietzsch, J. Insights into binding of S100 proteins to scavenger receptors: Class B scavenger receptor CD36 binds S100A12 with high affinity. Amino Acids 2017, 49, 183–191. [Google Scholar] [CrossRef]
- Park, L.; Zhou, J.; Zhou, P.; Pistick, R.; El Jamal, S.; Younkin, L.; Pierce, J.; Arreguin, A.; Anrather, J.; Younkin, S.G.; et al. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 3089–3094. [Google Scholar] [CrossRef] [PubMed]
- Rodrigue-Way, A.; Caron, V.; Bilodeau, S.; Keil, S.; Hassan, M.; Lévy, E.; Mitchell, G.A.; Tremblay, A. Scavenger receptor CD36 mediates inhibition of cholesterol synthesis via activation of the PPARγ/PGC-1α pathway and Insig1/2 expression in hepatocytes. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 1910–1923. [Google Scholar] [CrossRef]
- Marleau, S.; Harb, D.; Bujold, K.; Avallone, R.; Iken, K.; Wang, Y.; Demers, A.; Sirois, M.G.; Febbraio, M.; Silverstein, R.L.; et al. EP 80317, a ligand of the CD36 scavenger receptor, protects apolipoprotein E-deficient mice from developing atherosclerotic lesions. FASEB J. 2005, 19, 1869–1871. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Jay, A.; Brunaldi, K.; Huang, N.; Hamilton, J.A. CD36 enhances fatty acid uptake by increasing the rate of intracellular esterification but not transport across the plasma membrane. Biochemistry 2013, 52, 7254–7261. [Google Scholar] [CrossRef]
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99. [Google Scholar] [CrossRef]
- Savill, J.; Hogg, N.; Ren, Y.; Haslett, C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J. Clin. Investig. 1992, 90, 1513–1522. [Google Scholar] [CrossRef]
- Navazo, M.D.; Daviet, L.; Savill, J.; Ren, Y.; Leung, L.L.; McGregor, J.L. Identification of a domain (155-183) on CD36 implicated in the phagocytosis of apoptotic neutrophils. J. Biol. Chem. 1996, 271, 15381–15385. [Google Scholar] [CrossRef]
- Dawson, D.W.; Pearce, S.F.; Zhong, R.; Silverstein, R.L.; Frazier, W.A.; Bouck, N.P. CD36 mediates the In vitro inhibitory effects of thrombospondin-1 on endothelial cells. J. Cell Biol. 1997, 138, 707–717. [Google Scholar] [CrossRef]
- Baruch, D.I.; Ma, X.C.; Pasloske, B.; Howard, R.J.; Miller, L.H. CD36 peptides that block cytoadherence define the CD36 binding region for Plasmodium falciparum-infected erythrocytes. Blood 1999, 94, 2121–2127. [Google Scholar] [CrossRef]
- Puente Navazo, M.D.; Daviet, L.; Ninio, E.; McGregor, J.L. Identification on human CD36 of a domain (155-183) implicated in binding oxidized low-density lipoproteins (Ox-LDL). Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1033–1039. [Google Scholar] [CrossRef]
- Pepino, M.Y.; Kuda, O.; Samovski, D.; Abumrad, N.A. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu. Rev. Nutr. 2014, 34, 281–303. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, N.; Nagai, R.; Ikemoto, M.; Arai, H.; Kuniyasu, A.; Horiuchi, S.; Nakayama, H. CD36, a member of class B scavenger receptor family, is a receptor for advanced glycation end products. Ann. N. Y. Acad. Sci. 2001, 947, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Demers, A.; McNicoll, N.; Febbraio, M.; Servant, M.; Marleau, S.; Silverstein, R.; Ong, H. Identification of the growth hormone-releasing peptide binding site in CD36: A photoaffinity cross-linking study. Biochem J. 2004, 382, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.F.; Roy, P.; Nicholson, A.C.; Hajjar, D.P.; Febbraio, M.; Silverstein, R.L. Recombinant glutathione S-transferase/CD36 fusion proteins define an oxidized low density lipoprotein-binding domain. J. Biol. Chem. 1998, 273, 34875–34881. [Google Scholar] [CrossRef] [PubMed]
