

The Role of PPARs in Breast Cancer


Simple Summary
Abstract
1. Introduction
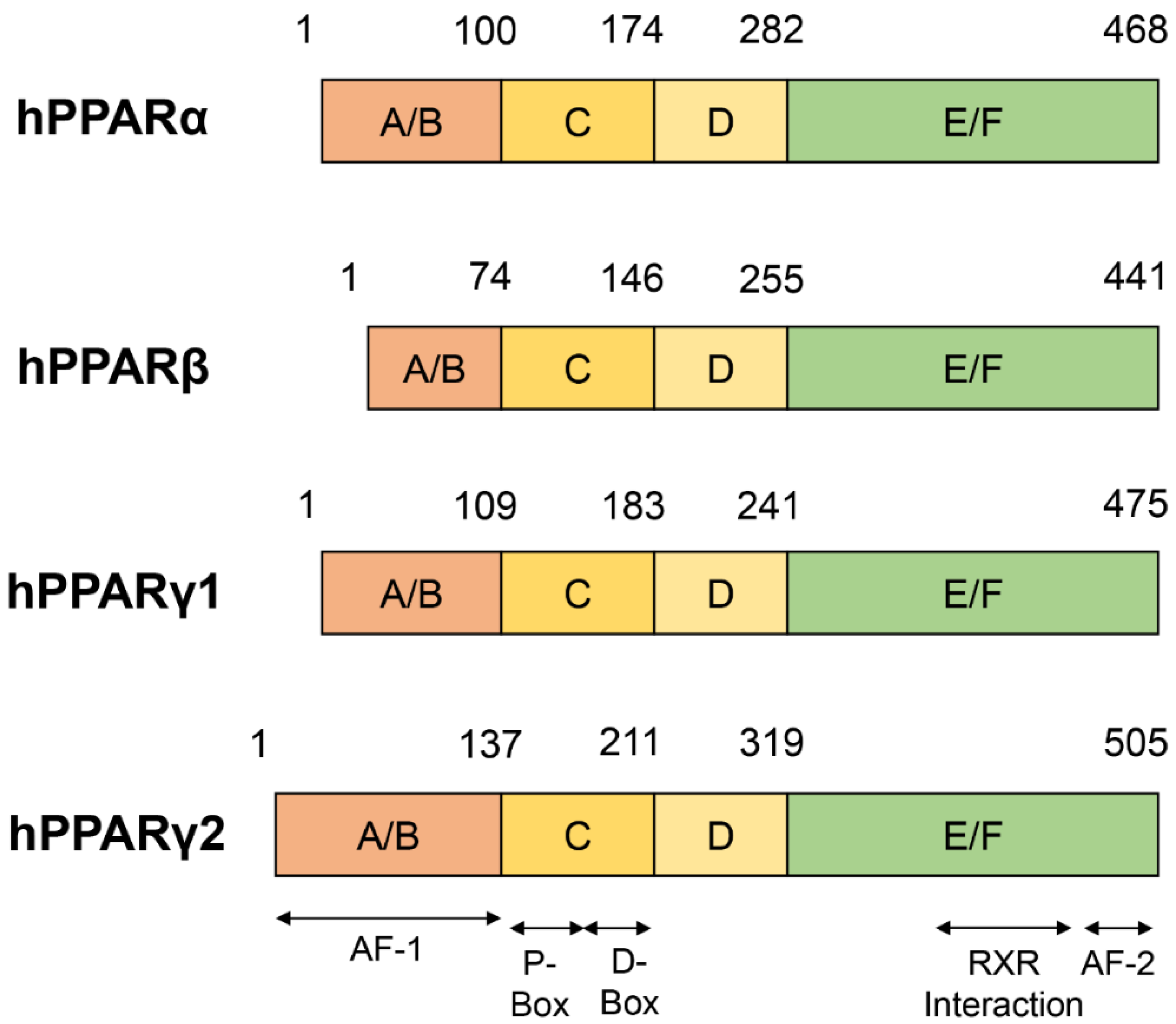
2. Structure of PPARs
3. Ligands for PPARs
3.1. Agonists and Antagonists of PPARα
3.2. Agonists and Antagonists of PPARβ/δ
3.3. Agonists and Antagonists of PPARγ

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PPARs | Agonists | Antagonists | |
|---|---|---|---|
| PPARα | fibrates | Bezafibrate [49] | GW6471 [56] |
| Fenofibrate [49] | L-663,536 [59] | ||
| Clofibrate [49] | |||
| Gemfibrozil [49] | |||
| Wy-14,643 [52] | |||
| GW9578 [54] | |||
| GW7647 [55] | |||
| PPARβ/δ | L-165,041 [60] | GW9662 [69] | |
| GW501,516 [62] | GSK0660 [29] | ||
| GW0742 [66] | SR13,904 [70] | ||
| MBX-8025/RWJ80,025 [67] | GSK3787 [71] | ||
| KD-3010 [68] | DG172 [72] | ||
| PT-S58 [73,74] | |||
| PPARγ | TZDs | rosiglitazone (RGZ) [75] | GW9662 [80,81] |
| pioglitazone(PGZ) [76] | T0,070,907 [82] | ||
| ciglitazone(CGZ) [76] | BADGE [83] | ||
| troglitazone(TGZ) [76] | |||
| englitazone(EGZ) [76] | |||
| balaglitazone(BGZ) [76] | |||
| L-764,406 [77] | |||
| GW0072 [78] | |||
| GW7845 [79] | |||
3.4. Structure of PPARs Ligands
4. Subtypes of PPARs and Breast Cancer
4.1. PPARα and Breast Cancer
4.2. PPARβ/δ and Breast Cancer
4.3. PPARγ and Breast Cancer
4.4. PPARs and TNBC
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Escher, P.; Wahli, W. Peroxisome proliferator-activated receptors: Insight into multiple cellular functions. Mutat. Res. 2000, 448, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef] [PubMed]
- Polvani, S.; Tarocchi, M.; Tempesti, S.; Bencini, L.; Galli, A. Peroxisome proliferator activated receptors at the crossroad of obesity, diabetes, and pancreatic cancer. World J. Gastroenterol. 2016, 22, 2441–2459. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-X. PPARs: Diverse regulators in energy metabolism and metabolic diseases. Cell Res. 2010, 20, 124–137. [Google Scholar] [CrossRef]
- Han, L.; Shen, W.-J.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-α. Future Cardiol. 2017, 13, 259–278. [Google Scholar] [CrossRef]
- Han, L.; Shen, W.-J.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part II: PPAR-β/δ and PPAR-γ. Future Cardiol. 2017, 13, 279–296. [Google Scholar] [CrossRef]
- Burns, K.A.; Vanden Heuvel, J.P. Modulation of PPAR activity via phosphorylation. Biochim. Biophys. Acta 2007, 1771, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Castillo, G.; Brun, R.P.; Rosenfield, J.K.; Hauser, S.; Park, C.W.; Troy, A.E.; Wright, M.E.; Spiegelman, B.M. An adipogenic cofactor bound by the differentiation domain of PPARgamma. EMBO J. 1999, 18, 3676–3687. [Google Scholar] [CrossRef] [PubMed]
- Hummasti, S.; Tontonoz, P. The peroxisome proliferator-activated receptor N-terminal domain controls isotype-selective gene expression and adipogenesis. Mol. Endocrinol. 2006, 20, 1261–1275. [Google Scholar] [CrossRef] [PubMed]
