
Sleep-Disturbance-Induced Microglial Activation Involves CRH-Mediated Galectin 3 and Autophagy Dysregulation

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal and Reagents
2.2. SF Procedure
2.3. Adult Microglia Isolation
2.4. Primary Microglia and BV2 Cell Culture
2.5. Microglia Treatment
2.6. RNA Extraction, Reverse Transcription, and Quantitative (Q) Polymerase Chain Reaction
2.7. Western Blots
2.8. Immunofluorescence Staining
2.9. Immunocytochemistry
2.10. Statistical Analysis
3. Results
3.1. Three-Weeks of SF Activates Microglia in the HP and Increases Peripheral Inflammation Levels
3.2. The Effects of Three Weeks of SF on Microglia, Astrocytes, and CRH Signaling in the HP
3.3. CRH Activates Mg and Upregulates Gal3 Levels In Vitro
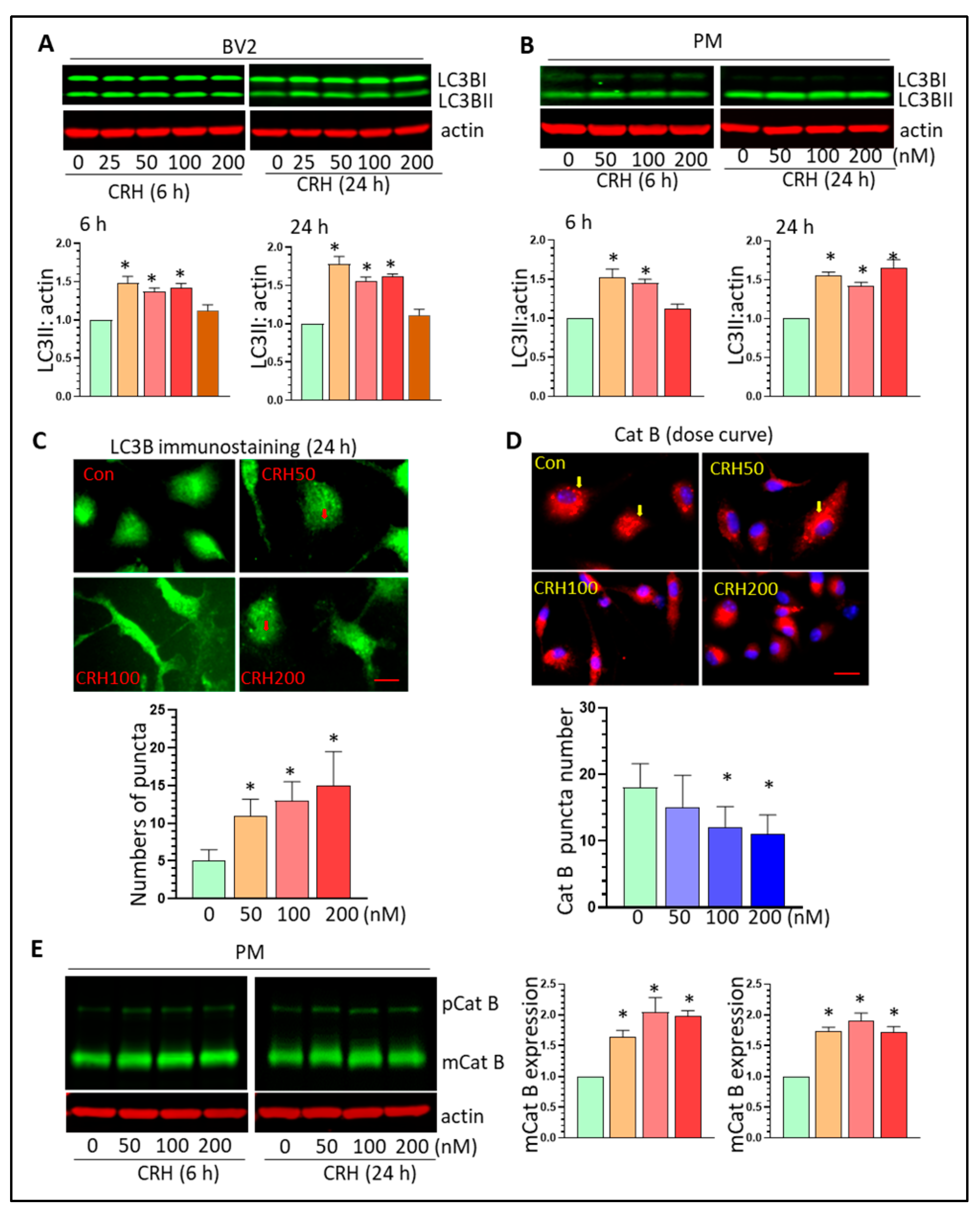
3.4. CRH Dysregulates Autophagy Processes and Induces Lysosome Damage In Vitro
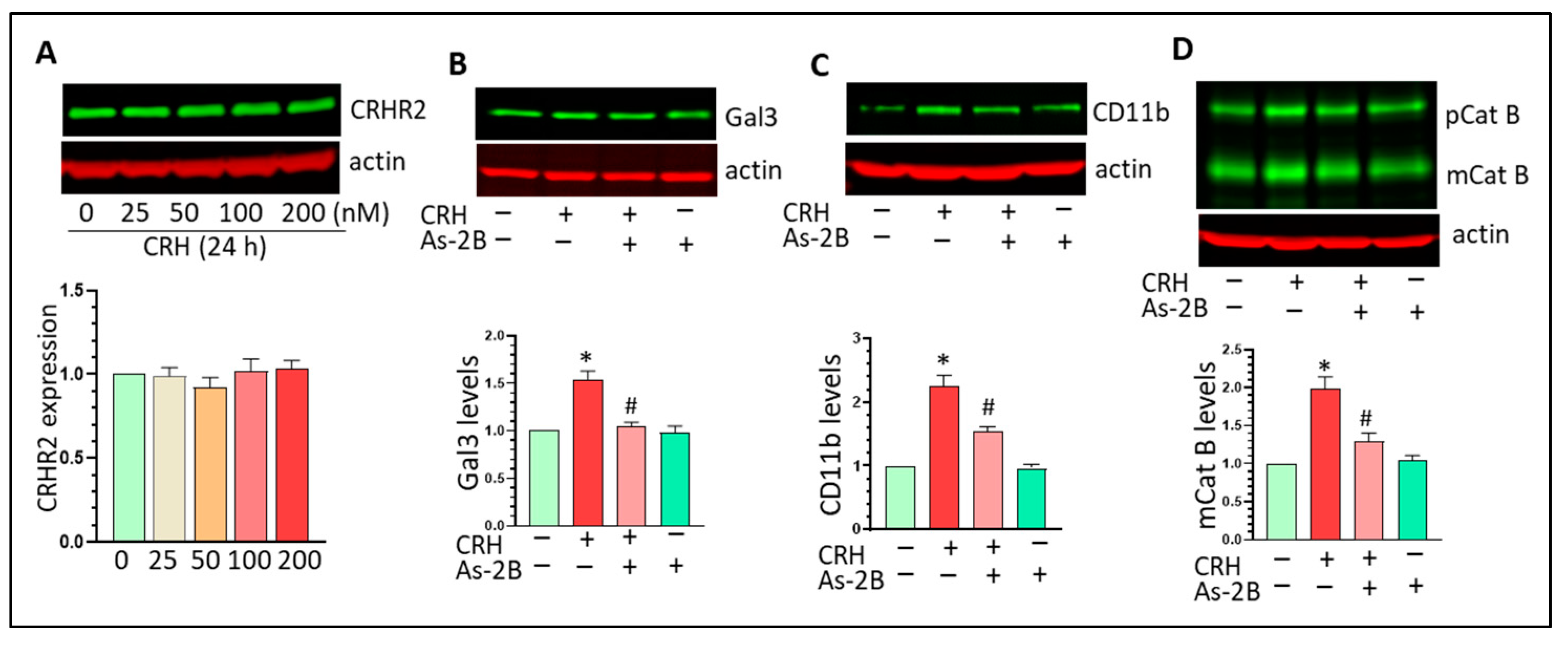
3.5. CRHR2 Blockage Mitigates CRH-Mediated Gal3 Upregulation and Mg Activation In Vitro
3.6. SF Mice Show Increased Gal3 Expression and Autophagy Dysregulation in the Brain HP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buysse, D.J. Sleep health: Can we define it? Does it matter? Sleep 2014, 37, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Gupta, C.C.; Vincent, G.E.; Coates, A.M.; Khalesi, S.; Irwin, C.; Dorrian, J.; Ferguson, S.A. A Time to Rest, a Time to Dine: Sleep, Time-Restricted Eating, and Cardiometabolic Health. Nutrients 2022, 14, 420. [Google Scholar] [CrossRef]
- Grandner, M.A.; Fernandez, F.X. The translational neuroscience of sleep: A contextual framework. Science 2021, 374, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Ohayon, M.M. Epidemiology of insomnia: What we know and what we still need to learn. Sleep Med. Rev. 2002, 6, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Minakawa, E.N. Bidirectional Relationship Between Sleep Disturbances and Parkinson’s Disease. Front. Neurol. 2022, 13, 927994. [Google Scholar] [CrossRef]
- Bubu, O.M.; Andrade, A.G.; Umasabor-Bubu, O.Q.; Hogan, M.M.; Turner, A.D.; de Leon, M.J.; Ogedegbe, G.; Ayappa, I.; Jean-Louis, G.G.; Jackson, M.L.; et al. Obstructive sleep apnea, cognition and Alzheimer’s disease: A systematic review integrating three decades of multidisciplinary research. Sleep Med. Rev. 2020, 50, 101250. [Google Scholar] [CrossRef]
- Sahu, M.; Tripathi, R.; Jha, N.K.; Jha, S.K.; Ambasta, R.K.; Kumar, P. Cross talk mechanism of disturbed sleep patterns in neurological and psychological disorders. Neurosci. Biobehav. Rev. 2022, 140, 104767. [Google Scholar] [CrossRef]
