

The Cytokine Network in Colorectal Cancer: Implications for New Treatment Strategies
 ,
,
Abstract
1. Introduction
1.1. Classical Classification of CRC
- (I)
- The conventional pathway that harbors CRCs that are CIMP-, CIN+++ and MSS/MSIlow.
- (II)
- The serrated pathway that contains two groups.
- Cancer cells that are MSS with a mutated KRAS and a low CIMP phenotype.
- MSS cancers with BRAF mutations and a high CIMP phenotype.
- (III)
- The MSI pathway, consisting of two groups.
- MSI, CIMP-high tumors with frequent BRAF mutations and MLH1 methylation.
- MSI, CIMP-negative tumors with KRAS mutations and no BRAF mutations.
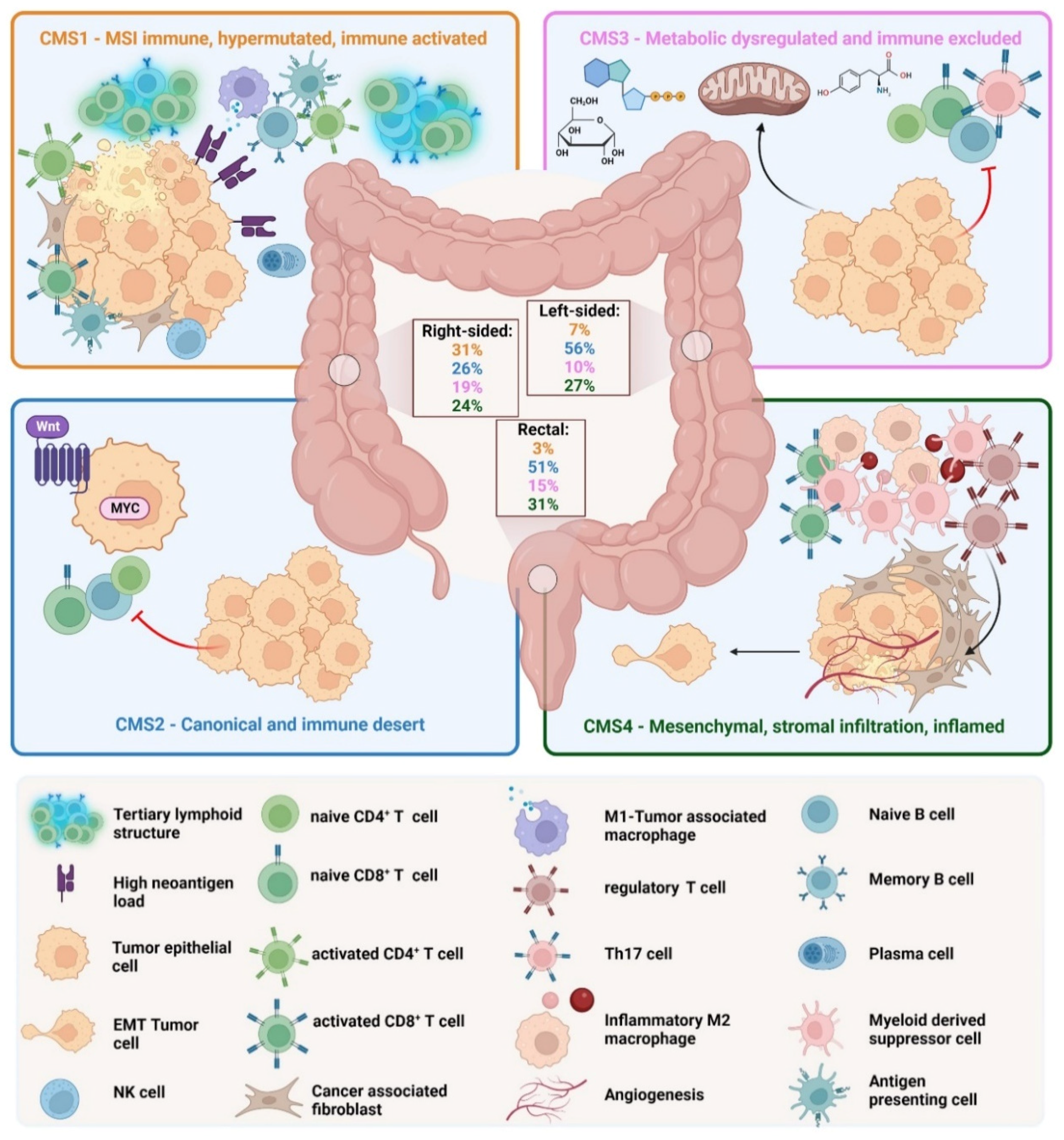
1.2. The Consensus Molecular Subtype Classification
- (I)
- CMS1 is the so-called MSI-like subtype, characterized by MSI and a hypermutated profile with mutations in the MLH1 and BRAF genes. CMS1 patients have a diffuse immune infiltrate of T helper 1 (TH1) cells, natural killer (NK) cells and cytotoxic T lymphocytes (CTL). This subtype shows also a CIMP phenotype. Approximately 14% of CRCs belong to the CMS1 subtype.
- (II)
- CMS2 is the so-called canonical subtype and includes CRCs with higher CIN and a high level of somatic copy number alterations. CMS2 CRCs show an upregulated WNT and MYC pathway and no dendritic cell (DC) recruitment. The CMS2 subtype is only poorly immunogenic with no immune infiltrate and no immune regulatory cytokines. Approximately 37% of CRCs belong to CMS2.
- (III)
- The CMS3 subtype is the metabolic subtype, characterized by the dysregulation of the glucose pentose, nitrogen, fatty acid and several other metabolic pathways. CMS3 shows CIN and CIMPlow status. The level of KRAS mutations is higher compared with other CMS phenotypes. CMS3 is, similar to CMS2, only poorly immunogenic with no immune infiltrate and no immune regulatory cytokines. Approximately 13% belong to the CMS3 subtype.
- (IV)
- CMS4 is the mesenchymal phenotype with high CIN and a strong expression of the epithelial–mesenchymal transition (EMT). CMS4 tumors show TGF-β signaling and a high C-X-C Chemokine Ligand 12 (CXCL-12) expression. The CMS4 phenotype shows high levels of infiltrating CTLs, macrophages and stromal cells [24]. Approximately 23% of CRC tumors fall into CMS4. Although CMS4 patients show high levels of leukocyte infiltration, patients with CMS4 tumors have the worst prognosis of the four subtypes [23].
1.3. Interleukins, Interferons and other Cytokines in CRC
1.4. Chemokines in CRC
2. Cytokine-Induced Inflammation
2.1. Cytokines in Gut Homeostasis and in Chronic Inflammation
2.2. Cytokines in Tumor-Elicited Inflammation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokines that Promote CRC | ||||
| Cytokine Family | Cytokine | Receptor | Functional effect in CRC | Reference |
| IL-1 Family | IL-1α | Interleukin 1 receptor type 1 (IL-1R1)-IL-1R3 | Promotes inflammatory carcinogenesis | [86] |
| IL-1β | IL-1R1-IL-1R3IL-1R2-IL-1R3 | Promotes the proliferation of colon cancer cells, promotes inflammation-induced carcinogenesis | [86,87] | |
| IL-2 (common γ-chain) Family | IL-4 | IL-4R | Promotes Th2-type inflammation and Th9 polarization | [88,89] |
| IL-6 Family | IL-6 | IL-6Rα-gp130 | Promotes carcinogenesis via upregulation of proliferation, mitosis, migration and angiogenesis | [90,91,92] |
| IL-11 | IL-11Rα-gp130 | Promotes inflammation-induced carcinogenesis, facilitates the proliferation of colon cancer cells | [93,94,95] | |
| IL-31 | IL-31Rα-OSMRb | Promotes Th2 cell polarization, evidently tumorigenic | [96] | |
| IL-8 Family | IL-8 | CXCR1, CXCR2 | Promotes colon cancer cell proliferation, attracts neutrophils, mediates a suppressive environment and chemoresistance | [97,98,99,100] |
| IL-10 Family | IL-19 | IL20Rα-IL20Rβ | Evidently tumorigenic | [101] |
| IL-20 | IL-20Rα-IL-20Rβ | Promotes carcinoma outgrowth, induces PD-1 | [102] | |
| IL-22 | IL-22Rα-IL-10Rβ | Promotes progression of carcinomas, stemness and proliferation | [103,104,105] | |
| IL-26 | IL-20Rα-IL-10Rβ | Promotes TH17 polarization, only expressed in humans, not in mice | [102] | |
| IL-12 Family | IL-23 | IL-23R-IL-12Rβ | Promotes pro-inflammatory cytokine secretion | [106,107] |
| IL-35 | IL-12Rβ-gp130 IL-12Rβ-IL-12Rβ | Promotes Treg-mediated suppression of T cell responses and exhaustion of T cells | [108] | |
| IL-17 Family | IL-17A | IL-17RA-IL-17RC | Promotes cell cycle progression of colon cancer cells and immunosuppression | [109,110,111] |
| Other cytokines | ||||
| IL-13 | IL-13Rα-IL-4Rα | Promotes Th2 cell polarization | [112] | |
| IL-16 | CD4 | Evidently pro-tumoral | [113] | |
| IL-34 | CSF1R | Promotes cancer progression and immune suppression and therapeutic resistance | [114,115,116] | |
| TNF | TNFR1, TNFR2 | promotes inflammation | [117,118,119] | |
| Chemokines | CXCL-1 | CXCR1, CXCR2 | Recruitment of tumor-associated macrophages (TAM) and TANs | [120,121] |
| CXC chemokines | CXCL-2 | CXCR2 | Recruitment of TAMs and TANs | [122] |
| CXCL-8 | CXCR1, CXCR2 | Recruitment of TANs, TAMs and cancer cells to the tumor site; promotion of angiogenesis and tumor stemness | [123,124,125] | |
| CXCL-12 | CXCR4, CXCR7 | Recruitment of TAMs at the invasive front, upregulation of IL-10 | [126,127] | |
| CXCL-16 | CXCR6 | Induces the polarization of macrophages toward a pro-tumoral M2 phenotype | [128] | |
| CC chemokines | CCL-2 | CCR2 | Recruitment of TAMs at the invasive front, recruitment of myeloid-derived suppressor cells (MDSC) into the tumor | [129,130] |
| CCL-3 | CCR1, CCR4 | Recruitment of MDSC into the tumor, promotes proliferation of colon cancer cells | [131] | |
| CCL-4 | CCR1, CCR3 | Recruitment of MDSC and TAMs into the tumor | [132] | |
| CCL-11 | CCR8 | Promotes tumor angiogenesis, inhibits apoptosis of endothelial cells | [133] | |
| CCL-16 | CCR1, CCR2, CCR3, CCR5, CCR8 | Promotes tumor angiogenesis, inhibits apoptosis of endothelial cells | [134] | |
| CCL-17 | CCR4 | Recruitment of Tregs and Th2 lymphocytes | [135] | |
| CCL-20 | CCR6 | Recruitment of Tregs and Th2 lymphocytes | [136,137] | |
| Cytokines with a more CRC-suppressive phenotype | ||||
| Cytokine Family | Cytokine | Receptor | Functional effect in CRC | Reference |
| IL-1 Family | IL-18 | IL-5R5-IL-1R7 | Activates lymphocytes to produce IFN-g, restricts ThH17 differentiation | [138,139] |
| IL-36 | IL-1R6-IL1R3 | Conservation of tertiary lymphoid structures | [34,140,141] | |
| IL-37 | IL-1R8-IL-1R5 | Inhibits β-catenin | [141,142] | |
| IL-2 (common γ-chain) Family | IL-2 | IL-2R | T and NK cell activation factor | [34,143] |
| IL-7 | IL-7Rα | Promotes the proliferation of T cells and NK cells | [144,145] | |
| IL-9 | IL-9Ra | Promotes the proliferation of CD8+ T cells | [146,147] | |
| IL-15 | IL-15-IL-15Rα | Promotes the proliferation and activation of NK cells and CD8+ T cells | [148,149,150] | |
| IL-21 | IL-21R | Promotes the proliferation and activation of NK cells and CD8+ T cells | [151,152,153] | |
| IL-10 Family | IL-24 | IL-20Rα-IL-20Rβ, IL-22Rα-IL-20Rβ | Induces apoptosis and autophagy of cancer cells | [154] |
| IL-12 Family | IL-12 | IL-12Rβ | Promotes T cell survival and proliferation and the proliferation of NK cells, enhances cytotoxic function | [155,156] |
| IL-17 Family | IL-17F | IL-17RA-IL-17RC | Inhibition of tumor angiogenesis, enhancing immune cell recruitment | [34,157] |
| Other cytokines | ||||
| Interferon Family | IFN-γ | IFNGR | Mediates anti-proliferative, anti-angiogenic and pro-apoptotic effects | [44,158,159] |
| Type I interferons | Interferon alpha and beta receptor subunit 1 (IFNAR1)-IFNRA2 | Recruitment of CD8+ lymphocytes | [160] | |
| TGF-β | TGF-β | TGFBR1-TGFBR2 | Inhibits cancer cell proliferation, regulates immune cell differentiation | [161,162] |
| Chemokines | ||||
| CXC chemokines | CXCL-9 | CXCR3 | Promotes the infiltration of NK cells, CD8+ and CD4+ T cells into the tumor, suppression of angiogenesis | [163,164] |
| CXCL-10 | CXCR3 | Promotes the infiltration of NK cells, CD8+ and CD4+ T cells into the tumor, suppression of angiogenesis | [160,163,165] | |
| CXCL-11 | CXCR3 | Promotes the infiltration of NK cells, CD8+ and CD4+ T cells into the tumor, suppression of angiogenesis | [166] | |
| Cytokines with a mixed phenotype | ||||
| Cytokine Family | Cytokine | Receptor | Functional effect in CRC | Reference |
| IL-1 Family | IL-33 | IL-1R4 | Alters TME, promotes angiogenesis, enhances colon cancer stemness but induces anti-tumoral IFN-γ responses | [167,168,169,170] |
| IL-2 (common γ-chain) Family | IL-9 | IL-9R | Inhibits tumor growth by enhancing immune responses and promotes tumor growth by suppressing immune responses | [112,171] |
| IL-10 Family | IL-10 | IL-10Rα-IL-10Rβ | Promotes cytotoxicity but inhibits anti-tumor responses | [172] |
| Chemokines | ||||
| CXC chemokines | CXCL-4 | Suppresses the activity of CD8+ T cells and inhibits tumor angiogenesis and endothelial cell proliferation | [173] | |