- Bugge, A.; Grøntved, L.; Aagaard, M.M.; Borup, R.; Mandrup, S. The PPARgamma2 A/B-domain plays a gene-specific role in transactivation and cofactor recruitment. Mol. Endocrinol. 2009, 23, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Gampe, R.T.J.; Montana, V.G.; Lambert, M.H.; Miller, A.B.; Bledsoe, R.K.; Milburn, M.V.; Kliewer, S.A.; Willson, T.M.; Xu, H.E. Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol. Cell 2000, 5, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Nolte, R.T.; Wisely, G.B.; Westin, S.; Cobb, J.E.; Lambert, M.H.; Kurokawa, R.; Rosenfeld, M.G.; Willson, T.M.; Glass, C.K.; Milburn, M.V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Sheu, S.-H.; Kaya, T.; Waxman, D.J.; Vajda, S. Exploring the binding site structure of the PPAR gamma ligand-binding domain by computational solvent mapping. Biochemistry 2005, 44, 1193–1209. [Google Scholar] [CrossRef]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Parks, D.J.; Blanchard, S.G.; Brown, P.J.; Sternbach, D.D.; Lehmann, J.M.; Wisely, G.B.; Willson, T.M.; et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell 1999, 3, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Rangwala, S.M.; Bailey, S.T.; Krakow, S.L.; Reginato, M.J.; Lazar, M.A. Interdomain communication regulating ligand binding by PPAR-gamma. Nature 1998, 396, 377–380. [Google Scholar] [CrossRef]
- Deeb, S.S.; Fajas, L.; Nemoto, M.; Pihlajamäki, J.; Mykkänen, L.; Kuusisto, J.; Laakso, M.; Fujimoto, W.; Auwerx, J. A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat. Genet. 1998, 20, 284–287. [Google Scholar] [CrossRef]
- Lee, C.-H.; Chawla, A.; Urbiztondo, N.; Liao, D.; Boisvert, W.A.; Evans, R.M.; Curtiss, L.K. Transcriptional repression of atherogenic inflammation: Modulation by PPARdelta. Science 2003, 302, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.M.; McDonnell, D.P. Coregulators in nuclear estrogen receptor action: From concept to therapeutic targeting. Mol. Interv. 2005, 5, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Göttlicher, M.; Widmark, E.; Li, Q.; Gustafsson, J.A. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 1992, 89, 4653–4657. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Ziouzenkova, O.; Perrey, S.; Asatryan, L.; Hwang, J.; MacNaul, K.L.; Moller, D.E.; Rader, D.J.; Sevanian, A.; Zechner, R.; Hoefler, G.; et al. Lipolysis of triglyceride-rich lipoproteins generates PPAR ligands: Evidence for an antiinflammatory role for lipoprotein lipase. Proc. Natl. Acad. Sci. USA 2003, 100, 2730–2735. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Lee, C.-H.; Barak, Y.; He, W.; Rosenfeld, J.; Liao, D.; Han, J.; Kang, H.; Evans, R.M. PPARdelta is a very low-density lipoprotein sensor in macrophages. Proc. Natl. Acad. Sci. USA 2003, 100, 1268–1273. [Google Scholar] [CrossRef]
- Yu, K.; Bayona, W.; Kallen, C.B.; Harding, H.P.; Ravera, C.P.; McMahon, G.; Brown, M.; Lazar, M.A. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J. Biol. Chem. 1995, 270, 23975–23983. [Google Scholar] [CrossRef]
- Willson, T.M.; Brown, P.J.; Sternbach, D.D.; Henke, B.R. The PPARs: From orphan receptors to drug discovery. J. Med. Chem. 2000, 43, 527–550. [Google Scholar] [CrossRef]
- Shearer, B.G.; Steger, D.J.; Way, J.M.; Stanley, T.B.; Lobe, D.C.; Grillot, D.A.; Iannone, M.A.; Lazar, M.A.; Willson, T.M.; Billin, A.N. Identification and characterization of a selective peroxisome proliferator-activated receptor beta/delta (NR1C2) antagonist. Mol. Endocrinol. 2008, 22, 523–529. [Google Scholar] [CrossRef]
- Palkar, P.S.; Borland, M.G.; Naruhn, S.; Ferry, C.H.; Lee, C.; Sk, U.H.; Sharma, A.K.; Amin, S.; Murray, I.A.; Anderson, C.R.; et al. Cellular and pharmacological selectivity of the peroxisome proliferator-activated receptor-beta/delta antagonist GSK3787. Mol. Pharmacol. 2010, 78, 419–430. [Google Scholar] [CrossRef]