- Mitter, P.; De Crescenzo, F.; Loo Yong Kee, K.; Xia, J.; Roberts, S.; Chi, W.; Kurtulumus, A.; Kyle, S.D.; Geddes, J.R.; Cipriani, A. Sleep deprivation as a treatment for major depressive episodes: A systematic review and meta-analysis. Sleep Med. Rev. 2022, 64, 101647. [Google Scholar] [CrossRef]
- Hatton, C.F.; Duncan, C.J.A. Microglia Are Essential to Protective Antiviral Immunity: Lessons From Mouse Models of Viral Encephalitis. Front. Immunol. 2019, 10, 2656. [Google Scholar] [CrossRef]
- Gipson, C.D.; Rawls, S.; Scofield, M.D.; Siemsen, B.M.; Bondy, E.O.; Maher, E.E. Interactions of neuroimmune signaling and glutamate plasticity in addiction. J. Neuroinflammation 2021, 18, 56. [Google Scholar] [CrossRef]
- Zamanian, M.Y.; Taheri, N.; Opulencia, M.J.C.; Bokov, D.O.; Abdullaev, S.Y.; Gholamrezapour, M.; Heidari, M.; Bazmandegan, G. Neuroprotective and Anti-inflammatory Effects of Pioglitazone on Traumatic Brain Injury. Mediat. Inflamm. 2022, 2022, 9860855. [Google Scholar] [CrossRef] [PubMed]
- Fathi, M.; Vakili, K.; Yaghoobpoor, S.; Qadirifard, M.S.; Kosari, M.; Naghsh, N.; Asgari Taei, A.; Klegeris, A.; Dehghani, M.; Bahrami, A.; et al. Pre-clinical Studies Identifying Molecular Pathways of Neuroinflammation in Parkinson’s Disease: A Systematic Review. Front. Aging Neurosci. 2022, 14, 855776. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N. Microglia in Parkinson’s Disease. J. Park. Dis. 2022, 12, S105–S112. [Google Scholar] [CrossRef]
- Yong, V.W. Microglia in multiple sclerosis: Protectors turn destroyers. Neuron 2022, 110, 3534–3548. [Google Scholar] [CrossRef]
- Althafar, Z.M. Targeting Microglia in Alzheimer’s Disease: From Molecular Mechanisms to Potential Therapeutic Targets for Small Molecules. Molecules 2022, 27, 4124. [Google Scholar] [CrossRef]
- Lewcock, J.W.; Schlepckow, K.; Di Paolo, G.; Tahirovic, S.; Monroe, K.M.; Haass, C. Emerging Microglia Biology Defines Novel Therapeutic Approaches for Alzheimer’s Disease. Neuron 2020, 108, 801–821. [Google Scholar] [CrossRef] [PubMed]
- Ngarka, L.; Siewe Fodjo, J.N.; Aly, E.; Masocha, W.; Njamnshi, A.K. The Interplay Between Neuroinfections, the Immune System and Neurological Disorders: A Focus on Africa. Front. Immunol. 2021, 12, 803475. [Google Scholar] [CrossRef] [PubMed]
- Dauvilliers, Y. Hypocretin/Orexin, Sleep and Alzheimer’s Disease. Front. Neurol. Neurosci. 2021, 45, 139–149. [Google Scholar] [CrossRef]
- Wadhwa, M.; Kumari, P.; Chauhan, G.; Roy, K.; Alam, S.; Kishore, K.; Ray, K.; Panjwani, U. Sleep deprivation induces spatial memory impairment by altered hippocampus neuroinflammatory responses and glial cells activation in rats. J. Neuroimmunol. 2017, 312, 38–48. [Google Scholar] [CrossRef]