| CCL-5 | CCR1, CCR3, CCR4, CCR5 | Recruitment of MDSC and fibroblasts into the tumor but also CD8+ T cells | [160,174,175] | |
2.2.1. Cytokines in the Early Stages of CRC
2.2.2. Cytokines in the Late Stages of CRC
2.3. Cytokines in Therapy-Induced Inflammation
- (I)
- alkylating agents that elicit DNA crosslinks and destabilize DNA during replication, such as cyclophosphamide and oxaliplatin.
- (II)
- antimetabolites that disrupt DNA and RNA synthesis, such as 5-fluorouracil and gemcitabine
- (III)
- topoisomerase inhibitors that interfere with DNA unwinding processes, such as irinotecan
- (IV)
- microtubule poisons that inhibit tubulin polymerization and depolimerization such as paclitaxel
- (V)
- antibiotics that kill via excessive ROS production and DNA intercalation, such as anthracyclines and bleomycin
3. Cytokine-Mediated Therapeutic Options and New Treatment Strategies
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Adenomatous Polyposis Coli |
| ATP | Adenosine Triphosphate |
| BRAF | B-Raf and v-Raf Murine Sarcoma Viral Oncogene Homolog B |
| Breg | Regulatory B Cell |
| BSA | Bovine Serum Albumin |
| CAC | Colitis-Associated Cancer |
| CAF | Cancer-Associated Fibroblast |
| CCL2 | CC-Chemokine Ligand 2 |
| CCR6 | CC Chemokine Receptor 6 |
| CD4 | Cluster of Differentiation Antigen 4 |
| CEA | Carcinoembryonic Antigen |
| CIMP | CpG Island Methylator Phenotype |
| CIN | Chromosomal Instability |
| CMS | Consensus Molecular Subtype |
| CRC | Colorectal Cancer |
| CTL | Cytotoxic T Lymphocyte |
| CTLA-4 | Cytotoxic T-Lymphocyte-Associated Protein 4 |
| CTNNB1 | Catenin Beta 1 |
| CXCL-12 | C-X-C Chemokine Ligand 12 |
| CXCR4 | C-X-C Chemokine Receptor Type 4 |
| DAMP | Damage-Associated Molecular Pattern |
| DC | Dendritic Cell |
| DCC | Deleted in Colorectal Carcinoma |
| DNA | Deoxyribonucleic Acid |
| EMT | Epithelial–Mesenchymal Transition |
| FOXP3 | Forkhead Box P3 |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| gp130 | Glycoprotein 130 |
| HRAS | Harvey Rat Sarcoma Virus |
| IBD | Inflammatory Bowel Disease |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| IEC | Intestinal Epithelial Cell |
| IEL | Intraepithelial Lymphocyte |
| IFN-γ | Interferon-Gamma |
| IFNAR1 | Interferon Alpha and Beta Receptor Subunit 1 |
| IGF1R | Insulin-Like Growth Factor-1 Receptor |
| IL-17 | Interleukin-17 |
| IL-1R1 | Interleukin 1 Receptor Type 1 |
| KRAS | Kirsten Rat Sarcoma Virus Oncogene |
| LAK | Lymphokine-Activated Killer Cell |
| LFA-3 | Lymphocyte Function-Associated Antigen 3 |
| LOH | Loss Of Heterozygosity |
| LPS | Lipopolysaccharide |
| M2 | Macrophage Type 2 |
| MAP | Mitogen-Activated Protein |
| MCC | Mutated in Colorectal Cancer |
| MDSC | Myeloid-Derived Suppressor Cell |
| MLH1 | MutL Homolog 1 |
| MMR | Mismatch Repair |
| MSH6 | MutS Homolog 6 |
| MSI | Microsatellite Instability |
| N1 | Neutrophil Type 1 |
| NFkB | Nuclear Factor Kappa-B |
| NK | Natural Killer Cell |
| NRAS | Neuroblastoma Rat Sarcoma Virus |
| OSMRb | Oncostatin-M-Specific Receptor b |
| PAMPs | Pathogen-Associated Molecular Patterns |
| PI3K | Phosphatidylinositol 3-Kinase |
| PIK3CA | Phosphatidylinositol-4,5-Bisphosphate 3-Kinase, Catalytic Subunit Alpha |
| PMS1 | Protein Homolog 1 |
| Poly(A).Poly(U) | Polyadenylic-Polyuridylic Acid |
| RCT | Randomized Controled Trial |
| RIP-Tag2 | Rat Insuline Promotor T Antigen 2 |
| ROS | Reactive Oxygen Species |
| SCFAs | Short-Chain Fatty Acids |
| SDF-1 | Stromal Cell-Derived Factor 1 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| TAM | Tumor-Associated Macrophage |
| TAN | Tumor-Associated Neutrophil |
| TGF-β | Transforming Growth Factor Beta |
| TGFBR1 | Transforming Growth Factor-Beta Receptor 1 |
| TH1 | T Helper 1 Cell |
| TIL | Tumor-Infiltrating Lymphocyte |
| TIM | Tumor-Infiltrating Myeloid Cell |
| TLR | Toll-Like Receptor |
| TME | Tumor Microenvironment |
| TNF | Tumor Necrosis Factor |
| TNFR1 | Tumor Necrosis Factor Receptor 1 |
| Treg | T Regulatory Cell |
| WHO | World Health Organization |
| WNT | Wingless Pathway |
References
- Xi, Y.; Xu, P. Global colorectal cancer burden in 2020 and projections to 2040. Transl. Oncol. 2021, 14, 101174. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Campos, F.; Figueiredo, M.N.; Monteiro, M.; Nahas, S.C.; Cecconello, I. Incidence of colorectal cancer in young patients. Rev. Col. Bras. Cir. 2017, 44, 208–215. [Google Scholar] [CrossRef]
- El Bali, M.; Bakkach, J.; Bennani Mechita, M. Colorectal Cancer: From Genetic Landscape to Targeted Therapy. J. Oncol. 2021, 2021, 9918116. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef]
- Power, D.G.; Gloglowski, E.; Lipkin, S.M. Clinical genetics of hereditary colorectal cancer. Hematol. Oncol. Clin. N. Am. 2010, 24, 837–859. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, E.M.; Kastrinos, F. Familial colorectal cancer, beyond Lynch syndrome. Clin. Gastroenterol. Hepatol. 2014, 12, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Powell, S.M.; Zilz, N.; Beazer-Barclay, Y.; Bryan, T.M.; Hamilton, S.R.; Thibodeau, S.N.; Vogelstein, B.; Kinzler, K.W. APC mutations occur early during colorectal tumorigenesis. Nature 1992, 359, 235–237. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef]
- Herr, R.; Kohler, M.; Andrlova, H.; Weinberg, F.; Moller, Y.; Halbach, S.; Lutz, L.; Mastroianni, J.; Klose, M.; Bittermann, N.; et al. B-Raf inhibitors induce epithelial differentiation in BRAF-mutant colorectal cancer cells. Cancer Res. 2015, 75, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.C.N.; Benndorf, R.A. Aspirin sensitivity of PIK3CA-mutated Colorectal Cancer: Potential mechanisms revisited. Cell Mol. Life Sci. 2022, 79, 393. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.Y.; Richardson, B.C. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005, 6, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Huang, W.; Liu, X.; Liu, X.; Chen, N.; Xu, Q.; Hu, Y.; Song, W.; Zhou, J. IMPDH2 promotes colorectal cancer progression through activation of the PI3K/AKT/mTOR and PI3K/AKT/FOXO1 signaling pathways. J. Exp. Clin. Cancer Res. 2018, 37, 304. [Google Scholar] [CrossRef]
- Woodford-Richens, K.L.; Rowan, A.J.; Gorman, P.; Halford, S.; Bicknell, D.C.; Wasan, H.S.; Roylance, R.R.; Bodmer, W.F.; Tomlinson, I.P. SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 9719–9723. [Google Scholar] [CrossRef]
- Tarafa, G.; Villanueva, A.; Farre, L.; Rodriguez, J.; Musulen, E.; Reyes, G.; Seminago, R.; Olmedo, E.; Paules, A.B.; Peinado, M.A.; et al. DCC and SMAD4 alterations in human colorectal and pancreatic tumor dissemination. Oncogene 2000, 19, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Wang, D.; Kok, S.Y.; Oshima, H.; Oshima, M. Genetic Alterations and Microenvironment that Drive Malignant Progression of Colorectal Cancer: Lessons from Mouse and Organoid Models. J. Cancer Prev. 2022, 27, 1–6. [Google Scholar] [CrossRef]
- Li, H.; Zhang, J.; Tong, J.H.M.; Chan, A.W.H.; Yu, J.; Kang, W.; To, K.F. Targeting the Oncogenic p53 Mutants in Colorectal Cancer and Other Solid Tumors. Int. J. Mol. Sci. 2019, 20, 5999. [Google Scholar] [CrossRef]
- Kim, H.; Jen, J.; Vogelstein, B.; Hamilton, S.R. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am. J. Pathol. 1994, 145, 148–156. [Google Scholar]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Wong, J.J.; Hawkins, N.J.; Ward, R.L. Colorectal cancer: A model for epigenetic tumorigenesis. Gut 2007, 56, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, N.; Tierling, S.; Es, H.A.; Varkiani, M.; Mojarad, E.N.; Aghdaei, H.A.; Walter, J.; Totonchi, M. DNA methylation biomarkers in colorectal cancer: Clinical applications for precision medicine. Int. J. Cancer 2022, 151, 2068–2081. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; de Reynies, A.; Giraldo, N.A.; Pilati, C.; Buttard, B.; Lacroix, L.; Selves, J.; Sautes-Fridman, C.; Laurent-Puig, P.; Fridman, W.H. Immune and Stromal Classification of Colorectal Cancer Is Associated with Molecular Subtypes and Relevant for Precision Immunotherapy. Clin. Cancer Res. 2016, 22, 4057–4066. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautes-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Lan, T.; Chen, L.; Wei, X. Inflammatory Cytokines in Cancer: Comprehensive Understanding and Clinical Progress in Gene Therapy. Cells 2021, 10, 100. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Mantovani, A. Cancer-related inflammation: Common themes and therapeutic opportunities. Semin. Cancer Biol. 2012, 22, 33–40. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Cominelli, F. Colitis-Associated and Sporadic Colon Cancers: Different Diseases, Different Mutations? Gastroenterology 2016, 150, 808–810. [Google Scholar] [CrossRef]