- Rieu, Q.; Bougoüin, A.; Zagar, Y.; Chatagnon, J.; Hamieh, A.; Enderlin, J.; Huby, T.; Nandrot, E.F. Pleiotropic Roles of Scavenger Receptors in Circadian Retinal Phagocytosis: A New Function for Lysosomal SR-B2/LIMP-2 at the RPE Cell Surface. Int. J. Mol. Sci. 2022, 23, 3445. [Google Scholar] [CrossRef]
- Nandrot, E.F.; Kim, Y.; Brodie, S.E.; Huang, X.; Sheppard, D.; Finnemann, S.C. Loss of synchronized retinal phagocytosis and age-related blindness in mice lacking alphavbeta5 integrin. J. Exp. Med. 2004, 200, 1539–1545. [Google Scholar] [CrossRef]
- Yang, C.; Shani, S.; Tahiri, H.; Ortiz, C.; Gu, M.; Lavoie, J.C.; Croteau, S.; Hardy, P. Extracellular microparticles exacerbate oxidative damage to retinal pigment epithelial cells. Exp. Cell Res. 2020, 390, 111957. [Google Scholar] [CrossRef]
- Vollrath, D.; Feng, W.; Duncan, J.L.; Yasumura, D.; D’Cruz, P.M.; Chappelow, A.; Matthes, M.T.; Kay, M.A.; LaVail, M.M. Correction of the retinal dystrophy phenotype of the RCS rat by viral gene transfer of Mertk. Proc. Natl. Acad. Sci. USA 2001, 98, 12584–12589. [Google Scholar] [CrossRef]
- Wu, Z.; Wang, S.; Sorenson, C.M.; Sheibani, N. Attenuation of retinal vascular development and neovascularization in transgenic mice over-expressing thrombospondin-1 in the lens. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2006, 235, 1908–1920. [Google Scholar] [CrossRef]
- Rocha, D.M.; Caldas, A.P.; Oliveira, L.L.; Bressan, J.; Hermsdorff, H.H. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis 2016, 244, 211–215. [Google Scholar] [CrossRef]
- Adamiec-Mroczek, J.; Oficjalska-Młyńczak, J.; Misiuk-Hojło, M. Roles of endothelin-1 and selected proinflammatory cytokines in the pathogenesis of proliferative diabetic retinopathy: Analysis of vitreous samples. Cytokine 2010, 49, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Fredrikson, G.N.; Anand, D.V.; Hopkins, D.; Corder, R.; Alm, R.; Bengtsson, E.; Shah, P.K.; Lahiri, A.; Nilsson, J. Associations between autoantibodies against apolipoprotein B-100 peptides and vascular complications in patients with type 2 diabetes. Diabetologia 2009, 52, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Hopkins, B.D.; Tsujikawa, K.; Perruzzi, C.; Adini, I.; Swerlick, R.; Bornstein, P.; Lawler, J.; Benjamin, L.E. Thrombospondin-1 modulates VEGF-A-mediated Akt signaling and capillary survival in the developing retina. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1344–H1351. [Google Scholar] [CrossRef] [PubMed]
- Howlett, D.R.; Bate, S.T.; Collier, S.; Lawman, A.; Chapman, T.; Ashmeade, T.; Marshall, I.; Anderson, P.J.; Philpott, K.L.; Richardson, J.C.; et al. Characterisation of amyloid-induced inflammatory responses in the rat retina. Exp. Brain Res. 2011, 214, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Mellal, K.; Omri, S.; Mulumba, M.; Tahiri, H.; Fortin, C.; Dorion, M.F.; Pham, H.; Garcia Ramos, Y.; Zhang, J.; Pundir, S.; et al. Immunometabolic modulation of retinal inflammation by CD36 ligand. Sci. Rep. 2019, 9, 12903. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.W.; Qi, X.; Jia, C.K.; Wang, Y.Q. Serum amyloid A and pairing formyl peptide receptor 2 are expressed in corneas and involved in inflammation-mediated neovascularization. Int. J. Ophthalmol. 2014, 7, 187–193. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jia, C.; Zhu, W.; Ren, S.; Xi, H.; Li, S.; Wang, Y. Comparison of genome-wide gene expression in suture- and alkali burn-induced murine corneal neovascularization. Mol. Vis. 2011, 17, 2386–2399. [Google Scholar] [PubMed]