- Wadhwa, M.; Prabhakar, A.; Ray, K.; Roy, K.; Kumari, P.; Jha, P.K.; Kishore, K.; Kumar, S.; Panjwani, U. Inhibiting the microglia activation improves the spatial memory and adult neurogenesis in rat hippocampus during 48 h of sleep deprivation. J. Neuroinflammation 2017, 14, 222. [Google Scholar] [CrossRef]
- Hurtado-Alvarado, G.; Becerril-Villanueva, E.; Contis-Montes de Oca, A.; Dominguez-Salazar, E.; Salinas-Jazmin, N.; Perez-Tapia, S.M.; Pavon, L.; Velazquez-Moctezuma, J.; Gomez-Gonzalez, B. The yin/yang of inflammatory status: Blood-brain barrier regulation during sleep. Brain Behav. Immun. 2018, 69, 154–166. [Google Scholar] [CrossRef]
- Kincheski, G.C.; Valentim, I.S.; Clarke, J.R.; Cozachenco, D.; Castelo-Branco, M.T.L.; Ramos-Lobo, A.M.; Rumjanek, V.; Donato, J., Jr.; De Felice, F.G.; Ferreira, S.T. Chronic sleep restriction promotes brain inflammation and synapse loss, and potentiates memory impairment induced by amyloid-beta oligomers in mice. Brain Behav. Immun. 2017, 64, 140–151. [Google Scholar] [CrossRef]
- Manchanda, S.; Singh, H.; Kaur, T.; Kaur, G. Low-grade neuroinflammation due to chronic sleep deprivation results in anxiety and learning and memory impairments. Mol. Cell. Biochem. 2018, 449, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Besedovsky, L.; Schmidt, E.M.; Linz, B.; Diekelmann, S.; Lange, T.; Born, J. Signs of enhanced sleep and sleep-associated memory processing following the anti-inflammatory antibiotic minocycline in men. J. Psychopharmacol. 2017, 31, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, K.H.; Khabour, O.F.; Albawaana, A.S.; Alhashimi, F.H.; Athamneh, R.Y. Tempol prevents chronic sleep-deprivation induced memory impairment. Brain Res. Bull. 2016, 120, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Wan, Y.; Sun, X.; Zhang, X.; Gao, W.; Wu, W. Nicotinic Mitigation of Neuroinflammation and Oxidative Stress After Chronic Sleep Deprivation. Front. Immunol. 2019, 10, 2546. [Google Scholar] [CrossRef]
- Smith, C.; Trageser, K.J.; Wu, H.; Herman, F.J.; Iqbal, U.H.; Sebastian-Valverde, M.; Frolinger, T.; Zeng, E.; Pasinetti, G.M. Anxiolytic effects of NLRP3 inflammasome inhibition in a model of chronic sleep deprivation. Transl. Psychiatry 2021, 11, 52. [Google Scholar] [CrossRef]
- Guyon, A.; Morselli, L.L.; Balbo, M.L.; Tasali, E.; Leproult, R.; L’Hermite-Baleriaux, M.; Van Cauter, E.; Spiegel, K. Effects of Insufficient Sleep on Pituitary-Adrenocortical Response to CRH Stimulation in Healthy Men. Sleep 2017, 40, zsx064. [Google Scholar] [CrossRef]