- Robles, A.I.; Traverso, G.; Zhang, M.; Roberts, N.J.; Khan, M.A.; Joseph, C.; Lauwers, G.Y.; Selaru, F.M.; Popoli, M.; Pittman, M.E.; et al. Whole-Exome Sequencing Analyses of Inflammatory Bowel Disease-Associated Colorectal Cancers. Gastroenterology 2016, 150, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Joanito, I.; Wirapati, P.; Zhao, N.; Nawaz, Z.; Yeo, G.; Lee, F.; Eng, C.L.P.; Macalinao, D.C.; Kahraman, M.; Srinivasan, H.; et al. Single-cell and bulk transcriptome sequencing identifies two epithelial tumor cell states and refines the consensus molecular classification of colorectal cancer. Nat. Genet. 2022, 54, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Briukhovetska, D.; Dorr, J.; Endres, S.; Libby, P.; Dinarello, C.A.; Kobold, S. Interleukins in cancer: From biology to therapy. Nat. Rev. Cancer 2021, 21, 481–499. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, L.; Zhao, H.; Yan, Y.; Lu, J. The Role of Interleukins in Colorectal Cancer. Int. J. Biol. Sci. 2020, 16, 2323–2339. [Google Scholar] [CrossRef]
- Leonard, W.J.; Lin, J.X.; O’Shea, J.J. The gamma(c) Family of Cytokines: Basic Biology to Therapeutic Ramifications. Immunity 2019, 50, 832–850. [Google Scholar] [CrossRef]
- Bhat, A.A.; Nisar, S.; Singh, M.; Ashraf, B.; Masoodi, T.; Prasad, C.P.; Sharma, A.; Maacha, S.; Karedath, T.; Hashem, S.; et al. Cytokine- and chemokine-induced inflammatory colorectal tumor microenvironment: Emerging avenue for targeted therapy. Cancer Commun. 2022, 42, 689–715. [Google Scholar] [CrossRef]
- Rebe, C.; Ghiringhelli, F. Interleukin-1beta and Cancer. Cancers 2020, 12, 1791. [Google Scholar] [CrossRef]
- Fridman, W.H.; Miller, I.; Sautes-Fridman, C.; Byrne, A.T. Therapeutic Targeting of the Colorectal Tumor Stroma. Gastroenterology 2020, 158, 303–321. [Google Scholar] [CrossRef]
- Boukhaled, G.M.; Harding, S.; Brooks, D.G. Opposing Roles of Type I Interferons in Cancer Immunity. Annu. Rev. Pathol. 2021, 16, 167–198. [Google Scholar] [CrossRef]
- Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Interferon-gamma: Teammate or opponent in the tumour microenvironment? Nat. Rev. Immunol. 2022, 22, 158–172. [Google Scholar] [CrossRef]
- Galon, J.; Lanzi, A. Immunoscore and its introduction in clinical practice. Q. J. Nucl. Med. Mol. Imaging 2020, 64, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Kojder, K.; Siminska, D.; Bohatyrewicz, R.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. CC Chemokines in a Tumor: A Review of Pro-Cancer and Anti-Cancer Properties of the Ligands of Receptors CCR1, CCR2, CCR3, and CCR4. Int. J. Mol. Sci. 2020, 21, 8412. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Zumwalt, T.J.; Arnold, M.; Goel, A.; Boland, C.R. Active secretion of CXCL10 and CCL5 from colorectal cancer microenvironments associates with GranzymeB+ CD8+ T-cell infiltration. Oncotarget 2015, 6, 2981–2991. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Lei, X.; Xu, A.; Li, Z.; He, Q.; Huang, X.; Xu, G.; Tian, F.; Ding, Y.; Zhu, W. Chemokines in progression, chemoresistance, diagnosis, and prognosis of colorectal cancer. Front. Immunol. 2022, 13, 724139. [Google Scholar] [CrossRef]
- Li, B.; Qi, Z.P.; He, D.L.; Chen, Z.H.; Liu, J.Y.; Wong, M.W.; Zhang, J.W.; Xu, E.P.; Shi, Q.; Cai, S.L.; et al. NLRP7 deubiquitination by USP10 promotes tumor progression and tumor-associated macrophage polarization in colorectal cancer. J. Exp. Clin. Cancer Res. 2021, 40, 126. [Google Scholar] [CrossRef]
- Do, H.T.T.; Lee, C.H.; Cho, J. Chemokines and their Receptors: Multifaceted Roles in Cancer Progression and Potential Value as Cancer Prognostic Markers. Cancers 2020, 12, 287. [Google Scholar] [CrossRef]
- Khare, T.; Bissonnette, M.; Khare, S. CXCL12-CXCR4/CXCR7 Axis in Colorectal Cancer: Therapeutic Target in Preclinical and Clinical Studies. Int. J. Mol. Sci. 2021, 22, 7371. [Google Scholar] [CrossRef]
- Strater, J.; Koretz, K.; Gunthert, A.R.; Moller, P. In situ detection of enterocytic apoptosis in normal colonic mucosa and in familial adenomatous polyposis. Gut 1995, 37, 819–825. [Google Scholar] [CrossRef]
- Ngo, P.A.; Neurath, M.F.; Lopez-Posadas, R. Impact of Epithelial Cell Shedding on Intestinal Homeostasis. Int. J. Mol. Sci. 2022, 23, 4160. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M.; Kessler, S.; Sadler, T.; West, G.; Homer, C.; McDonald, C.; de la Motte, C.; Fiocchi, C.; Stylianou, E. The epithelial danger signal IL-1alpha is a potent activator of fibroblasts and reactivator of intestinal inflammation. Am. J. Pathol. 2015, 185, 1624–1637. [Google Scholar] [CrossRef] [PubMed]
- Nunberg, M.; Werbner, N.; Neuman, H.; Bersudsky, M.; Braiman, A.; Ben-Shoshan, M.; Ben Izhak, M.; Louzoun, Y.; Apte, R.N.; Voronov, E.; et al. Interleukin 1alpha-Deficient Mice Have an Altered Gut Microbiota Leading to Protection from Dextran Sodium Sulfate-Induced Colitis. mSystems 2018, 3, e00213–e00217. [Google Scholar] [CrossRef] [PubMed]
- Menghini, P.; Corridoni, D.; Butto, L.F.; Osme, A.; Shivaswamy, S.; Lam, M.; Bamias, G.; Pizarro, T.T.; Rodriguez-Palacios, A.; Dinarello, C.A.; et al. Neutralization of IL-1alpha ameliorates Crohn’s disease-like ileitis by functional alterations of the gut microbiome. Proc. Natl. Acad. Sci. USA 2019, 116, 26717–26726. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Siracusa, R.; Cordaro, M.; Peritore, A.F.; Gugliandolo, E.; Mancuso, G.; Midiri, A.; Di Paola, R.; Cuzzocrea, S. Therapeutic potential of dinitrobenzene sulfonic acid (DNBS)-induced colitis in mice by targeting IL-1beta and IL-18. Biochem. Pharmacol. 2018, 155, 150–161. [Google Scholar] [CrossRef]
- Bersudsky, M.; Luski, L.; Fishman, D.; White, R.M.; Ziv-Sokolovskaya, N.; Dotan, S.; Rider, P.; Kaplanov, I.; Aychek, T.; Dinarello, C.A.; et al. Non-redundant properties of IL-1alpha and IL-1beta during acute colon inflammation in mice. Gut 2014, 63, 598–609. [Google Scholar] [CrossRef]
- Matarazzo, L.; Hernandez Santana, Y.E.; Walsh, P.T.; Fallon, P.G. The IL-1 cytokine family as custodians of barrier immunity. Cytokine 2022, 154, 155890. [Google Scholar] [CrossRef]
- Coccia, M.; Harrison, O.J.; Schiering, C.; Asquith, M.J.; Becher, B.; Powrie, F.; Maloy, K.J. IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J. Exp. Med. 2012, 209, 1595–1609. [Google Scholar] [CrossRef]
- Lopetuso, L.R.; Chowdhry, S.; Pizarro, T.T. Opposing Functions of Classic and Novel IL-1 Family Members in Gut Health and Disease. Front. Immunol. 2013, 4, 181. [Google Scholar] [CrossRef]
- Bradford, E.M.; Ryu, S.H.; Singh, A.P.; Lee, G.; Goretsky, T.; Sinh, P.; Williams, D.B.; Cloud, A.L.; Gounaris, E.; Patel, V.; et al. Epithelial TNF Receptor Signaling Promotes Mucosal Repair in Inflammatory Bowel Disease. J. Immunol. 2017, 199, 1886–1897. [Google Scholar] [CrossRef]
- Jeffery, V.; Goldson, A.J.; Dainty, J.R.; Chieppa, M.; Sobolewski, A. IL-6 Signaling Regulates Small Intestinal Crypt Homeostasis. J. Immunol. 2017, 199, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Gu, H.D.; He, Y.; Wang, J.W. Role of IL-6 polymorphism on the development of cardiovascular events and coronary artery disease in patients receiving hemodialysis. Genet. Mol. Res. 2015, 14, 2631–2637. [Google Scholar] [CrossRef] [PubMed]
- Lindemans, C.A.; Calafiore, M.; Mertelsmann, A.M.; O’Connor, M.H.; Dudakov, J.A.; Jenq, R.R.; Velardi, E.; Young, L.F.; Smith, O.M.; Lawrence, G.; et al. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 2015, 528, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Aparicio-Domingo, P.; Romera-Hernandez, M.; Karrich, J.J.; Cornelissen, F.; Papazian, N.; Lindenbergh-Kortleve, D.J.; Butler, J.A.; Boon, L.; Coles, M.C.; Samsom, J.N.; et al. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J. Exp. Med. 2015, 212, 1783–1791. [Google Scholar] [CrossRef]
- Scheibe, K.; Backert, I.; Wirtz, S.; Hueber, A.; Schett, G.; Vieth, M.; Probst, H.C.; Bopp, T.; Neurath, M.F.; Neufert, C. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut 2017, 66, 823–838. [Google Scholar] [CrossRef]
- von Moltke, J.; Ji, M.; Liang, H.E.; Locksley, R.M. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 2016, 529, 221–225. [Google Scholar] [CrossRef]
- Mahapatro, M.; Erkert, L.; Becker, C. Cytokine-Mediated Crosstalk between Immune Cells and Epithelial Cells in the Gut. Cells 2021, 10, 111. [Google Scholar] [CrossRef]
- Hirano, T.; Hirayama, D.; Wagatsuma, K.; Yamakawa, T.; Yokoyama, Y.; Nakase, H. Immunological Mechanisms in Inflammation-Associated Colon Carcinogenesis. Int. J. Mol. Sci. 2020, 21, 3062. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Varga, J.; Greten, F.R. Cell plasticity in epithelial homeostasis and tumorigenesis. Nat. Cell Biol. 2017, 19, 1133–1141. [Google Scholar] [CrossRef]
- Ritter, B.; Greten, F.R. Modulating inflammation for cancer therapy. J. Exp. Med. 2019, 216, 1234–1243. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.K.; Williams, S.M. Helicobacter pylori infection causes both protective and deleterious effects in human health and disease. Genes Immun. 2021, 22, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Senchukova, M.A.; Tomchuk, O.; Shurygina, E.I. Helicobacter pylori in gastric cancer: Features of infection and their correlations with long-term results of treatment. World J. Gastroenterol. 2021, 27, 6290–6305. [Google Scholar] [CrossRef] [PubMed]