- Dowell, P.; Peterson, V.J.; Zabriskie, T.M.; Leid, M. Ligand-induced peroxisome proliferator-activated receptor alpha conformational change. J. Biol. Chem. 1997, 272, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- Tien, E.S.; Hannon, D.B.; Thompson, J.T.; Vanden Heuvel, J.P. Examination of Ligand-Dependent Coactivator Recruitment by Peroxisome Proliferator-Activated Receptor-alpha (PPARalpha). PPAR Res. 2006, 2006, 69612. [Google Scholar] [CrossRef] [PubMed]
- Sumanasekera, W.K.; Tien, E.S.; Turpey, R.; Vanden Heuvel, J.P.; Perdew, G.H. Evidence that peroxisome proliferator-activated receptor alpha is complexed with the 90-kDa heat shock protein and the hepatitis virus B X-associated protein 2. J. Biol. Chem. 2003, 278, 4467–4473. [Google Scholar] [CrossRef] [PubMed]
- Sumanasekera, W.K.; Tien, E.S.; Davis, J.W., 2nd; Turpey, R.; Perdew, G.H.; Vanden Heuvel, J.P. Heat shock protein-90 (Hsp90) acts as a repressor of peroxisome proliferator-activated receptor-alpha (PPARalpha) and PPARbeta activity. Biochemistry 2003, 42, 10726–10735. [Google Scholar] [CrossRef]
- Akiyama, T.E.; Baumann, C.T.; Sakai, S.; Hager, G.L.; Gonzalez, F.J. Selective intranuclear redistribution of PPAR isoforms by RXR alpha. Mol. Endocrinol. 2002, 16, 707–721. [Google Scholar] [CrossRef]
- Patel, H.; Truant, R.; Rachubinski, R.A.; Capone, J.P. Activity and subcellular compartmentalization of peroxisome proliferator-activated receptor alpha are altered by the centrosome-associated protein CAP350. J. Cell Sci. 2005, 118, 175–186. [Google Scholar] [CrossRef]
- Malek, M.A.; Hoang, M.-H.; Jia, Y.; Lee, J.H.; Jun, H.J.; Lee, D.-H.; Lee, H.J.; Lee, C.; Lee, M.K.; Hwang, B.Y.; et al. Ombuin-3-O-β-D-glucopyranoside from Gynostemma pentaphyllum is a dual agonistic ligand of peroxisome proliferator-activated receptors α and δ/β. Biochem. Biophys. Res. Commun. 2013, 430, 1322–1328. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z. Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: The bezafibrate lessons. Cardiovasc. Diabetol. 2005, 4, 14. [Google Scholar] [CrossRef]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.; Chen, H.; Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef]
- Shureiqi, I.; Jiang, W.; Zuo, X.; Wu, Y.; Stimmel, J.B.; Leesnitzer, L.M.; Morris, J.S.; Fan, H.-Z.; Fischer, S.M.; Lippman, S.M. The 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid down-regulates PPAR-delta to induce apoptosis in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9968–9973. [Google Scholar] [CrossRef]
- Coleman, J.D.; Prabhu, K.S.; Thompson, J.T.; Reddy, P.S.; Peters, J.M.; Peterson, B.R.; Reddy, C.C.; Vanden Heuvel, J.P. The oxidative stress mediator 4-hydroxynonenal is an intracellular agonist of the nuclear receptor peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta). Free Radic. Biol. Med. 2007, 42, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Rubenstrunk, A.; Hanf, R.; Hum, D.W.; Fruchart, J.-C.; Staels, B. Safety issues and prospects for future generations of PPAR modulators. Biochim. Biophys. Acta 2007, 1771, 1065–1081. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, A.; Boyko, V.; Fisman, E.Z.; Goldenberg, I.; Adler, Y.; Feinberg, M.S.; Motro, M.; Tanne, D.; Shemesh, J.; Schwammenthal, E.; et al. Does the lipid-lowering peroxisome proliferator-activated receptors ligand bezafibrate prevent colon cancer in patients with coronary artery disease? Cardiovasc. Diabetol. 2008, 7, 18. [Google Scholar] [CrossRef]