- Klocke, J.; Barcia, R.N.; Heimer, S.; Cario, E.; Zieske, J.; Gilmore, M.S.; Ksander, B.R.; Gregory, M.S. Spontaneous bacterial keratitis in CD36 knockout mice. Investig. Ophthalmol. Vis. Sci. 2011, 52, 256–263. [Google Scholar] [CrossRef][Green Version]
- Zhao, L.; Varghese, Z.; Moorhead, J.F.; Chen, Y.; Ruan, X.Z. CD36 and lipid metabolism in the evolution of atherosclerosis. Br. Med. Bull. 2018, 126, 101–112. [Google Scholar] [CrossRef]
- Elsøe, S.; Ahnström, J.; Christoffersen, C.; Hoofnagle, A.N.; Plomgaard, P.; Heinecke, J.W.; Binder, C.J.; Björkbacka, H.; Dahlbäck, B.; Nielsen, L.B. Apolipoprotein M binds oxidized phospholipids and increases the antioxidant effect of HDL. Atherosclerosis 2012, 221, 91–97. [Google Scholar] [CrossRef]
- Dhungana, H.; Huuskonen, M.T.; Jaronen, M.; Lemarchant, S.; Ali, H.; Keksa-Goldsteine, V.; Goldsteins, G.; Kanninen, K.M.; Koistinaho, J.; Malm, T. Sulfosuccinimidyl oleate sodium is neuroprotective and alleviates stroke-induced neuroinflammation. J. Neuroinflammation 2017, 14, 237. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; D’Amico, E.; Farilla, L.; Hui, H.; Wawrowsky, K.A.; Mlynarski, W.; Doria, A.; Abumrad, N.A.; Perfetti, R. Fatty acid translocase (FAT/CD36) is localized on insulin-containing granules in human pancreatic beta-cells and mediates fatty acid effects on insulin secretion. Diabetes 2005, 54, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jung, E.; Lee, J.; Huh, S.; Kim, Y.S.; Kim, Y.W.; Kim, Y.S.; Park, D. Anti-adipogenesis by 6-thioinosine is mediated by downregulation of PPAR gamma through JNK-dependent upregulation of iNOS. Cell. Mol. Life Sci. 2010, 67, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Daquinag, A.C.; Gao, Z.; Fussell, C.; Immaraj, L.; Pasqualini, R.; Arap, W.; Akimzhanov, A.M.; Febbraio, M.; Kolonin, M.G. Fatty acid mobilization from adipose tissue is mediated by CD36 posttranslational modifications and intracellular trafficking. JCI Insight 2021, 6, e147057. [Google Scholar] [CrossRef]
- Son, N.H.; Basu, D.; Samovski, D.; Pietka, T.A.; Peche, V.S.; Willecke, F.; Fang, X.; Yu, S.Q.; Scerbo, D.; Chang, H.R.; et al. Endothelial cell CD36 optimizes tissue fatty acid uptake. J. Clin. Investig. 2018, 128, 4329–4342. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.L.; Pearce, S.F.; Francisco, L.M.; Sauter, B.; Roy, P.; Silverstein, R.L.; Bhardwaj, N. Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J. Exp. Med. 1998, 188, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, J.P.; Lukens, J.R.; Wilhelm, A.J.; Moore, J.L.; Mendez-Fernandez, Y.; Kanneganti, T.D.; Major, A.S. Oxidized Low-Density Lipoprotein Immune Complex Priming of the Nlrp3 Inflammasome Involves TLR and FcγR Cooperation and Is Dependent on CARD9. J. Immunol. 2017, 198, 2105–2114. [Google Scholar] [CrossRef]
- Ekici, M.; Kisa, U.; Arikan Durmaz, S.; Ugur, E.; Nergiz-Unal, R. Fatty acid transport receptor soluble CD36 and dietary fatty acid pattern in type 2 diabetic patients: A comparative study. Br. J. Nutr. 2018, 119, 153–162. [Google Scholar] [CrossRef]
- Lawler, J. The functions of thrombospondin-1 and-2. Curr. Opin. Cell Biol. 2000, 12, 634–640. [Google Scholar] [CrossRef]
- Jiménez, B.; Volpert, O.V.; Crawford, S.E.; Febbraio, M.; Silverstein, R.L.; Bouck, N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000, 6, 41–48. [Google Scholar] [CrossRef]