- Vgontzas, A.N.; Fernandez-Mendoza, J.; Lenker, K.P.; Basta, M.; Bixler, E.O.; Chrousos, G.P. Hypothalamic-pituitary-adrenal (HPA) axis response to exogenous corticotropin-releasing hormone (CRH) is attenuated in men with chronic insomnia. J. Sleep Res. 2022, 31, e13526. [Google Scholar] [CrossRef]
- Makino, S.; Gold, P.W.; Schulkin, J. Corticosterone effects on corticotropin-releasing hormone mRNA in the central nucleus of the amygdala and the parvocellular region of the paraventricular nucleus of the hypothalamus. Brain Res. 1994, 640, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Itoga, C.A.; Chen, Y.; Fateri, C.; Echeverry, P.A.; Lai, J.M.; Delgado, J.; Badhon, S.; Short, A.; Baram, T.Z.; Xu, X. New viral-genetic mapping uncovers an enrichment of corticotropin-releasing hormone-expressing neuronal inputs to the nucleus accumbens from stress-related brain regions. J. Comp. Neurol. 2019, 527, 2474–2487. [Google Scholar] [CrossRef] [PubMed]
- Pomrenze, M.B.; Millan, E.Z.; Hopf, F.W.; Keiflin, R.; Maiya, R.; Blasio, A.; Dadgar, J.; Kharazia, V.; De Guglielmo, G.; Crawford, E.; et al. A Transgenic Rat for Investigating the Anatomy and Function of Corticotrophin Releasing Factor Circuits. Front. Neurosci. 2015, 9, 487. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, H.M.; Schulkin, J.; Berridge, K.C. Activating Corticotropin-Releasing Factor Systems in the Nucleus Accumbens, Amygdala, and Bed Nucleus of Stria Terminalis: Incentive Motivation or Aversive Motivation? Biol. Psychiatry 2021, 89, 1162–1175. [Google Scholar] [CrossRef]
- Piskunov, A.; Stepanichev, M.; Tishkina, A.; Novikova, M.; Levshina, I.; Gulyaeva, N. Chronic combined stress induces selective and long-lasting inflammatory response evoked by changes in corticosterone accumulation and signaling in rat hippocampus. Metab. Brain Dis. 2016, 31, 445–454. [Google Scholar] [CrossRef]
- Gai, Z.; Su, D.; Wang, Y.; Li, W.; Cui, B.; Li, K.; She, X.; Wang, R. Effects of chronic noise on the corticotropin-releasing factor system in the rat hippocampus: Relevance to Alzheimer’s disease-like tau hyperphosphorylation. Environ. Health Prev. Med. 2017, 22, 79. [Google Scholar] [CrossRef]
- Sakamoto, K.; Higo-Yamamoto, S.; Egi, Y.; Miyazaki, K.; Oishi, K. Memory dysfunction and anxiety-like behavior in a mouse model of chronic sleep disorders. Biochem. Biophys. Res. Commun. 2020, 529, 175–179. [Google Scholar] [CrossRef]
- Wang, W.; Ji, P.; Dow, K.E. Corticotropin-releasing hormone induces proliferation and TNF-alpha release in cultured rat microglia via MAP kinase signalling pathways. J. Neurochem. 2003, 84, 189–195. [Google Scholar] [CrossRef]