- Yaturu, S.; Bryant, B.; Jain, S.K. Thiazolidinedione treatment decreases bone mineral density in type 2 diabetic men. Diabetes Care 2007, 30, 1574–1576. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grey, A.; Bolland, M.; Gamble, G.; Wattie, D.; Horne, A.; Davidson, J.; Reid, I.R. The peroxisome proliferator-activated receptor-gamma agonist rosiglitazone decreases bone formation and bone mineral density in healthy postmenopausal women: A randomized, controlled trial. J. Clin. Endocrinol. Metab. 2007, 92, 1305–1310. [Google Scholar] [CrossRef]
- Schwartz, A.V.; Sellmeyer, D.E. Thiazolidinedione therapy gets complicated: Is bone loss the price of improved insulin resistance? Diabetes Care 2007, 30, 1670–1671. [Google Scholar] [CrossRef]
- Schwartz, A.V.; Sellmeyer, D.E.; Vittinghoff, E.; Palermo, L.; Lecka-Czernik, B.; Feingold, K.R.; Strotmeyer, E.S.; Resnick, H.E.; Carbone, L.; Beamer, B.A.; et al. Thiazolidinedione use and bone loss in older diabetic adults. J. Clin. Endocrinol. Metab. 2006, 91, 3349–3354. [Google Scholar] [CrossRef]
- Still, K.; Grabowski, P.; Mackie, I.; Perry, M.; Bishop, N. The peroxisome proliferator activator receptor alpha/delta agonists linoleic acid and bezafibrate upregulate osteoblast differentiation and induce periosteal bone formation in vivo. Calcif. Tissue Int. 2008, 83, 285–292. [Google Scholar] [CrossRef]
- Rakhshandehroo, M.; Knoch, B.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 2010, 612089. [Google Scholar] [CrossRef]
- Santilli, A.A.; Scotese, A.C.; Tomarelli, R.M. A potent antihypercholesterolemic agent: (4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio) acetic acid (Wy-14643). Experientia 1974, 30, 1110–1111. [Google Scholar] [CrossRef]
- Reddy, J.K.; Moody, D.E.; Azarnoff, D.L.; Tomarelli, R.M. Hepatic effects of some [4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio] acetic acid (WY-14,643) analogs in the mouse. Arch. Int. Pharmacodyn. Ther. 1977, 225, 51–57. [Google Scholar]
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 7355–7359. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Gao, Y.; Qu, A.; Jiang, Y.; Li, H.; Xie, G.; Yao, X.; Yang, X.; Zhu, S.; Yagai, T.; et al. YAP-TEAD mediates PPAR α-induced hepatomegaly and liver regeneration in mice. Hepatology 2022, 75, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.J.; Winegar, D.A.; Plunket, K.D.; Moore, L.B.; Lewis, M.C.; Wilson, J.G.; Sundseth, S.S.; Koble, C.S.; Wu, Z.; Chapman, J.M.; et al. A ureido-thioisobutyric acid (GW9578) is a subtype-selective PPARalpha agonist with potent lipid-lowering activity. J. Med. Chem. 1999, 42, 3785–3788. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.J.; Stuart, L.W.; Hurley, K.P.; Lewis, M.C.; Winegar, D.A.; Wilson, J.G.; Wilkison, W.O.; Ittoop, O.R.; Willson, T.M. Identification of a subtype selective human PPARα agonist through parallel-array synthesis. Bioorg. Med. Chem. Lett. 2001, 11, 1225–1227. [Google Scholar] [CrossRef]