- Blasiak, J. Senescence in the pathogenesis of age-related macular degeneration. Cell. Mol. Life Sci. 2020, 77, 789–805. [Google Scholar] [CrossRef] [PubMed]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Segawa, K.; Nagata, S. An Apoptotic ’Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015, 25, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Penberthy, K.K.; Lysiak, J.J.; Ravichandran, K.S. Rethinking Phagocytes: Clues from the Retina and Testes. Trends Cell Biol. 2018, 28, 317–327. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Sinha, D.; Blasiak, J.; Kauppinen, A.; Veréb, Z.; Salminen, A.; Boulton, M.E.; Petrovski, G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy 2013, 9, 973–984. [Google Scholar] [CrossRef]
- Ryeom, S.W.; Silverstein, R.L.; Scotto, A.; Sparrow, J.R. Binding of anionic phospholipids to retinal pigment epithelium may be mediated by the scavenger receptor CD36. J. Biol. Chem. 1996, 271, 20536–20539. [Google Scholar] [CrossRef][Green Version]
- Ren, Y.; Silverstein, R.L.; Allen, J.; Savill, J. CD36 gene transfer confers capacity for phagocytosis of cells undergoing apoptosis. J. Exp. Med. 1995, 181, 1857–1862. [Google Scholar] [CrossRef]
- Lin, H.; Clegg, D.O. Integrin alphavbeta5 participates in the binding of photoreceptor rod outer segments during phagocytosis by cultured human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1703–1712. [Google Scholar]
- Duncan, K.G.; Bailey, K.R.; Kane, J.P.; Schwartz, D.M. Human retinal pigment epithelial cells express scavenger receptors BI and BII. Biochem. Biophys. Res. Commun. 2002, 292, 1017–1022. [Google Scholar] [CrossRef]
- Westenskow, P.D.; Moreno, S.K.; Krohne, T.U.; Kurihara, T.; Zhu, S.; Zhang, Z.N.; Zhao, T.; Xu, Y.; Ding, S.; Friedlander, M. Using flow cytometry to compare the dynamics of photoreceptor outer segment phagocytosis in iPS-derived RPE cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6282–6290. [Google Scholar] [CrossRef]
- Finnemann, S.C.; Silverstein, R.L. Differential roles of CD36 and alphavbeta5 integrin in photoreceptor phagocytosis by the retinal pigment epithelium. J. Exp. Med. 2001, 194, 1289–1298. [Google Scholar] [CrossRef]
- Chang, Y.; Finnemann, S.C. Tetraspanin CD81 is required for the alpha v beta5-integrin-dependent particle-binding step of RPE phagocytosis. J. Cell Sci. 2007, 120, 3053–3063. [Google Scholar] [CrossRef]
- Sun, M.; Finnemann, S.C.; Febbraio, M.; Shan, L.; Annangudi, S.P.; Podrez, E.A.; Hoppe, G.; Darrow, R.; Organisciak, D.T.; Salomon, R.G.; et al. Light-induced oxidation of photoreceptor outer segment phospholipids generates ligands for CD36-mediated phagocytosis by retinal pigment epithelium: A potential mechanism for modulating outer segment phagocytosis under oxidant stress conditions. J. Biol. Chem. 2006, 281, 4222–4230. [Google Scholar] [CrossRef] [PubMed]
- Podrez, E.A.; Poliakov, E.; Shen, Z.; Zhang, R.; Deng, Y.; Sun, M.; Finton, P.J.; Shan, L.; Febbraio, M.; Hajjar, D.P.; et al. A novel family of atherogenic oxidized phospholipids promotes macrophage foam cell formation via the scavenger receptor CD36 and is enriched in atherosclerotic lesions. J. Biol. Chem. 2002, 277, 38517–38523. [Google Scholar] [CrossRef] [PubMed]
- Gordiyenko, N.; Campos, M.; Lee, J.W.; Fariss, R.N.; Sztein, J.; Rodriguez, I.R. RPE cells internalize low-density lipoprotein (LDL) and oxidized LDL (oxLDL) in large quantities in vitro and in vivo. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2822–2829. [Google Scholar] [CrossRef] [PubMed]