- Wang, W.; Solc, M.; Ji, P.; Dow, K.E. Corticotropin-releasing hormone potentiates neural injury induced by oxygen-glucose deprivation: A possible involvement of microglia. Neurosci. Lett. 2004, 371, 133–137. [Google Scholar] [CrossRef]
- Hakim, F.; Wang, Y.; Zhang, S.X.; Zheng, J.; Yolcu, E.S.; Carreras, A.; Khalyfa, A.; Shirwan, H.; Almendros, I.; Gozal, D. Fragmented sleep accelerates tumor growth and progression through recruitment of tumor-associated macrophages and TLR4 signaling. Cancer Res. 2014, 74, 1329–1337. [Google Scholar] [CrossRef]
- Nair, D.; Zhang, S.X.; Ramesh, V.; Hakim, F.; Kaushal, N.; Wang, Y.; Gozal, D. Sleep fragmentation induces cognitive deficits via nicotinamide adenine dinucleotide phosphate oxidase-dependent pathways in mouse. Am. J. Respir. Crit. Care Med. 2011, 184, 1305–1312. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, V.; Nair, D.; Zhang, S.X.; Hakim, F.; Kaushal, N.; Kayali, F.; Wang, Y.; Li, R.C.; Carreras, A.; Gozal, D. Disrupted sleep without sleep curtailment induces sleepiness and cognitive dysfunction via the tumor necrosis factor-alpha pathway. J. Neuroinflammation 2012, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Ba, L.; Wang, M.; Deng, S.Y.; Chen, S.M.; Huang, L.F.; Zhang, M.; Wang, W.; Ding, F.F. Chronic sleep fragmentation shares similar pathogenesis with neurodegenerative diseases: Endosome-autophagosome-lysosome pathway dysfunction and microglia-mediated neuroinflammation. CNS Neurosci. Ther. 2020, 26, 215–227. [Google Scholar] [CrossRef]
- Bellesi, M.; de Vivo, L.; Chini, M.; Gilli, F.; Tononi, G.; Cirelli, C. Sleep Loss Promotes Astrocytic Phagocytosis and Microglial Activation in Mouse Cerebral Cortex. J. Neurosci. 2017, 37, 5263–5273. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, J.H.; Cho, Y.E.; Baek, M.C.; Jung, J.Y.; Lee, M.G.; Jang, I.S.; Lee, H.W.; Suk, K. Chronic sleep deprivation-induced proteome changes in astrocytes of the rat hypothalamus. J. Proteome Res. 2014, 13, 4047–4061. [Google Scholar] [CrossRef] [PubMed]
- Borgonetti, V.; Governa, P.; Biagi, M.; Dalia, P.; Corsi, L. Rhodiola rosea L. modulates inflammatory processes in a CRH-activated BV2 cell model. Phytomedicine 2020, 68, 153143. [Google Scholar] [CrossRef]
- Garcia-Revilla, J.; Boza-Serrano, A.; Espinosa-Oliva, A.M.; Soto, M.S.; Deierborg, T.; Ruiz, R.; de Pablos, R.M.; Burguillos, M.A.; Venero, J.L. Galectin-3, a rising star in modulating microglia activation under conditions of neurodegeneration. Cell Death Dis. 2022, 13, 628. [Google Scholar] [CrossRef]