- Xu, H.E.; Stanley, T.B.; Montana, V.G.; Lambert, M.H.; Shearer, B.G.; Cobb, J.E.; McKee, D.D.; Galardi, C.M.; Plunket, K.D.; Nolte, R.T.; et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature 2002, 415, 813–817. [Google Scholar] [CrossRef]
- Guhlmann, A.; Keppler, A.; Kästner, S.; Krieter, H.; Brückner, U.B.; Messmer, K.; Keppler, D. Prevention of endogenous leukotriene production during anaphylaxis in the guinea pig by an inhibitor of leukotriene biosynthesis (MK-886) but not by dexamethasone. J. Exp. Med. 1989, 170, 1905–1918. [Google Scholar] [CrossRef]
- Datta, K.; Biswal, S.S.; Kehrer, J.P. The 5-lipoxygenase-activating protein (FLAP) inhibitor, MK886, induces apoptosis independently of FLAP. Biochem. J. 1999, 340, 371–375. [Google Scholar] [CrossRef]
- Kehrer, J.P.; Biswal, S.S.; La, E.; Thuillier, P.; Datta, K.; Fischer, S.M.; Vanden Heuvel, J.P. Inhibition of peroxisome-proliferator-activated receptor (PPAR)alpha by MK886. Biochem. J. 2001, 356, 899–906. [Google Scholar] [CrossRef]
- Leibowitz, M.D.; Fiévet, C.; Hennuyer, N.; Peinado-Onsurbe, J.; Duez, H.; Bergera, J.; Cullinan, C.A.; Sparrow, C.P.; Baffic, J.; Berger, G.D.; et al. Activation of PPARdelta alters lipid metabolism in db/db mice. FEBS Lett. 2000, 473, 333–336. [Google Scholar] [CrossRef]
- Westergaard, M.; Henningsen, J.; Svendsen, M.L.; Johansen, C.; Jensen, U.B.; Schrøder, H.D.; Kratchmarova, I.; Berge, R.K.; Iversen, L.; Bolund, L.; et al. Modulation of keratinocyte gene expression and differentiation by PPAR-selective ligands and tetradecylthioacetic acid. J. Investig. Dermatol. 2001, 116, 702–712. [Google Scholar] [CrossRef]
- Oliver, W.R.J.; Shenk, J.L.; Snaith, M.R.; Russell, C.S.; Plunket, K.D.; Bodkin, N.L.; Lewis, M.C.; Winegar, D.A.; Sznaidman, M.L.; Lambert, M.H.; et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. USA 2001, 98, 5306–5311. [Google Scholar] [CrossRef] [PubMed]
- Pelton, P. GW-501516 GlaxoSmithKline/Ligand. Curr. Opin. Investig. Drugs 2006, 7, 360–370. [Google Scholar] [PubMed]
- Iwaisako, K.; Haimerl, M.; Paik, Y.-H.; Taura, K.; Kodama, Y.; Sirlin, C.; Yu, E.; Yu, R.T.; Downes, M.; Evans, R.M.; et al. Protection from liver fibrosis by a peroxisome proliferator-activated receptor δ agonist. Proc. Natl. Acad. Sci. USA 2012, 109, E1369–E1376. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.; Schubert-Zsilavecz, M.; Merk, D. Therapeutic modulators of peroxisome proliferator-activated receptors (PPAR): A patent review (2008-present). Expert Opin. Ther. Pat. 2012, 22, 803–841. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Zhu, X.; McLntee, F.L.; Xiao, H.; Zhang, J.; Fu, M.; Chen, Y.E. Interferon regulatory factor-1 mediates PPARgamma-induced apoptosis in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-J.; Roberts, B.K.; Wang, X.; Geaney, J.C.; Naim, S.; Wojnoonski, K.; Karpf, D.B.; Krauss, R.M. Effects of the PPAR-δ agonist MBX-8025 on atherogenic dyslipidemia. Atherosclerosis 2012, 220, 470–476. [Google Scholar] [CrossRef]
- Billin, A.N. PPAR-beta/delta agonists for Type 2 diabetes and dyslipidemia: An adopted orphan still looking for a home. Expert Opin. Investig. Drugs 2008, 17, 1465–1471. [Google Scholar] [CrossRef] [PubMed]
- Seimandi, M.; Lemaire, G.; Pillon, A.; Perrin, A.; Carlavan, I.; Voegel, J.J.; Vignon, F.; Nicolas, J.-C.; Balaguer, P. Differential responses of PPARalpha, PPARdelta, and PPARgamma reporter cell lines to selective PPAR synthetic ligands. Anal. Biochem. 2005, 344, 8–15. [Google Scholar] [CrossRef]