- Courtois, Y. The role of CD36 receptor in the phagocytosis of oxidized lipids and AMD. Aging 2010, 2, 888–889. [Google Scholar] [CrossRef]
- Rigotti, A.; Acton, S.L.; Krieger, M. The class B scavenger receptors SR-BI and CD36 are receptors for anionic phospholipids. J. Biol. Chem. 1995, 270, 16221–16224. [Google Scholar] [CrossRef]
- Martini, C.; DeNichilo, M.; King, D.P.; Cockshell, M.P.; Ebert, B.; Dale, B.; Ebert, L.M.; Woods, A.; Bonder, C.S. CD36 promotes vasculogenic mimicry in melanoma by mediating adhesion to the extracellular matrix. BMC Cancer 2021, 21, 765. [Google Scholar] [CrossRef]
- Kondo, N.; Honda, S.; Kuno, S.; Negi, A. Positive association of common variants in CD36 with neovascular age-related macular degeneration. Aging 2009, 1, 266–274. [Google Scholar] [CrossRef][Green Version]
- Honda, S.; Bessho, H.; Kondo, N.; Kusuhara, S.; Tsukahara, Y.; Negi, A. Positive association of CD36 gene variants with the visual outcome of photodynamic therapy in polypoidal choroidal vasculopathy. Mol. Vis. 2012, 18, 2796–2804. [Google Scholar]
- Yanagi, Y.; Foo, V.H.X.; Yoshida, A. Asian age-related macular degeneration: From basic science research perspective. Eye (Lond. Engl.) 2019, 33, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Bowers, C.Y. Growth hormone-releasing peptide (GHRP). Cell. Mol. Life Sci. 1998, 54, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Picard, E.; Houssier, M.; Bujold, K.; Sapieha, P.; Lubell, W.; Dorfman, A.; Racine, J.; Hardy, P.; Febbraio, M.; Lachapelle, P.; et al. CD36 plays an important role in the clearance of oxLDL and associated age-dependent sub-retinal deposits. Aging 2010, 2, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Proulx, C.; Picard, É.; Boeglin, D.; Pohankova, P.; Chemtob, S.; Ong, H.; Lubell, W.D. Azapeptide analogues of the growth hormone releasing peptide 6 as cluster of differentiation 36 receptor ligands with reduced affinity for the growth hormone secretagogue receptor 1a. J. Med. Chem. 2012, 55, 6502–6511. [Google Scholar] [CrossRef]
- Cogan, D.G.; Toussaint, D.; Kuwabara, T. Retinal vascular patterns. IV. Diabetic retinopathy. Arch. Ophthalmol. 1961, 66, 366–378. [Google Scholar] [CrossRef]
- Adamis, A.P.; Berman, A.J. Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin. Immunopathol. 2008, 30, 65–84. [Google Scholar] [CrossRef]
- Sennlaub, F.; Valamanesh, F.; Vazquez-Tello, A.; El-Asrar, A.M.; Checchin, D.; Brault, S.; Gobeil, F.; Beauchamp, M.H.; Mwaikambo, B.; Courtois, Y.; et al. Cyclooxygenase-2 in human and experimental ischemic proliferative retinopathy. Circulation 2003, 108, 198–204. [Google Scholar] [CrossRef]
- Truman, J.P.; Al Gadban, M.M.; Smith, K.J.; Jenkins, R.W.; Mayroo, N.; Virella, G.; Lopes-Virella, M.F.; Bielawska, A.; Hannun, Y.A.; Hammad, S.M. Differential regulation of acid sphingomyelinase in macrophages stimulated with oxidized low-density lipoprotein (LDL) and oxidized LDL immune complexes: Role in phagocytosis and cytokine release. Immunology 2012, 136, 30–45. [Google Scholar] [CrossRef]
- Lopes-Virella, M.F.; Baker, N.L.; Hunt, K.J.; Lyons, T.J.; Jenkins, A.J.; Virella, G. High concentrations of AGE-LDL and oxidized LDL in circulating immune complexes are associated with progression of retinopathy in type 1 diabetes. Diabetes Care 2012, 35, 1333–1340. [Google Scholar] [CrossRef]
- Fu, D.; Yu, J.Y.; Wu, M.; Du, M.; Chen, Y.; Abdelsamie, S.A.; Li, Y.; Chen, J.; Boulton, M.E.; Ma, J.X.; et al. Immune complex formation in human diabetic retina enhances toxicity of oxidized LDL towards retinal capillary pericytes. J. Lipid Res. 2014, 55, 860–869. [Google Scholar] [CrossRef]