- Puigdellivol, M.; Allendorf, D.H.; Brown, G.C. Sialylation and Galectin-3 in Microglia-Mediated Neuroinflammation and Neurodegeneration. Front. Cell. Neurosci. 2020, 14, 162. [Google Scholar] [CrossRef]
- Aits, S.; Kricker, J.; Liu, B.; Ellegaard, A.M.; Hamalisto, S.; Tvingsholm, S.; Corcelle-Termeau, E.; Hogh, S.; Farkas, T.; Holm Jonassen, A.; et al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy 2015, 11, 1408–1424. [Google Scholar] [CrossRef]
- Cursano, S.; Battaglia, C.R.; Urrutia-Ruiz, C.; Grabrucker, S.; Schon, M.; Bockmann, J.; Braumuller, S.; Radermacher, P.; Roselli, F.; Huber-Lang, M.; et al. A CRHR1 antagonist prevents synaptic loss and memory deficits in a trauma-induced delirium-like syndrome. Mol. Psychiatry 2021, 26, 3778–3794. [Google Scholar] [CrossRef]
- Hori, H.; Teraishi, T.; Sasayama, D.; Ozeki, Y.; Matsuo, J.; Kawamoto, Y.; Kinoshita, Y.; Hattori, K.; Higuchi, T.; Kunugi, H. Poor sleep is associated with exaggerated cortisol response to the combined dexamethasone/CRH test in a non-clinical population. J. Psychiatr. Res. 2011, 45, 1257–1263. [Google Scholar] [CrossRef] [PubMed]
- Galvao Mde, O.; Sinigaglia-Coimbra, R.; Kawakami, S.E.; Tufik, S.; Suchecki, D. Paradoxical sleep deprivation activates hypothalamic nuclei that regulate food intake and stress response. Psychoneuroendocrinology 2009, 34, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Chrousos, G.; Vgontzas, A.N.; Kritikou, I. HPA Axis and Sleep. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Han, K.S.; Kim, L.; Shim, I. Stress and sleep disorder. Exp. Neurobiol. 2012, 21, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Tang, X.; Wellman, L.L.; Liu, X.; Sanford, L.D. Corticotropin releasing factor (CRF) modulates fear-induced alterations in sleep in mice. Brain Res. 2009, 1276, 112–122. [Google Scholar] [CrossRef][Green Version]
- Liu, X.; Wellman, L.L.; Yang, L.; Ambrozewicz, M.A.; Tang, X.; Sanford, L.D. Antagonizing corticotropin-releasing factor in the central nucleus of the amygdala attenuates fear-induced reductions in sleep but not freezing. Sleep 2011, 34, 1539–1549. [Google Scholar] [CrossRef] [PubMed]
- Wellman, L.L.; Yang, L.; Ambrozewicz, M.A.; Machida, M.; Sanford, L.D. Basolateral amygdala and the regulation of fear-conditioned changes in sleep: Role of corticotropin-releasing factor. Sleep 2013, 36, 471–480. [Google Scholar] [CrossRef]
- Wang, M.J.; Lin, S.Z.; Kuo, J.S.; Huang, H.Y.; Tzeng, S.F.; Liao, C.H.; Chen, D.C.; Chen, W.F. Urocortin modulates inflammatory response and neurotoxicity induced by microglial activation. J. Immunol. 2007, 179, 6204–6214. [Google Scholar] [CrossRef]