- Zaveri, N.T.; Sato, B.G.; Jiang, F.; Calaoagan, J.; Laderoute, K.R.; Murphy, B.J. A novel peroxisome proliferator-activated receptor delta antagonist, SR13904, has anti-proliferative activity in human cancer cells. Cancer Biol. Ther. 2009, 8, 1252–1261. [Google Scholar] [CrossRef]
- Shearer, B.G.; Wiethe, R.W.; Ashe, A.; Billin, A.N.; Way, J.M.; Stanley, T.B.; Wagner, C.D.; Xu, R.X.; Leesnitzer, L.M.; Merrihew, R.V.; et al. Identification and characterization of 4-chloro-N-(2-{[5-trifluoromethyl)-2-pyridyl]sulfonyl}ethyl)benzamide (GSK3787), a selective and irreversible peroxisome proliferator-activated receptor delta (PPARdelta) antagonist. J. Med. Chem. 2010, 53, 1857–1861. [Google Scholar] [CrossRef] [PubMed]
- Lieber, S.; Scheer, F.; Meissner, W.; Naruhn, S.; Adhikary, T.; Müller-Brüsselbach, S.; Diederich, W.E.; Müller, R. (Z)-2-(2-bromophenyl)-3-{[4-(1-methyl-piperazine)amino]phenyl}acrylonitrile (DG172): An orally bioavailable PPARβ/δ-selective ligand with inverse agonistic properties. J. Med. Chem. 2012, 55, 2858–2868. [Google Scholar] [CrossRef] [PubMed]
- Levi, L.; Lobo, G.; Doud, M.K.; von Lintig, J.; Seachrist, D.; Tochtrop, G.P.; Noy, N. Genetic ablation of the fatty acid-binding protein FABP5 suppresses HER2-induced mammary tumorigenesis. Cancer Res. 2013, 73, 4770–4780. [Google Scholar] [CrossRef] [PubMed]
- Naruhn, S.; Toth, P.M.; Adhikary, T.; Kaddatz, K.; Pape, V.; Dörr, S.; Klebe, G.; Müller-Brüsselbach, S.; Diederich, W.E.; Müller, R. High-affinity peroxisome proliferator-activated receptor β/δ-specific ligands with pure antagonistic or inverse agonistic properties. Mol. Pharmacol. 2011, 80, 828–838. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.Z.; Althagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef]
- Elbrecht, A.; Chen, Y.; Adams, A.; Berger, J.; Griffin, P.; Klatt, T.; Zhang, B.; Menke, J.; Zhou, G.; Smith, R.G.; et al. L-764406 is a partial agonist of human peroxisome proliferator-activated receptor gamma. The role of Cys313 in ligand binding. J. Biol. Chem. 1999, 274, 7913–7922. [Google Scholar] [CrossRef]
- Oberfield, J.L.; Collins, J.L.; Holmes, C.P.; Goreham, D.M.; Cooper, J.P.; Cobb, J.E.; Lenhard, J.M.; Hull-Ryde, E.A.; Mohr, C.P.; Blanchard, S.G.; et al. A peroxisome proliferator-activated receptor gamma ligand inhibits adipocyte differentiation. Proc. Natl. Acad. Sci. USA 1999, 96, 6102–6106. [Google Scholar] [CrossRef]
- Suh, N.; Wang, Y.; Williams, C.R.; Risingsong, R.; Gilmer, T.; Willson, T.M.; Sporn, M.B. A new ligand for the peroxisome proliferator-activated receptor-gamma (PPAR-gamma), GW7845, inhibits rat mammary carcinogenesis. Cancer Res. 1999, 59, 5671–5673. [Google Scholar]
- Miyahara, T.; Schrum, L.; Rippe, R.; Xiong, S.; Yee, H.F.J.; Motomura, K.; Anania, F.A.; Willson, T.M.; Tsukamoto, H. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J. Biol. Chem. 2000, 275, 35715–35722. [Google Scholar] [CrossRef]
- Leesnitzer, L.M.; Parks, D.J.; Bledsoe, R.K.; Cobb, J.E.; Collins, J.L.; Consler, T.G.; Davis, R.G.; Hull-Ryde, E.A.; Lenhard, J.M.; Patel, L.; et al. Functional consequences of cysteine modification in the ligand binding sites of peroxisome proliferator activated receptors by GW9662. Biochemistry 2002, 41, 6640–6650. [Google Scholar] [CrossRef]