- Abdelsamie, S.A.; Li, Y.; Huang, Y.; Lee, M.H.; Klein, R.L.; Virella, G.; Lopes-Virella, M.F. Oxidized LDL immune complexes stimulate collagen IV production in mesangial cells via Fc gamma receptors I and III. Clin. Immunol. 2011, 139, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Ralston, J.C.; Metherel, A.H.; Stark, K.D.; Mutch, D.M. SCD1 mediates the influence of exogenous saturated and monounsaturated fatty acids in adipocytes: Effects on cellular stress, inflammatory markers and fatty acid elongation. J. Nutr. Biochem. 2016, 27, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Kawasaki, R.; Rogers, S.; Man, R.E.; Itakura, K.; Xie, J.; Flood, V.; Tsubota, K.; Lamoureux, E.; Wang, J.J. The Associations of Dietary Intake of Polyunsaturated Fatty Acids With Diabetic Retinopathy in Well-Controlled Diabetes. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7473–7479. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Chakravarty, K.; Kong, X.; Tuy, T.T.; Arinze, I.J.; Bone, F.; Massillon, D. Several transcription factors are recruited to the glucose-6-phosphatase gene promoter in response to palmitate in rat hepatocytes and H4IIE cells. J. Nutr. 2007, 137, 554–559. [Google Scholar] [CrossRef][Green Version]
- Baranova, I.N.; Kurlander, R.; Bocharov, A.V.; Vishnyakova, T.G.; Chen, Z.; Remaley, A.T.; Csako, G.; Patterson, A.P.; Eggerman, T.L. Role of human CD36 in bacterial recognition, phagocytosis, and pathogen-induced JNK-mediated signaling. J. Immunol. 2008, 181, 7147–7156. [Google Scholar] [CrossRef]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef]
- Wilkinson, K.; Boyd, J.D.; Glicksman, M.; Moore, K.J.; El Khoury, J. A high content drug screen identifies ursolic acid as an inhibitor of amyloid beta protein interactions with its receptor CD36. J. Biol. Chem. 2011, 286, 34914–34922. [Google Scholar] [CrossRef]
- Simons, E.S.; Smith, M.A.; Dengler-Crish, C.M.; Crish, S.D. Retinal ganglion cell loss and gliosis in the retinofugal projection following intravitreal exposure to amyloid-beta. Neurobiol. Dis. 2021, 147, 105146. [Google Scholar] [CrossRef]
- Chen, H.; Herndon, M.E.; Lawler, J. The cell biology of thrombospondin-1. Matrix Biol. 2000, 19, 597–614. [Google Scholar] [CrossRef]
- Tian, R.; Deng, A.; Pang, X.; Chen, Y.; Gao, Y.; Liu, H.; Hu, Z. VR-10 polypeptide interacts with CD36 to induce cell apoptosis and autophagy in choroid-retinal endothelial cells: Identification of VR-10 as putative novel therapeutic agent for choroid neovascularization (CNV) treatment. Peptides 2022, 157, 170868. [Google Scholar] [CrossRef]
- Upalakalin, J.N.; Hemo, I.; Dehio, C.; Keshet, E.; Benjamin, L.E. Survival mechanisms of VEGF and PlGF during microvascular remodeling. Cold Spring Harb. Symp. Quant. Biol. 2002, 67, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.Y.; Ramakrishnan, D.P.; Silverstein, R.L. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood 2013, 122, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Cai, X.; Wu, Y.; Liu, Y.; Deng, L.; Chen, H. Insights from Genetic Model Systems of Retinal Degeneration: Role of Epsins in Retinal Angiogenesis and VEGFR2 Signaling. J. Nat. Sci. 2017, 3. [Google Scholar]
- Whitcup, S.M.; Nussenblatt, R.B.; Lightman, S.L.; Hollander, D.A. Inflammation in retinal disease. Int. J. Inflamm. 2013, 2013, 724648. [Google Scholar] [CrossRef]