- Drewes, J.L.; Chen, J.; Markham, N.O.; Knippel, R.J.; Domingue, J.C.; Tam, A.J.; Chan, J.L.; Kim, L.; McMann, M.; Stevens, C.; et al. Human Colon Cancer-Derived Clostridioides difficile Strains Drive Colonic Tumorigenesis in Mice. Cancer Discov. 2022, 12, 1873–1885. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Hua, X.; Phipps, A.I.; Burnett-Hartman, A.N.; Adams, S.V.; Hardikar, S.; Cohen, S.A.; Kocarnik, J.M.; Ahnen, D.J.; Lindor, N.M.; Baron, J.A.; et al. Timing of Aspirin and Other Nonsteroidal Anti-Inflammatory Drug Use Among Patients With Colorectal Cancer in Relation to Tumor Markers and Survival. J. Clin. Oncol. 2017, 35, 2806–2813. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef]
- Leppkes, M.; Neurath, M.F. Cytokines in inflammatory bowel diseases—Update 2020. Pharmacol. Res. 2020, 158, 104835. [Google Scholar] [CrossRef]
- Sommer, K.; Wiendl, M.; Muller, T.M.; Heidbreder, K.; Voskens, C.; Neurath, M.F.; Zundler, S. Intestinal Mucosal Wound Healing and Barrier Integrity in IBD-Crosstalk and Trafficking of Cellular Players. Front. Med. 2021, 8, 643973. [Google Scholar] [CrossRef]
- Chen, B.; Scurrah, C.R.; McKinley, E.T.; Simmons, A.J.; Ramirez-Solano, M.A.; Zhu, X.; Markham, N.O.; Heiser, C.N.; Vega, P.N.; Rolong, A.; et al. Differential pre-malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell 2021, 184, 6262–6280.e26. [Google Scholar] [CrossRef]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Busque, L.; Patel, J.P.; Figueroa, M.E.; Vasanthakumar, A.; Provost, S.; Hamilou, Z.; Mollica, L.; Li, J.; Viale, A.; Heguy, A.; et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet. 2012, 44, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 2019, 574, 532–537. [Google Scholar] [CrossRef]
- Garlanda, C.; Mantovani, A. Interleukin-1 in tumor progression, therapy, and prevention. Cancer Cell 2021, 39, 1023–1027. [Google Scholar] [CrossRef]
- Pastille, E.; Wasmer, M.H.; Adamczyk, A.; Vu, V.P.; Mager, L.F.; Phuong, N.N.T.; Palmieri, V.; Simillion, C.; Hansen, W.; Kasper, S.; et al. The IL-33/ST2 pathway shapes the regulatory T cell phenotype to promote intestinal cancer. Mucosal Immunol. 2019, 12, 990–1003. [Google Scholar] [CrossRef]
- Bergmann, H.; Roth, S.; Pechloff, K.; Kiss, E.A.; Kuhn, S.; Heikenwalder, M.; Diefenbach, A.; Greten, F.R.; Ruland, J. Card9-dependent IL-1beta regulates IL-22 production from group 3 innate lymphoid cells and promotes colitis-associated cancer. Eur. J. Immunol. 2017, 47, 1342–1353. [Google Scholar] [CrossRef]
- Chen, J.; Gong, C.; Mao, H.; Li, Z.; Fang, Z.; Chen, Q.; Lin, M.; Jiang, X.; Hu, Y.; Wang, W.; et al. E2F1/SP3/STAT6 axis is required for IL-4-induced epithelial-mesenchymal transition of colorectal cancer cells. Int. J. Oncol. 2018, 53, 567–578. [Google Scholar] [CrossRef]
- Lin, X.; Wang, S.; Sun, M.; Zhang, C.; Wei, C.; Yang, C.; Dou, R.; Liu, Q.; Xiong, B. miR-195-5p/NOTCH2-mediated EMT modulates IL-4 secretion in colorectal cancer to affect M2-like TAM polarization. J. Hematol. Oncol. 2019, 12, 20. [Google Scholar] [CrossRef]
- Ohno, Y.; Kitamura, H.; Takahashi, N.; Ohtake, J.; Kaneumi, S.; Sumida, K.; Homma, S.; Kawamura, H.; Minagawa, N.; Shibasaki, S.; et al. IL-6 down-regulates HLA class II expression and IL-12 production of human dendritic cells to impair activation of antigen-specific CD4(+) T cells. Cancer Immunol. Immunother. 2016, 65, 193–204. [Google Scholar] [CrossRef]
- Zhou, L.; Ivanov, I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef]
- Heichler, C.; Scheibe, K.; Schmied, A.; Geppert, C.I.; Schmid, B.; Wirtz, S.; Thoma, O.M.; Kramer, V.; Waldner, M.J.; Buttner, C.; et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut 2020, 69, 1269–1282. [Google Scholar] [CrossRef]
- Grivennikov, S.I. Inflammation and colorectal cancer: Colitis-associated neoplasia. Semin. Immunopathol. 2013, 35, 229–244. [Google Scholar] [CrossRef]
- Grivennikov, S.I. IL-11: A prominent pro-tumorigenic member of the IL-6 family. Cancer Cell 2013, 24, 145–147. [Google Scholar] [CrossRef]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef]
- Lee, Y.S.; Choi, I.; Ning, Y.; Kim, N.Y.; Khatchadourian, V.; Yang, D.; Chung, H.K.; Choi, D.; LaBonte, M.J.; Ladner, R.D.; et al. Interleukin-8 and its receptor CXCR2 in the tumour microenvironment promote colon cancer growth, progression and metastasis. Br. J. Cancer 2012, 106, 1833–1841. [Google Scholar] [CrossRef]
- Kern, L.; Mittenbuhler, M.J.; Vesting, A.J.; Ostermann, A.L.; Wunderlich, C.M.; Wunderlich, F.T. Obesity-Induced TNFalpha and IL-6 Signaling: The Missing Link between Obesity and Inflammation-Driven Liver and Colorectal Cancers. Cancers 2018, 11, 24. [Google Scholar] [CrossRef]
- Najdaghi, S.; Razi, S.; Rezaei, N. An overview of the role of interleukin-8 in colorectal cancer. Cytokine 2020, 135, 155205. [Google Scholar] [CrossRef]
- O’Hara, A.M.; Bhattacharyya, A.; Bai, J.; Mifflin, R.C.; Ernst, P.B.; Mitra, S.; Crowe, S.E. Tumor necrosis factor (TNF)-alpha-induced IL-8 expression in gastric epithelial cells: Role of reactive oxygen species and AP endonuclease-1/redox factor (Ref)-1. Cytokine 2009, 46, 359–369. [Google Scholar] [CrossRef]
- Amin, M.N.; Siddiqui, S.A.; Ibrahim, M.; Hakim, M.L.; Ahammed, M.S.; Kabir, A.; Sultana, F. Inflammatory cytokines in the pathogenesis of cardiovascular disease and cancer. SAGE Open Med. 2020, 8, 2050312120965752. [Google Scholar] [CrossRef] [PubMed]
- Niess, J.H.; Hruz, P.; Kaymak, T. The Interleukin-20 Cytokines in Intestinal Diseases. Front. Immunol. 2018, 9, 1373. [Google Scholar] [CrossRef]
- Kantola, T.; Klintrup, K.; Vayrynen, J.P.; Vornanen, J.; Bloigu, R.; Karhu, T.; Herzig, K.H.; Napankangas, J.; Makela, J.; Karttunen, T.J.; et al. Stage-dependent alterations of the serum cytokine pattern in colorectal carcinoma. Br. J. Cancer 2012, 107, 1729–1736. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Lin, Y.; Nagarsheth, N.; Peng, D.; Zhao, L.; Zhao, E.; Vatan, L.; Szeliga, W.; Dou, Y.; Owens, S.; et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity 2014, 40, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Gronke, K.; Hernandez, P.P.; Zimmermann, J.; Klose, C.S.N.; Kofoed-Branzk, M.; Guendel, F.; Witkowski, M.; Tizian, C.; Amann, L.; Schumacher, F.; et al. Triantafyllopoulou, A.; Diefenbach, A. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature 2019, 566, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Hue, S.; Ahern, P.; Buonocore, S.; Kullberg, M.C.; Cua, D.J.; McKenzie, B.S.; Powrie, F.; Maloy, K.J. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 2006, 203, 2473–2483. [Google Scholar] [CrossRef]
- Yen, D.; Cheung, J.; Scheerens, H.; Poulet, F.; McClanahan, T.; McKenzie, B.; Kleinschek, M.A.; Owyang, A.; Mattson, J.; Blumenschein, W.; et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Investig. 2006, 116, 1310–1316. [Google Scholar] [CrossRef]
- Teymouri, M.; Pirro, M.; Fallarino, F.; Gargaro, M.; Sahebkar, A. IL-35, a hallmark of immune-regulation in cancer progression, chronic infections and inflammatory diseases. Int. J. Cancer 2018, 143, 2105–2115. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef]
- Zepp, J.A.; Zhao, J.; Liu, C.; Bulek, K.; Wu, L.; Chen, X.; Hao, Y.; Wang, Z.; Wang, X.; Ouyang, W.; et al. IL-17A-Induced PLET1 Expression Contributes to Tissue Repair and Colon Tumorigenesis. J. Immunol. 2017, 199, 3849–3857. [Google Scholar] [CrossRef]
- Chung, A.S.; Wu, X.; Zhuang, G.; Ngu, H.; Kasman, I.; Zhang, J.; Vernes, J.M.; Jiang, Z.; Meng, Y.G.; Peale, F.V.; et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat. Med. 2013, 19, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pages, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- Richmond, J.; Tuzova, M.; Cruikshank, W.; Center, D. Regulation of cellular processes by interleukin-16 in homeostasis and cancer. J. Cell Physiol. 2014, 229, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Baghdadi, M.; Umeyama, Y.; Hama, N.; Kobayashi, T.; Han, N.; Wada, H.; Seino, K.I. Interleukin-34, a comprehensive review. J. Leukoc. Biol. 2018, 104, 931–951. [Google Scholar] [CrossRef]
- Franze, E.; Di Grazia, A.; Sica, G.S.; Biancone, L.; Laudisi, F.; Monteleone, G. Interleukin-34 Enhances the Tumor Promoting Function of Colorectal Cancer-Associated Fibroblasts. Cancers 2020, 12, 3537. [Google Scholar] [CrossRef]
- Franze, E.; Stolfi, C.; Troncone, E.; Scarozza, P.; Monteleone, G. Role of Interleukin-34 in Cancer. Cancers 2020, 12, 252. [Google Scholar] [CrossRef]
- Kobelt, D.; Zhang, C.; Clayton-Lucey, I.A.; Glauben, R.; Voss, C.; Siegmund, B.; Stein, U. Pro-inflammatory TNF-alpha and IFN-gamma Promote Tumor Growth and Metastasis via Induction of MACC1. Front. Immunol. 2020, 11, 980. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 2008, 118, 560–570. [Google Scholar]