- Siew, J.J.; Chern, Y. Microglial Lectins in Health and Neurological Diseases. Front. Mol. Neurosci. 2018, 11, 158. [Google Scholar] [CrossRef]
- Burguillos, M.A.; Svensson, M.; Schulte, T.; Boza-Serrano, A.; Garcia-Quintanilla, A.; Kavanagh, E.; Santiago, M.; Viceconte, N.; Oliva-Martin, M.J.; Osman, A.M.; et al. Microglia-Secreted Galectin-3 Acts as a Toll-like Receptor 4 Ligand and Contributes to Microglial Activation. Cell Rep. 2015, 10, 1626–1638. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, C.; Meng, J.; Li, N.; Xu, Z.; Liu, X.; Hou, S. Galectin-3 regulates microglial activation and promotes inflammation through TLR4/MyD88/NF-kB in experimental autoimmune uveitis. Clin. Immunol. 2022, 236, 108939. [Google Scholar] [CrossRef]
- Boza-Serrano, A.; Ruiz, R.; Sanchez-Varo, R.; Garcia-Revilla, J.; Yang, Y.; Jimenez-Ferrer, I.; Paulus, A.; Wennstrom, M.; Vilalta, A.; Allendorf, D.; et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. 2019, 138, 251–273. [Google Scholar] [CrossRef] [PubMed]
- Boza-Serrano, A.; Vrillon, A.; Minta, K.; Paulus, A.; Camprubi-Ferrer, L.; Garcia, M.; Andreasson, U.; Antonell, A.; Wennstrom, M.; Gouras, G.; et al. Galectin-3 is elevated in CSF and is associated with Abeta deposits and tau aggregates in brain tissue in Alzheimer’s disease. Acta Neuropathol. 2022, 144, 843–859. [Google Scholar] [CrossRef]
- Fermino, M.L.; Polli, C.D.; Toledo, K.A.; Liu, F.T.; Hsu, D.K.; Roque-Barreira, M.C.; Pereira-da-Silva, G.; Bernardes, E.S.; Halbwachs-Mecarelli, L. LPS-induced galectin-3 oligomerization results in enhancement of neutrophil activation. PLoS ONE 2011, 6, e26004. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, E.; Sanchez-Maldonado, C.; Mayoral, M.A.; Mendieta, L.; Alatriste, V.; Patricio-Martinez, A.; Limon, I.D. Neuroinflammation induced by the peptide amyloid-beta (25–35) increase the presence of galectin-3 in astrocytes and microglia and impairs spatial memory. Neuropeptides 2019, 74, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Guo, Y.; Chen, Y.Y.; Liu, Q.; Chen, Y.; Tan, L.; Zhang, S.H.; Gao, Z.R.; Zhou, Y.H.; Zhang, G.Y.; et al. miR-124-3p increases in high glucose induced osteocyte-derived exosomes and regulates galectin-3 expression: A possible mechanism in bone remodeling alteration in diabetic periodontitis. FASEB J. 2020, 34, 14234–14249. [Google Scholar] [CrossRef]
- Zhao, J.; He, Z.; Wang, J. MicroRNA-124: A Key Player in Microglia-Mediated Inflammation in Neurological Diseases. Front. Cell. Neurosci. 2021, 15, 771898. [Google Scholar] [CrossRef]