- Ji, J.; Xue, T.-F.; Guo, X.-D.; Yang, J.; Guo, R.-B.; Wang, J.; Huang, J.-Y.; Zhao, X.-J.; Sun, X.-L. Antagonizing peroxisome proliferator-activated receptor γ facilitates M1-to-M2 shift of microglia by enhancing autophagy via the LKB1-AMPK signaling pathway. Aging Cell 2018, 17, e12774. [Google Scholar] [CrossRef] [PubMed]
- Ghoochani, A.; Shabani, K.; Peymani, M.; Ghaedi, K.; Karamali, F.; Karbalaei, K.; Tanhaie, S.; Salamian, A.; Esmaeili, A.; Valian-Borujeni, S.; et al. The influence of peroxisome proliferator-activated receptor γ(1) during differentiation of mouse embryonic stem cells to neural cells. Differentiation 2012, 83, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.M.; Clish, C.B.; Mikami, T.; Hauser, S.; Yanagi, K.; Hiramatsu, R.; Serhan, C.N.; Spiegelman, B.M. A synthetic antagonist for the peroxisome proliferator-activated receptor gamma inhibits adipocyte differentiation. J. Biol. Chem. 2000, 275, 1873–1877. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Annu. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Zhang, M.; Li, Y.; Liu, Y.; Cui, Q.; Wang, N. PPARgene: A Database of Experimentally Verified and Computationally Predicted PPAR Target Genes. PPAR Res. 2016, 2016, 6042162. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Foreman, J.E.; Gonzalez, F.J. Dissecting the role of peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) in colon, breast, and lung carcinogenesis. Cancer Metastasis Rev. 2011, 30, 619–640. [Google Scholar] [CrossRef]
- Tontonoz, P.; Spiegelman, B.M. Fat and beyond: The diverse biology of PPARgamma. Annu. Rev. Biochem. 2008, 77, 289–312. [Google Scholar] [CrossRef]
- Escher, P.; Braissant, O.; Basu-Modak, S.; Michalik, L.; Wahli, W.; Desvergne, B. Rat PPARs: Quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology 2001, 142, 4195–4202. [Google Scholar] [CrossRef]
- Pyper, S.R.; Viswakarma, N.; Yu, S.; Reddy, J.K. PPARalpha: Energy combustion, hypolipidemia, inflammation and cancer. Nucl. Recept. Signal. 2010, 8, e002. [Google Scholar] [CrossRef]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Balfour, J.A.; McTavish, D.; Heel, R.C. Fenofibrate. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in dyslipidaemia. Drugs 1990, 40, 260–290. [Google Scholar] [CrossRef] [PubMed]
- Reddy, J.K.; Azarnoff, D.L.; Hignite, C.E. Hypolipidaemic hepatic peroxisome proliferators form a novel class of chemical carcinogens. Nature 1980, 283, 397–398. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Cattley, R.C.; Gonzalez, F.J. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis 1997, 18, 2029–2033. [Google Scholar] [CrossRef]
- Hays, T.; Rusyn, I.; Burns, A.M.; Kennett, M.J.; Ward, J.M.; Gonzalez, F.J.; Peters, J.M. Role of peroxisome proliferator-activated receptor-alpha (PPARalpha) in bezafibrate-induced hepatocarcinogenesis and cholestasis. Carcinogenesis 2005, 26, 219–227. [Google Scholar] [CrossRef]
- Peters, J.M.; Cheung, C.; Gonzalez, F.J. Peroxisome proliferator-activated receptor-alpha and liver cancer: Where do we stand? J. Mol. Med. 2005, 83, 774–785. [Google Scholar] [CrossRef]
- Shah, Y.M.; Morimura, K.; Yang, Q.; Tanabe, T.; Takagi, M.; Gonzalez, F.J. Peroxisome proliferator-activated receptor alpha regulates a microRNA-mediated signaling cascade responsible for hepatocellular proliferation. Mol. Cell. Biol. 2007, 27, 4238–4247. [Google Scholar] [CrossRef]