- Ogawa, R.; Yamamoto, T.; Hirai, H.; Hanada, K.; Kiyasu, Y.; Nishikawa, G.; Mizuno, R.; Inamoto, S.; Itatani, Y.; Sakai, Y.; et al. Loss of SMAD4 Promotes Colorectal Cancer Progression by Recruiting Tumor-Associated Neutrophils via the CXCL1/8-CXCR2 Axis. Clin. Cancer Res. 2019, 25, 2887–2899. [Google Scholar] [CrossRef]
- Xia, C.; He, L.; Sun, Y. Expression and Prognostic Role of CXCL1 Gene in Colorectal Adenocarcinoma. Comput. Intell. Neurosci. 2022, 2022, 5504731. [Google Scholar] [CrossRef]
- Chen, M.C.; Baskaran, R.; Lee, N.H.; Hsu, H.H.; Ho, T.J.; Tu, C.C.; Lin, Y.M.; Viswanadha, V.P.; Kuo, W.W.; Huang, C.Y. CXCL2/CXCR2 axis induces cancer stem cell characteristics in CPT-11-resistant LoVo colon cancer cells via Galphai-2 and Galphaq/11. J. Cell Physiol. 2019, 234, 11822–11834. [Google Scholar] [CrossRef]
- Teijeira, A.; Garasa, S.; Gato, M.; Alfaro, C.; Migueliz, I.; Cirella, A.; de Andrea, C.; Ochoa, M.C.; Otano, I.; Etxeberria, I.; et al. CXCR1 and CXCR2 Chemokine Receptor Agonists Produced by Tumors Induce Neutrophil Extracellular Traps that Interfere with Immune Cytotoxicity. Immunity 2020, 52, 856–871.e8. [Google Scholar] [CrossRef]
- Fisher, R.C.; Bellamkonda, K.; Alex Molina, L.; Xiang, S.; Liska, D.; Sarvestani, S.K.; Chakrabarti, S.; Berg, A.; Jorgensen, M.L.; Hatala, D.; et al. Disrupting Inflammation-Associated CXCL8-CXCR1 Signaling Inhibits Tumorigenicity Initiated by Sporadic- and Colitis-Colon Cancer Stem Cells. Neoplasia 2019, 21, 269–281. [Google Scholar] [CrossRef]
- Pennel, K.A.; Quinn, J.A.; Nixon, C.; Inthagard, J.; van Wyk, H.C.; Chang, D.; Rebus, S.; Group, G.; Hay, J.; Maka, N.N.; et al. CXCL8 expression is associated with advanced stage, right sidedness, and distinct histological features of colorectal cancer. J Pathol. Clin. Res. 2022, 8, 509–520. [Google Scholar] [CrossRef]
- Biasci, D.; Smoragiewicz, M.; Connell, C.M.; Wang, Z.; Gao, Y.; Thaventhiran, J.E.D.; Basu, B.; Magiera, L.; Johnson, T.I.; Bax, L.; et al. CXCR4 inhibition in human pancreatic and colorectal cancers induces an integrated immune response. Proc. Natl. Acad. Sci. USA 2020, 117, 28960–28970. [Google Scholar] [CrossRef]
- Karin, M.; Shalapour, S. Regulation of antitumor immunity by inflammation-induced epigenetic alterations. Cell Mol. Immunol. 2022, 19, 59–66. [Google Scholar] [CrossRef]
- Wang, W.; Ding, C.L.; Wu, M.X.; Guo, W.; Hu, R.; Liu, Y.; Qi, Z.T.; Jia, X.M. RAI16 maintains intestinal homeostasis and inhibits NLRP3-dependent IL-18/CXCL16-induced colitis and the progression of colitis-associated colorectal cancer. Clin. Transl. Med. 2022, 12, e993. [Google Scholar] [CrossRef]
- Hao, Q.; Vadgama, J.V.; Wang, P. CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun. Signal. 2020, 18, 82. [Google Scholar] [CrossRef]
- Xu, M.; Wang, Y.; Xia, R.; Wei, Y.; Wei, X. Role of the CCL2-CCR2 signalling axis in cancer: Mechanisms and therapeutic targeting. Cell Prolif. 2021, 54, e13115. [Google Scholar] [CrossRef]
- Ma, X.; Su, J.; Zhao, S.; He, Y.; Li, S.; Yang, X.; Zhai, S.; Rong, S.; Zhang, X.; Xu, G.; et al. CCL3 Promotes Proliferation of Colorectal Cancer Related with TRAF6/NF-kappaB Molecular Pathway. Contrast. Media Mol. Imaging 2022, 2022, 2387192. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente Lopez, M.; Landskron, G.; Parada, D.; Dubois-Camacho, K.; Simian, D.; Martinez, M.; Romero, D.; Roa, J.C.; Chahuan, I.; Gutierrez, R.; et al. The relationship between chemokines CCL2, CCL3, and CCL4 with the tumor microenvironment and tumor-associated macrophage markers in colorectal cancer. Tumour Biol. 2018, 40, 1010428318810059. [Google Scholar] [CrossRef] [PubMed]
- Braoudaki, M.; Ahmad, M.S.; Mustafov, D.; Seriah, S.; Siddiqui, M.N.; Siddiqui, S.S. Chemokines and chemokine receptors in colorectal cancer; multifarious roles and clinical impact. Semin. Cancer Biol. 2022, 86, 436–449. [Google Scholar] [CrossRef]
- Strasly, M.; Doronzo, G.; Cappello, P.; Valdembri, D.; Arese, M.; Mitola, S.; Moore, P.; Alessandri, G.; Giovarelli, M.; Bussolino, F. CCL16 activates an angiogenic program in vascular endothelial cells. Blood 2004, 103, 40–49. [Google Scholar] [CrossRef]
- Wagsater, D.; Dienus, O.; Lofgren, S.; Hugander, A.; Dimberg, J. Quantification of the chemokines CCL17 and CCL22 in human colorectal adenocarcinomas. Mol. Med. Rep. 2008, 1, 211–217. [Google Scholar]
- Frick, V.O.; Rubie, C.; Keilholz, U.; Ghadjar, P. Chemokine/chemokine receptor pair CCL20/CCR6 in human colorectal malignancy: An overview. World J. Gastroenterol. 2016, 22, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Vicinus, B.; Rubie, C.; Stegmaier, N.; Frick, V.O.; Kolsch, K.; Kauffels, A.; Ghadjar, P.; Wagner, M.; Glanemann, M. miR-21 and its target gene CCL20 are both highly overexpressed in the microenvironment of colorectal tumors: Significance of their regulation. Oncol. Rep. 2013, 30, 1285–1292. [Google Scholar] [CrossRef]
- Fung, K.Y.; Nguyen, P.M.; Putoczki, T.L. Emerging Roles for Interleukin-18 in the Gastrointestinal Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1240, 59–72. [Google Scholar]
- Salcedo, R.; Worschech, A.; Cardone, M.; Jones, Y.; Gyulai, Z.; Dai, R.M.; Wang, E.; Ma, W.; Haines, D.; O’hUigin, C.; et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: Role of interleukin 18. J. Exp. Med. 2010, 207, 1625–1636. [Google Scholar] [CrossRef]
- Weinstein, A.M.; Giraldo, N.A.; Petitprez, F.; Julie, C.; Lacroix, L.; Peschaud, F.; Emile, J.F.; Marisa, L.; Fridman, W.H.; Storkus, W.J.; et al. Association of IL-36gamma with tertiary lymphoid structures and inflammatory immune infiltrates in human colorectal cancer. Cancer Immunol. Immunother. 2019, 68, 109–120. [Google Scholar] [CrossRef]
- Elias, M.; Zhao, S.; Le, H.T.; Wang, J.; Neurath, M.F.; Neufert, C.; Fiocchi, C.; Rieder, F. IL-36 in chronic inflammation and fibrosis—bridging the gap? J. Clin. Investig. 2021, 131, 4336. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zheng, R.; Wang, P.; Yang, H.; He, X.; Ji, Q.; Bai, W.; Chen, H.; Chen, J.; Peng, W.; et al. IL-37 Confers Protection against Mycobacterial Infection Involving Suppressing Inflammation and Modulating T Cell Activation. PLoS ONE 2017, 12, e0169922. [Google Scholar] [CrossRef] [PubMed]
- Dimberg, J.; Shamoun, L.; Landerholm, K.; Andersson, R.E.; Kolodziej, B.; Wagsater, D. Genetic Variants of the IL2 Gene Related to Risk and Survival in Patients With Colorectal Cancer. Anticancer Res. 2019, 39, 4933–4940. [Google Scholar] [CrossRef] [PubMed]
- Morre, M.; Beq, S. Interleukin-7 and immune reconstitution in cancer patients: A new paradigm for dramatically increasing overall survival. Target Oncol. 2012, 7, 55–68. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gao, J.; Zhao, L.; Wan, Y.Y.; Zhu, B. Mechanism of Action of IL-7 and Its Potential Applications and Limitations in Cancer Immunotherapy. Int. J. Mol. Sci. 2015, 16, 10267–10280. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Wu, Y.; Ji, X.; Huang, L.; Cai, W.; Su, Z.; Wang, S.; Xu, H. IL-9 and IL-9-producing cells in tumor immunity. Cell Commun. Signal. 2020, 18, 50. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, M.; Zhao, H.; Huang, Y.; Li, D.; Mao, D.; Zhang, Z.; Zhu, X.; Dong, X.; Zhao, X. IL-9 Exerts Antitumor Effects in Colon Cancer and Transforms the Tumor Microenvironment In Vivo. Technol. Cancer Res. Treat. 2019, 18, 1533033819857737. [Google Scholar] [CrossRef]
- Mishra, A.; Sullivan, L.; Caligiuri, M.A. Molecular pathways: Interleukin-15 signaling in health and in cancer. Clin. Cancer Res. 2014, 20, 2044–2050. [Google Scholar] [CrossRef]
- Jabri, B.; Abadie, V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat. Rev. Immunol. 2015, 15, 771–783. [Google Scholar] [CrossRef]
- Ciszewski, C.; Discepolo, V.; Pacis, A.; Doerr, N.; Tastet, O.; Mayassi, T.; Maglio, M.; Basheer, A.; Al-Mawsawi, L.Q.; Green, P.H.R.; et al. Identification of a gammac Receptor Antagonist That Prevents Reprogramming of Human Tissue-resident Cytotoxic T Cells by IL15 and IL21. Gastroenterology 2020, 158, 625–637.e13. [Google Scholar] [CrossRef]
- Ong, C.Y.; Abdalkareem, E.A.; Khoo, B.Y. Functional roles of cytokines in infectious disease associated colorectal carcinogenesis. Mol. Biol. Rep. 2022, 49, 1529–1535. [Google Scholar] [CrossRef]
- Li, Y.; Cong, Y.; Jia, M.; He, Q.; Zhong, H.; Zhao, Y.; Li, H.; Yan, M.; You, J.; Liu, J.; et al. Targeting IL-21 to tumor-reactive T cells enhances memory T cell responses and anti-PD-1 antibody therapy. Nat. Commun. 2021, 12, 951. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Jeon, I.; Kim, B.S.; Park, M.; Bae, E.A.; Song, B.; Koh, C.H.; Shin, K.S.; Kim, I.K.; Choi, K.; et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat. Commun. 2017, 8, 15776. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.E.; Bhatia, S.; Bhoopathi, P.; Das, S.K.; Emdad, L.; Dasgupta, S.; Dent, P.; Wang, X.Y.; Sarkar, D.; Fisher, P.B. MDA-7/IL-24: Multifunctional cancer killing cytokine. Adv. Exp. Med. Biol. 2014, 818, 127–153. [Google Scholar]