- Periyasamy, P.; Liao, K.; Kook, Y.H.; Niu, F.; Callen, S.E.; Guo, M.L.; Buch, S. Cocaine-Mediated Downregulation of miR-124 Activates Microglia by Targeting KLF4 and TLR4 Signaling. Mol. Neurobiol. 2018, 55, 3196–3210. [Google Scholar] [CrossRef]
- Giannogonas, P.; Apostolou, A.; Manousopoulou, A.; Theocharis, S.; Macari, S.A.; Psarras, S.; Garbis, S.D.; Pothoulakis, C.; Karalis, K.P. Identification of a novel interaction between corticotropin releasing hormone (Crh) and macroautophagy. Sci. Rep. 2016, 6, 23342. [Google Scholar] [CrossRef][Green Version]
- Jin, L.; Qian, Y.; Zhou, J.; Dai, L.; Cao, C.; Zhu, C.; Li, S. Activated CRH receptors inhibit autophagy by repressing conversion of LC3BI to LC3BII. Cell. Signal. 2019, 58, 119–130. [Google Scholar] [CrossRef]
- Zhao, S.B.; Wu, J.Y.; He, Z.X.; Song, Y.H.; Chang, X.; Xia, T.; Fang, X.; Li, Z.S.; Xu, C.; Wang, S.L.; et al. Corticotropin releasing hormone promotes inflammatory bowel disease via inducing intestinal macrophage autophagy. Cell Death Discov. 2021, 7, 377. [Google Scholar] [CrossRef]
- Zubova, S.G.; Suvorova, I.I.; Karpenko, M.N. Macrophage and microglia polarization: Focus on autophagy-dependent reprogramming. Front. Biosci. Sch. 2022, 14, 3. [Google Scholar] [CrossRef]
- Mo, Y.; Sun, Y.Y.; Liu, K.Y. Autophagy and inflammation in ischemic stroke. Neural Regen. Res. 2020, 15, 1388–1396. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, L.; Reed, K.M.; Carter, A.; Cheng, Y.; Roodsari, S.K.; Martinez Pineda, D.; Wellman, L.L.; Sanford, L.D.; Guo, M.-L. Sleep-Disturbance-Induced Microglial Activation Involves CRH-Mediated Galectin 3 and Autophagy Dysregulation. Cells 2023, 12, 160. https://doi.org/10.3390/cells12010160
Guo L, Reed KM, Carter A, Cheng Y, Roodsari SK, Martinez Pineda D, Wellman LL, Sanford LD, Guo M-L. Sleep-Disturbance-Induced Microglial Activation Involves CRH-Mediated Galectin 3 and Autophagy Dysregulation. Cells. 2023; 12(1):160. https://doi.org/10.3390/cells12010160
Chicago/Turabian StyleGuo, Liyang, Kirstin M. Reed, Ashley Carter, Yan Cheng, Soheil Kazemi Roodsari, Damian Martinez Pineda, Laurie L. Wellman, Larry D. Sanford, and Ming-Lei Guo. 2023. "Sleep-Disturbance-Induced Microglial Activation Involves CRH-Mediated Galectin 3 and Autophagy Dysregulation" Cells 12, no. 1: 160. https://doi.org/10.3390/cells12010160
APA StyleGuo, L., Reed, K. M., Carter, A., Cheng, Y., Roodsari, S. K., Martinez Pineda, D., Wellman, L. L., Sanford, L. D., & Guo, M.-L. (2023). Sleep-Disturbance-Induced Microglial Activation Involves CRH-Mediated Galectin 3 and Autophagy Dysregulation. Cells, 12(1), 160. https://doi.org/10.3390/cells12010160