- Abe, T.; Shimamura, M.; Jackman, K.; Kurinami, H.; Anrather, J.; Zhou, P.; Iadecola, C. Key role of CD36 in Toll-like receptor 2 signaling in cerebral ischemia. Stroke 2010, 41, 898–904. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Lavalette, S.; Conart, J.B.; Touhami, S.; Roubeix, C.; Houssier, M.; Augustin, S.; Raoul, W.; Combadière, C.; Febbraio, M.; Ong, H.; et al. CD36 Deficiency Inhibits Retinal Inflammation and Retinal Degeneration in Cx3cr1 Knockout Mice. Front. Immunol. 2019, 10, 3032. [Google Scholar] [CrossRef]
- Nicholas, M.P.; Mysore, N. Corneal neovascularization. Exp. Eye Res. 2021, 202, 108363. [Google Scholar] [CrossRef]
- Mwaikambo, B.R.; Yang, C.; Ong, H.; Chemtob, S.; Hardy, P. Emerging roles for the CD36 scavenger receptor as a potential therapeutic target for corneal neovascularization. Endocr. Metab. Immune Disord. Drug Targets 2008, 8, 255–272. [Google Scholar] [CrossRef]
- Ricciuto, J.; Heimer, S.R.; Gilmore, M.S.; Argüeso, P. Cell surface O-glycans limit Staphylococcus aureus adherence to corneal epithelial cells. Infect. Immun. 2008, 76, 5215–5220. [Google Scholar] [CrossRef]
- Mwaikambo, B.R.; Sennlaub, F.; Ong, H.; Chemtob, S.; Hardy, P. Activation of CD36 inhibits and induces regression of inflammatory corneal neovascularization. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4356–4364. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Romaní, L.; García-Posadas, L.; López-García, A.; Paraoan, L.; Diebold, Y. Thrombospondin-1 induces differential response in human corneal and conjunctival epithelial cells lines under in vitro inflammatory and apoptotic conditions. Exp. Eye Res. 2015, 134, 1–14. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eye Disease | Cell Type | CD36 Ligands | Pathological Damage | Refs. |
|---|---|---|---|---|
| AMD | POS(PtdSer) | Metabolic abnormalities, oxidative stress injury | [16,95,96] | |
| POS(oxPCs) | Metabolic abnormalities, oxidative stress injury | [97] | ||
| Retinal pigment epithelium cells | POS(PS, PI) | Metabolic abnormalities | [11] | |
| ox-LDL, EP80317 | Metabolic abnormalities | [98] | ||
| RMPs | Metabolic abnormalities, oxidative stress injury | [99] | ||
| MPE-001 | Oxidative stress injury, autophagy | [100] | ||
| DR | Retinal microvascular endothelial cells | SFA, LPS, TSP-1 | Apoptosis | [14,101] |
| Periretinal cells | ox-LDL-ICS | Oxidative stress injury, apoptosis, inflammation | [102] | |
| Retinal neovascularization | Retinal microvascular endothelial cells | TSP-1 | Apoptosis | [103] |
| RGC degenerative injury | Retinal ganglion cells | Aβ peptide | Inflammation, oxidative stress injury | [104] |
| Subretinal inflammation | Mononuclear phagocytes | MPE-001 | Inflammation | [105] |
| CNV | Corneal epithelial cell | SAA, Fpr2 | Inflammation | [106,107] |
| Keratitis | Corneal epithelial cell | TSP-1 | Inflammation | [108,109] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, R.; Liu, Q.; Zhang, M. The Past and Present Lives of the Intraocular Transmembrane Protein CD36. Cells 2023, 12, 171. https://doi.org/10.3390/cells12010171
Yang R, Liu Q, Zhang M. The Past and Present Lives of the Intraocular Transmembrane Protein CD36. Cells. 2023; 12(1):171. https://doi.org/10.3390/cells12010171
Chicago/Turabian StyleYang, Rucui, Qingping Liu, and Mingzhi Zhang. 2023. "The Past and Present Lives of the Intraocular Transmembrane Protein CD36" Cells 12, no. 1: 171. https://doi.org/10.3390/cells12010171
APA StyleYang, R., Liu, Q., & Zhang, M. (2023). The Past and Present Lives of the Intraocular Transmembrane Protein CD36. Cells, 12(1), 171. https://doi.org/10.3390/cells12010171