- Malvicini, M.; Rizzo, M.; Alaniz, L.; Pinero, F.; Garcia, M.; Atorrasagasti, C.; Aquino, J.B.; Rozados, V.; Scharovsky, O.G.; Matar, P.; et al. A novel synergistic combination of cyclophosphamide and gene transfer of interleukin-12 eradicates colorectal carcinoma in mice. Clin. Cancer Res 2009, 15, 7256–7265. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Smyth, M.J.; Teng, M.W.L. Interleukin (IL)-12 and IL-23 and Their Conflicting Roles in Cancer. Cold Spring Harb. Perspect. Biol. 2018, 10, a028530. [Google Scholar] [CrossRef] [PubMed]
- Razi, S.; Baradaran Noveiry, B.; Keshavarz-Fathi, M.; Rezaei, N. IL-17 and colorectal cancer: From carcinogenesis to treatment. Cytokine 2019, 116, 7–12. [Google Scholar] [CrossRef]
- Zhang, C.; Hou, D.; Wei, H.; Zhao, M.; Yang, L.; Liu, Q.; Zhang, X.; Gong, Y.; Shao, C. Lack of interferon-gamma receptor results in a microenvironment favorable for intestinal tumorigenesis. Oncotarget 2016, 7, 42099–42109. [Google Scholar] [CrossRef]
- Simpson, J.A.; Al-Attar, A.; Watson, N.F.; Scholefield, J.H.; Ilyas, M.; Durrant, L.G. Intratumoral T cell infiltration, MHC class I and STAT1 as biomarkers of good prognosis in colorectal cancer. Gut 2010, 59, 926–933. [Google Scholar] [CrossRef]
- Mowat, C.; Mosley, S.R.; Namdar, A.; Schiller, D.; Baker, K. Anti-tumor immunity in mismatch repair-deficient colorectal cancers requires type I IFN-driven CCL5 and CXCL10. J. Exp. Med. 2021, 218, e20210108. [Google Scholar] [CrossRef]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B.; et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Sancho, E.; Batlle, E. Overcoming TGFbeta-mediated immune evasion in cancer. Nat. Rev. Cancer 2022, 22, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, C.; Pandit, H.; Nagy, B.A.; Stellas, D.; Jensen, S.M.; Bear, J.; Cam, M.; Valentin, A.; Fox, B.A.; Felber, B.K.; et al. Heterodimeric IL-15 delays tumor growth and promotes intratumoral CTL and dendritic cell accumulation by a cytokine network involving XCL1, IFN-gamma, CXCL9 and CXCL10. J. Immunother. Cancer 2020, 8, e000599. [Google Scholar] [CrossRef] [PubMed]
- Akeus, P.; Szeponik, L.; Ahlmanner, F.; Sundstrom, P.; Alsen, S.; Gustavsson, B.; Sparwasser, T.; Raghavan, S.; Quiding-Jarbrink, M. Regulatory T cells control endothelial chemokine production and migration of T cells into intestinal tumors of APC(min/+) mice. Cancer Immunol. Immunother. 2018, 67, 1067–1077. [Google Scholar] [CrossRef]
- Shang, S.; Yang, Y.W.; Chen, F.; Yu, L.; Shen, S.H.; Li, K.; Cui, B.; Lv, X.X.; Zhang, C.; Yang, C.; et al. TRIB3 reduces CD8(+) T cell infiltration and induces immune evasion by repressing the STAT1-CXCL10 axis in colorectal cancer. Sci. Transl. Med. 2022, 14, eabf0992. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Jiao, N.; Sun, T.; Ma, Y.; Zhang, X.; Chen, H.; Hong, J.; Zhang, Y. CXCL11 Correlates With Antitumor Immunity and an Improved Prognosis in Colon Cancer. Front. Cell Dev. Biol. 2021, 9, 646252. [Google Scholar] [CrossRef]
- Zhang, Y.; Davis, C.; Shah, S.; Hughes, D.; Ryan, J.C.; Altomare, D.; Pena, M.M. IL-33 promotes growth and liver metastasis of colorectal cancer in mice by remodeling the tumor microenvironment and inducing angiogenesis. Mol. Carcinog. 2017, 56, 272–287. [Google Scholar] [CrossRef]
- Zhou, Y.; Ji, Y.; Wang, H.; Zhang, H.; Zhou, H. IL-33 Promotes the Development of Colorectal Cancer Through Inducing Tumor-Infiltrating ST2L(+) Regulatory T Cells in Mice. Technol. Cancer Res. Treat 2018, 17, 1533033818780091. [Google Scholar] [CrossRef]
- Luo, P.; Deng, S.; Ye, H.; Yu, X.; Deng, Q.; Zhang, Y.; Jiang, L.; Li, J.; Yu, Y.; Han, W. The IL-33/ST2 pathway suppresses murine colon cancer growth and metastasis by upregulating CD40 L signaling. Biomed. Pharmacother. 2020, 127, 110232. [Google Scholar] [CrossRef]
- Eissmann, M.F.; Dijkstra, C.; Wouters, M.A.; Baloyan, D.; Mouradov, D.; Nguyen, P.M.; Davalos-Salas, M.; Putoczki, T.L.; Sieber, O.M.; Mariadason, J.M.; et al. Interleukin 33 Signaling Restrains Sporadic Colon Cancer in an Interferon-gamma-Dependent Manner. Cancer Immunol. Res. 2018, 6, 409–421. [Google Scholar] [CrossRef]
- Cui, G. TH9, TH17, and TH22 Cell Subsets and Their Main Cytokine Products in the Pathogenesis of Colorectal Cancer. Front. Oncol. 2019, 9, 1002. [Google Scholar] [CrossRef] [PubMed]
- Borowczak, J.; Szczerbowski, K.; Maniewski, M.; Kowalewski, A.; Janiczek-Polewska, M.; Szylberg, A.; Marszalek, A.; Szylberg, L. The Role of Inflammatory Cytokines in the Pathogenesis of Colorectal Carcinoma-Recent Findings and Review. Biomedicines 2022, 10, 1670. [Google Scholar] [CrossRef]
- Deng, S.; Deng, Q.; Zhang, Y.; Ye, H.; Yu, X.; Zhang, Y.; Han, G.Y.; Luo, P.; Wu, M.; Yu, Y.; et al. Non-platelet-derived CXCL4 differentially regulates cytotoxic and regulatory T cells through CXCR3 to suppress the immune response to colon cancer. Cancer Lett. 2019, 443, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.F.; Zhong, Y.; Long, T.; Wang, X.; Zhu, J.X.; Wang, X.Y.; Hu, Z.Y.; Li, Z.G. Tumor bud-derived CCL5 recruits fibroblasts and promotes colorectal cancer progression via CCR5-SLC25A24 signaling. J. Exp. Clin. Cancer Res. 2022, 41, 81. [Google Scholar] [CrossRef] [PubMed]
- Suenaga, M.; Zhang, W.U.; Mashima, T.; Schirripa, M.; Cao, S.; Okazaki, S.; Berger, M.D.; Miyamoto, Y.; Barzi, A.; Yamaguchi, T.; et al. Potential Molecular Cross Talk Among CCR5 Pathway Predicts Regorafenib Responsiveness in Metastatic Colorectal Cancer Patients. Cancer Genom. Proteom. 2021, 18, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Blacher, E.; Elinav, E. Microbiome, metabolites and host immunity. Curr. Opin. Microbiol. 2017, 35, 8–15. [Google Scholar] [CrossRef]
- McBurney, M.I.; Davis, C.; Fraser, C.M.; Schneeman, B.O.; Huttenhower, C.; Verbeke, K.; Walter, J.; Latulippe, M.E. Establishing What Constitutes a Healthy Human Gut Microbiome: State of the Science, Regulatory Considerations, and Future Directions. J. Nutr. 2019, 149, 1882–1895. [Google Scholar] [CrossRef]
- Maukonen, J.; Saarela, M. Human gut microbiota: Does diet matter? Proc. Nutr. Soc. 2015, 74, 23–36. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Khalyfa, A.A.; Punatar, S.; Aslam, R.; Yarbrough, A. Exploring the Inflammatory Pathogenesis of Colorectal Cancer. Diseases 2021, 9, 79. [Google Scholar] [CrossRef]
- Hanus, M.; Parada-Venegas, D.; Landskron, G.; Wielandt, A.M.; Hurtado, C.; Alvarez, K.; Hermoso, M.A.; Lopez-Kostner, F.; De la Fuente, M. Immune System, Microbiota, and Microbial Metabolites: The Unresolved Triad in Colorectal Cancer Microenvironment. Front. Immunol. 2021, 12, 612826. [Google Scholar] [CrossRef] [PubMed]
- Solis, A.G.; Klapholz, M.; Zhao, J.; Levy, M. The bidirectional nature of microbiome-epithelial cell interactions. Curr. Opin. Microbiol. 2020, 56, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Nastasi, C.; Candela, M.; Bonefeld, C.M.; Geisler, C.; Hansen, M.; Krejsgaard, T.; Biagi, E.; Andersen, M.H.; Brigidi, P.; Odum, N.; et al. The effect of short-chain fatty acids on human monocyte-derived dendritic cells. Sci. Rep. 2015, 5, 16148. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef]
- Boleij, A.; Hechenbleikner, E.M.; Goodwin, A.C.; Badani, R.; Stein, E.M.; Lazarev, M.G.; Ellis, B.; Carroll, K.C.; Albesiano, E.; Wick, E.C.; et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin. Infect. Dis. 2015, 60, 208–215. [Google Scholar] [CrossRef]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carra, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef]
- Atreya, I.; Neurath, M.F. How the Tumor Micromilieu Modulates the Recruitment and Activation of Colorectal Cancer-Infiltrating Lymphocytes. Biomedicines. 2022, 10, 2940. [Google Scholar] [CrossRef]
- Chinnaiyan, A.M.; Tepper, C.G.; Seldin, M.F.; O’Rourke, K.; Kischkel, F.C.; Hellbardt, S.; Krammer, P.H.; Peter, M.E.; Dixit, V.M. FADD/MORT1 is a common mediator of CD95 (Fas/APO-1) and tumor necrosis factor receptor-induced apoptosis. J. Biol. Chem. 1996, 271, 4961–4965. [Google Scholar] [CrossRef]
- Lin, Y.; He, Z.; Ye, J.; Liu, Z.; She, X.; Gao, X.; Liang, R. Progress in Understanding the IL-6/STAT3 Pathway in Colorectal Cancer. OncoTargets Ther. 2020, 13, 13023–13032. [Google Scholar] [CrossRef]
- McFarlane, A.J.; Fercoq, F.; Coffelt, S.B.; Carlin, L.M. Neutrophil dynamics in the tumor microenvironment. J. Clin. Investig. 2021, 131, 3759. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. The dual role of neutrophils in cancer. Semin. Immunol. 2021, 57, 101582. [Google Scholar] [CrossRef] [PubMed]
- Andzinski, L.; Kasnitz, N.; Stahnke, S.; Wu, C.F.; Gereke, M.; von Kockritz-Blickwede, M.; Schilling, B.; Brandau, S.; Weiss, S.; Jablonska, J. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int. J. Cancer 2016, 138, 1982–1993. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.M.; Pesic, M.; Engel, E.; Ziegler, P.K.; Diefenhardt, M.; Kennel, K.B.; Buettner, F.; Conche, C.; Petrocelli, V.; Elwakeel, E.; et al. Inflammatory fibroblasts mediate resistance to neoadjuvant therapy in rectal cancer. Cancer Cell 2022, 40, 168–184.e13. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.; Turovskaya, O.; Kim, G.; Madan, R.; Karp, C.L.; Cheroutre, H.; Kronenberg, M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 2009, 10, 1178–1184. [Google Scholar] [CrossRef]
- Ruder, B.; Becker, C. At the Forefront of the Mucosal Barrier: The Role of Macrophages in the Intestine. Cells 2020, 9, 2162. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Song, Z.; Chu, J.; Qu, X. Deficiency of interferon-gamma or its receptor promotes colorectal cancer development. J. Interferon. Cytokine Res. 2015, 35, 273–280. [Google Scholar] [CrossRef]
- Zhong, X.; Chen, B.; Yang, Z. The Role of Tumor-Associated Macrophages in Colorectal Carcinoma Progression. Cell Physiol. Biochem. 2018, 45, 356–365. [Google Scholar] [CrossRef]
- Herbeuval, J.P.; Lelievre, E.; Lambert, C.; Dy, M.; Genin, C. Recruitment of STAT3 for production of IL-10 by colon carcinoma cells induced by macrophage-derived IL-6. J. Immunol. 2004, 172, 4630–4636. [Google Scholar] [CrossRef]
- Dufait, I.; Schwarze, J.K.; Liechtenstein, T.; Leonard, W.; Jiang, H.; Escors, D.; De Ridder, M.; Breckpot, K. Ex vivo generation of myeloid-derived suppressor cells that model the tumor immunosuppressive environment in colorectal cancer. Oncotarget 2015, 6, 12369–12382. [Google Scholar] [CrossRef]
- Sasidharan Nair, V.; Saleh, R.; Taha, R.Z.; Toor, S.M.; Murshed, K.; Ahmed, A.A.; Kurer, M.A.; Abu Nada, M.; Al Ejeh, F.; Elkord, E. Differential gene expression of tumor-infiltrating CD4(+) T cells in advanced versus early stage colorectal cancer and identification of a gene signature of poor prognosis. Oncoimmunology 2020, 9, 1825178. [Google Scholar] [CrossRef]
- Bai, Z.; Zhou, Y.; Ye, Z.; Xiong, J.; Lan, H.; Wang, F. Tumor-Infiltrating Lymphocytes in Colorectal Cancer: The Fundamental Indication and Application on Immunotherapy. Front. Immunol. 2021, 12, 808964. [Google Scholar] [CrossRef] [PubMed]
- Kistner, L.; Doll, D.; Holtorf, A.; Nitsche, U.; Janssen, K.P. Interferon-inducible CXC-chemokines are crucial immune modulators and survival predictors in colorectal cancer. Oncotarget 2017, 8, 89998–90012. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.S.; Yang, Y.Z.; Jiang, C.; Quan, Q.; Xie, Q.K.; Wang, X.P.; He, W.Z.; Rong, Y.M.; Chen, P.; Yang, Q.; et al. Comparison of immunological characteristics between paired mismatch repair-proficient and -deficient colorectal cancer patients. J. Transl. Med. 2018, 16, 195. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F. Heterogeneity of Human CD4(+) T Cells Against Microbes. Annu. Rev. Immunol. 2016, 34, 317–334. [Google Scholar] [CrossRef]
- Bule, P.; Aguiar, S.I.; Aires-Da-Silva, F.; Dias, J.N.R. Chemokine-Directed Tumor Microenvironment Modulation in Cancer Immunotherapy. Int. J. Mol. Sci. 2021, 22, 9804. [Google Scholar] [CrossRef]
- Koller, F.L.; Hwang, D.G.; Dozier, E.A.; Fingleton, B. Epithelial interleukin-4 receptor expression promotes colon tumor growth. Carcinogenesis 2010, 31, 1010–1017. [Google Scholar] [CrossRef]
- Gerlach, K.; Hwang, Y.; Nikolaev, A.; Atreya, R.; Dornhoff, H.; Steiner, S.; Lehr, H.A.; Wirtz, S.; Vieth, M.; Waisman, A.; et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat. Immunol. 2014, 15, 676–686. [Google Scholar] [CrossRef]
- Huang, Y.; Cao, Y.; Zhang, S.; Gao, F. Association between low expression levels of interleukin-9 and colon cancer progression. Exp. Ther. Med. 2015, 10, 942–946. [Google Scholar] [CrossRef]
- De Simone, V.; Franze, E.; Ronchetti, G.; Colantoni, A.; Fantini, M.C.; Di Fusco, D.; Sica, G.S.; Sileri, P.; MacDonald, T.T.; Pallone, F.; et al. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 2015, 34, 3493–3503. [Google Scholar] [CrossRef]
- Perez, L.G.; Kempski, J.; McGee, H.M.; Pelzcar, P.; Agalioti, T.; Giannou, A.; Konczalla, L.; Brockmann, L.; Wahib, R.; Xu, H.; et al. TGF-beta signaling in Th17 cells promotes IL-22 production and colitis-associated colon cancer. Nat. Commun. 2020, 11, 2608. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Luber, B.; Tam, A.J.; Smith, K.N.; Siegel, N.; Awan, A.H.; Fan, H.; Oke, T.; Zhang, J.; Domingue, J.; et al. Intratumoral Adaptive Immunosuppression and Type 17 Immunity in Mismatch Repair Proficient Colorectal Tumors. Clin. Cancer Res. 2019, 25, 5250–5259. [Google Scholar] [CrossRef] [PubMed]
- Fantini, M.C.; Favale, A.; Onali, S.; Facciotti, F. Tumor Infiltrating Regulatory T Cells in Sporadic and Colitis-Associated Colorectal Cancer: The Red Little Riding Hood and the Wolf. Int. J. Mol. Sci. 2020, 21, 6744. [Google Scholar] [CrossRef] [PubMed]
- Akkaya, B.; Shevach, E.M. Regulatory T cells: Master thieves of the immune system. Cell Immunol. 2020, 355, 104160. [Google Scholar] [CrossRef]
- Akkaya, B.; Oya, Y.; Akkaya, M.; Al Souz, J.; Holstein, A.H.; Kamenyeva, O.; Kabat, J.; Matsumura, R.; Dorward, D.W.; Glass, D.D.; et al. Regulatory T cells mediate specific suppression by depleting peptide-MHC class II from dendritic cells. Nat. Immunol. 2019, 20, 218–231. [Google Scholar] [CrossRef]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef]
- Argon, A.; Vardar, E.; Kebat, T.; Erdinc, O.; Erkan, N. The Prognostic Significance of FoxP3+ T Cells and CD8+ T Cells in Colorectal Carcinomas. J. Environ. Pathol. Toxicol. Oncol. 2016, 35, 121–131. [Google Scholar] [CrossRef]
- Reimers, M.S.; Engels, C.C.; Putter, H.; Morreau, H.; Liefers, G.J.; van de Velde, C.J.; Kuppen, P.J. Prognostic value of HLA class I, HLA-E, HLA-G and Tregs in rectal cancer: A retrospective cohort study. BMC Cancer 2014, 14, 486. [Google Scholar] [CrossRef][Green Version]
- Fionda, C.; Scarno, G.; Stabile, H.; Molfetta, R.; Di Censo, C.; Gismondi, A.; Paolini, R.; Sozzani, S.; Santoni, A.; Sciume, G. NK Cells and Other Cytotoxic Innate Lymphocytes in Colorectal Cancer Progression and Metastasis. Int. J. Mol. Sci. 2022, 23, 7859. [Google Scholar] [CrossRef]
- Rosser, E.C.; Mauri, C. Regulatory B cells: Origin, phenotype, and function. Immunity 2015, 42, 607–612. [Google Scholar] [CrossRef]
- Melcher, C.; Yu, J.; Duong, V.H.H.; Westphal, K.; Helmi Siasi Farimany, N.; Shaverskyi, A.; Zhao, B.; Strowig, T.; Glage, S.; Brand, K.; et al. B cell-mediated regulatory mechanisms control tumor-promoting intestinal inflammation. Cell Rep. 2022, 40, 111051. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro-Vornhagen, A.; Schlosser, H.A.; Gryschok, L.; Malcher, J.; Wennhold, K.; Garcia-Marquez, M.; Herbold, T.; Neuhaus, L.S.; Becker, H.J.; Fiedler, A.; et al. Characterization of tumor-associated B-cell subsets in patients with colorectal cancer. Oncotarget 2014, 5, 4651–4664. [Google Scholar] [CrossRef] [PubMed]
- Subtil, B.; Cambi, A.; Tauriello, D.V.F.; de Vries, I.J.M. The Therapeutic Potential of Tackling Tumor-Induced Dendritic Cell Dysfunction in Colorectal Cancer. Front. Immunol. 2021, 12, 724883. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.; Keskinov, A.A.; Shurin, G.V.; Shurin, M.R. Tumor-derived factors modulating dendritic cell function. Cancer Immunol. Immunother. 2016, 65, 821–833. [Google Scholar] [CrossRef]
- Chow, M.T.; Ozga, A.J.; Servis, R.L.; Frederick, D.T.; Lo, J.A.; Fisher, D.E.; Freeman, G.J.; Boland, G.M.; Luster, A.D. Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity 2019, 50, 1498–1512.e5. [Google Scholar] [CrossRef]
- Pelka, K.; Hofree, M.; Chen, J.H.; Sarkizova, S.; Pirl, J.D.; Jorgji, V.; Bejnood, A.; Dionne, D.; Ge, W.H.; Xu, K.H.; et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell 2021, 184, 4734–4752.e20. [Google Scholar] [CrossRef]
- Wang, Q.; Shen, X.; Chen, G.; Du, J. Drug Resistance in Colorectal Cancer: From Mechanism to Clinic. Cancers 2022, 14, 2928. [Google Scholar] [CrossRef]
- Shaked, Y. The pro-tumorigenic host response to cancer therapies. Nat. Rev. Cancer 2019, 19, 667–685. [Google Scholar] [CrossRef]
- Wang, Y.J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef]
- Karagiannis, G.S.; Condeelis, J.S.; Oktay, M.H. Chemotherapy-Induced Metastasis: Molecular Mechanisms, Clinical Manifestations, Therapeutic Interventions. Cancer Res. 2019, 79, 4567–4576. [Google Scholar] [CrossRef]
- Silva, V.R.; Santos, L.S.; Dias, R.B.; Quadros, C.A.; Bezerra, D.P. Emerging agents that target signaling pathways to eradicate colorectal cancer stem cells. Cancer Commun. 2021, 41, 1275–1313. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal. Transduct. Target Ther. 2021, 6, 263. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Homann, L.; Rentschler, M.; Brenner, E.; Bohm, K.; Rocken, M.; Wieder, T. IFN-gamma and TNF Induce Senescence and a Distinct Senescence-Associated Secretory Phenotype in Melanoma. Cells 2022, 11, 1514. [Google Scholar] [CrossRef]
- Rentschler, M.; Braumuller, H.; Briquez, P.S.; Wieder, T. Cytokine-Induced Senescence in the Tumor Microenvironment and Its Effects on Anti-Tumor Immune Responses. Cancers 2022, 14, 1364. [Google Scholar] [CrossRef]
- Braumuller, H.; Mauerer, B.; Berlin, C.; Plundrich, D.; Marbach, P.; Cauchy, P.; Laessle, C.; Biesel, E.; Holzner, P.A.; Kesselring, R. Senescent Tumor Cells in the Peritoneal Carcinomatosis Drive Immunosenescence in the Tumor Microenvironment. Front. Immunol. 2022, 13, 908449. [Google Scholar] [CrossRef]
- Rodriguez-Ruiz, M.E.; Vitale, I.; Harrington, K.J.; Melero, I.; Galluzzi, L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat. Immunol. 2020, 21, 120–134. [Google Scholar] [CrossRef]
- Chan Wah Hak, C.M.L.; Rullan, A.; Patin, E.C.; Pedersen, M.; Melcher, A.A.; Harrington, K.J. Enhancing anti-tumour innate immunity by targeting the DNA damage response and pattern recognition receptors in combination with radiotherapy. Front. Oncol. 2022, 12, 971959. [Google Scholar] [CrossRef]
- Yamazaki, T.; Vanpouille-Box, C.; Demaria, S.; Galluzzi, L. Immunogenic Cell Death Driven by Radiation-Impact on the Tumor Microenvironment. Cancer Treat. Res. 2020, 180, 281–296. [Google Scholar]
- Zhang, X.; Zhang, H.; Zhang, J.; Yang, M.; Zhu, M.; Yin, Y.; Fan, X.; Yu, F. The paradoxical role of radiation-induced cGAS-STING signalling network in tumour immunity. Immunology 2022, in press. [Google Scholar] [CrossRef]
- Chiang, C.S.; Fu, S.Y.; Wang, S.C.; Yu, C.F.; Chen, F.H.; Lin, C.M.; Hong, J.H. Irradiation promotes an m2 macrophage phenotype in tumor hypoxia. Front. Oncol. 2012, 2, 89. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, M.; Patin, E.C.; Pedersen, M.; Wilkins, A.; Dillon, M.T.; Melcher, A.A.; Harrington, K.J. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 2020, 20, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Crucitti, A.; Corbi, M.; Tomaiuolo, P.M.; Fanali, C.; Mazzari, A.; Lucchetti, D.; Migaldi, M.; Sgambato, A. Laparoscopic surgery for colorectal cancer is not associated with an increase in the circulating levels of several inflammation-related factors. Cancer Biol. Ther. 2015, 16, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Predina, J.; Eruslanov, E.; Judy, B.; Kapoor, V.; Cheng, G.; Wang, L.C.; Sun, J.; Moon, E.K.; Fridlender, Z.G.; Albelda, S.; et al. Changes in the local tumor microenvironment in recurrent cancers may explain the failure of vaccines after surgery. Proc. Natl. Acad. Sci. USA 2013, 110, E415–E424. [Google Scholar] [CrossRef] [PubMed]
- Tartter, P.I.; Steinberg, B.; Barron, D.M.; Martinelli, G. The prognostic significance of natural killer cytotoxicity in patients with colorectal cancer. Arch. Surg. 1987, 122, 1264–1268. [Google Scholar] [CrossRef]
- De Assis, J.V.; Coutinho, L.A.; Oyeyemi, I.T.; Oyeyemi, O.T.; Grenfell, R. Diagnostic and therapeutic biomarkers in colorectal cancer: A review. Am. J. Cancer Res. 2022, 12, 661–680. [Google Scholar]
- Janney, A.; Powrie, F.; Mann, E.H. Host-microbiota maladaptation in colorectal cancer. Nature 2020, 585, 509–517. [Google Scholar] [CrossRef]
- Percario, R.; Panaccio, P.; di Mola, F.F.; Grottola, T.; Di Sebastiano, P. The Complex Network between Inflammation and Colorectal Cancer: A Systematic Review of the Literature. Cancers 2021, 13, 6237. [Google Scholar] [CrossRef]
- Wieder, T.; Eigentler, T.; Brenner, E.; Rocken, M. Immune checkpoint blockade therapy. J. Allergy Clin. Immunol. 2018, 142, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Rabin, H.; Adamson, R.H.; Neubauer, R.H.; Cicmanec, J.L.; Wallen, W.C. Pilot studies with human interferon in Herpesvirus saimiri-induced lymphoma in owl monkeys. Cancer Res. 1976, 36, 715–719. [Google Scholar] [PubMed]
- Youn, J.K.; Hovanessian, A.G.; Riviere, Y.; Hue, G.; Lacour, F. Enhancement of natural killer cell activity and 2-5A synthetase in mice treated with polyadenylic.polyuridylic acid. Cell Immunol. 1983, 79, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Lacour, J.; Lacour, F.; Spira, A.; Michelson, M.; Petit, J.Y.; Delage, G.; Sarrazin, D.; Contesso, G.; Viguier, J. Adjuvant treatment with polyadenylic-polyuridylic acid (Polya.Polyu) in operable breast cancer. Lancet 1980, 2, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Quesada, J.R.; Gutterman, J.U.; Hersh, E.M. Clinical and immunological study of beta interferon by intramuscular route in patients with metastatic breast cancer. J. Interferon. Res. 1982, 2, 593–599. [Google Scholar] [CrossRef]
- Chaplinski, T.; Laszlo, J.; Moore, J.; Silverman, P. Phase II trial of lymphoblastoid interferon in metastatic colon carcinoma. Cancer Treat. Rep. 1983, 67, 1009–1012. [Google Scholar]
- Lienard, D.; Ewalenko, P.; Delmotte, J.J.; Renard, N.; Lejeune, F.J. High-dose recombinant tumor necrosis factor alpha in combination with interferon gamma and melphalan in isolation perfusion of the limbs for melanoma and sarcoma. J. Clin. Oncol. 1992, 10, 52–60. [Google Scholar] [CrossRef]
- Ruegg, C.; Yilmaz, A.; Bieler, G.; Bamat, J.; Chaubert, P.; Lejeune, F.J. Evidence for the involvement of endothelial cell integrin alphaVbeta3 in the disruption of the tumor vasculature induced by TNF and IFN-gamma. Nat. Med. 1998, 4, 408–414. [Google Scholar] [CrossRef]
- Braumuller, H.; Wieder, T.; Brenner, E.; Assmann, S.; Hahn, M.; Alkhaled, M.; Schilbach, K.; Essmann, F.; Kneilling, M.; Griessinger, C.; et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013, 494, 361–365. [Google Scholar] [CrossRef]
- Duggan, M.C.; Jochems, C.; Donahue, R.N.; Richards, J.; Karpa, V.; Foust, E.; Paul, B.; Brooks, T.; Tridandapani, S.; Olencki, T.; et al. A phase I study of recombinant (r) vaccinia-CEA(6D)-TRICOM and rFowlpox-CEA(6D)-TRICOM vaccines with GM-CSF and IFN-alpha-2b in patients with CEA-expressing carcinomas. Cancer Immunol. Immunother. 2016, 65, 1353–1364. [Google Scholar] [CrossRef]
- Pawelec, G.; Borowitz, A.; Krammer, P.H.; Wernet, P. Constitutive interleukin 2 production by the JURKAT human leukemic T cell line. Eur. J. Immunol. 1982, 12, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, A.; Rosenberg, S.A. Successful immunotherapy of natural killer-resistant established pulmonary melanoma metastases by the intravenous adoptive transfer of syngeneic lymphocytes activated in vitro by interleukin 2. J. Exp. Med. 1984, 159, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Wolf, I.G.; Finke, S.; Trojaneck, B.; Denkena, A.; Lefterova, P.; Schwella, N.; Heuft, H.G.; Prange, G.; Korte, M.; Takeya, M.; et al. Phase I clinical study applying autologous immunological effector cells transfected with the interleukin-2 gene in patients with metastatic renal cancer, colorectal cancer and lymphoma. Br. J. Cancer 1999, 81, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Wittig, B.; Marten, A.; Dorbic, T.; Weineck, S.; Min, H.; Niemitz, S.; Trojaneck, B.; Flieger, D.; Kruopis, S.; Albers, A.; et al. Therapeutic vaccination against metastatic carcinoma by expression-modulated and immunomodified autologous tumor cells: A first clinical phase I/II trial. Hum. Gene Ther. 2001, 12, 267–278. [Google Scholar] [CrossRef]
- Caporale, A.; Brescia, A.; Galati, G.; Castelli, M.; Saputo, S.; Terrenato, I.; Cucina, A.; Liverani, A.; Gasparrini, M.; Ciardi, A.; et al. Locoregional IL-2 therapy in the treatment of colon cancer. Cell-induced lesions of a murine model. Anticancer Res. 2007, 27, 985–989. [Google Scholar]
- Hutmacher, C.; Gonzalo Nunez, N.; Liuzzi, A.R.; Becher, B.; Neri, D. Targeted Delivery of IL2 to the Tumor Stroma Potentiates the Action of Immune Checkpoint Inhibitors by Preferential Activation of NK and CD8(+) T Cells. Cancer Immunol. Res. 2019, 7, 572–583. [Google Scholar] [CrossRef]
- Campian, J.L.; Ghosh, S.; Kapoor, V.; Yan, R.; Thotala, S.; Jash, A.; Hu, T.; Mahadevan, A.; Rifai, K.; Page, L.; et al. Long-Acting Recombinant Human Interleukin-7, NT-I7, Increases Cytotoxic CD8 T Cells and Enhances Survival in Mouse Glioma Models. Clin. Cancer Res. 2022, 28, 1229–1239. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braumüller, H.; Mauerer, B.; Andris, J.; Berlin, C.; Wieder, T.; Kesselring, R. The Cytokine Network in Colorectal Cancer: Implications for New Treatment Strategies. Cells 2023, 12, 138. https://doi.org/10.3390/cells12010138
Braumüller H, Mauerer B, Andris J, Berlin C, Wieder T, Kesselring R. The Cytokine Network in Colorectal Cancer: Implications for New Treatment Strategies. Cells. 2023; 12(1):138. https://doi.org/10.3390/cells12010138
Chicago/Turabian StyleBraumüller, Heidi, Bernhard Mauerer, Johanna Andris, Christopher Berlin, Thomas Wieder, and Rebecca Kesselring. 2023. "The Cytokine Network in Colorectal Cancer: Implications for New Treatment Strategies" Cells 12, no. 1: 138. https://doi.org/10.3390/cells12010138
APA StyleBraumüller, H., Mauerer, B., Andris, J., Berlin, C., Wieder, T., & Kesselring, R. (2023). The Cytokine Network in Colorectal Cancer: Implications for New Treatment Strategies. Cells, 12(1), 138. https://doi.org/10.3390/cells12010138