- Cheung, C.; Akiyama, T.E.; Ward, J.M.; Nicol, C.J.; Feigenbaum, L.; Vinson, C.; Gonzalez, F.J. Diminished hepatocellular proliferation in mice humanized for the nuclear receptor peroxisome proliferator-activated receptor alpha. Cancer Res. 2004, 64, 3849–3854. [Google Scholar] [CrossRef]
- Morimura, K.; Cheung, C.; Ward, J.M.; Reddy, J.K.; Gonzalez, F.J. Differential susceptibility of mice humanized for peroxisome proliferator-activated receptor alpha to Wy-14,643-induced liver tumorigenesis. Carcinogenesis 2006, 27, 1074–1080. [Google Scholar] [CrossRef]
- Holland, C.M.; Saidi, S.A.; Evans, A.L.; Sharkey, A.M.; Latimer, J.A.; Crawford, R.A.F.; Charnock-Jones, D.S.; Print, C.G.; Smith, S.K. Transcriptome analysis of endometrial cancer identifies peroxisome proliferator-activated receptors as potential therapeutic targets. Mol. Cancer Ther. 2004, 3, 993–1001. [Google Scholar] [CrossRef]
- Nickkho-Amiry, M.; McVey, R.; Holland, C. Peroxisome proliferator-activated receptors modulate proliferation and angiogenesis in human endometrial carcinoma. Mol. Cancer Res. 2012, 10, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Zhao, B. Cytotoxic effect of peroxisome proliferator fenofibrate on human HepG2 hepatoma cell line and relevant mechanisms. Toxicol. Appl. Pharmacol. 2002, 185, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.; Plonka, P.M.; Urbanska, K.; Reiss, K. Peroxisome proliferator-activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin. Cancer Res. 2006, 12, 3028–3036. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.-W.; Wu, C.-T.; Chen, D.-R.; Yeh, C.-Y.; Lin, C. High levels of arachidonic acid and peroxisome proliferator-activated receptor-alpha in breast cancer tissues are associated with promoting cancer cell proliferation. J. Nutr. Biochem. 2013, 24, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Meng, Q.; Zhang, X.; Cui, Q.; Zhou, W.; Li, S. Design and Synthesis of New α-Naphthoflavones as Cytochrome P450 (CYP) 1B1 Inhibitors To Overcome Docetaxel-Resistance Associated with CYP1B1 Overexpression. J. Med. Chem. 2015, 58, 3534–3547. [Google Scholar] [CrossRef]
- Hwang, Y.P.; Won, S.S.; Jin, S.W.; Lee, G.H.; Pham, T.H.; Choi, J.H.; Kang, K.W.; Jeong, H.G. WY-14643 Regulates CYP1B1 Expression through Peroxisome Proliferator-Activated Receptor α-Mediated Signaling in Human Breast Cancer Cells. Int. J. Mol. Sci. 2019, 20, 5928. [Google Scholar] [CrossRef]
- Castelli, V.; Catanesi, M.; Alfonsetti, M.; Laezza, C.; Lombardi, F.; Cinque, B.; Cifone, M.G.; Ippoliti, R.; Benedetti, E.; Cimini, A.; et al. PPARα-Selective Antagonist GW6471 Inhibits Cell Growth in Breast Cancer Stem Cells Inducing Energy Imbalance and Metabolic Stress. Biomedicines 2021, 9, 127. [Google Scholar] [CrossRef]
- Pighetti, G.M.; Novosad, W.; Nicholson, C.; Hitt, D.C.; Hansens, C.; Hollingsworth, A.B.; Lerner, M.L.; Brackett, D.; Lightfoot, S.A.; Gimble, J.M. Therapeutic treatment of DMBA-induced mammary tumors with PPAR ligands. Anticancer Res. 2001, 21, 825–829. [Google Scholar]
- Chandran, K.; Goswami, S.; Sharma-Walia, N. Implications of a peroxisome proliferator-activated receptor alpha (PPARα) ligand clofibrate in breast cancer. Oncotarget 2016, 7, 15577–15599. [Google Scholar] [CrossRef]
- Yin, X.; Teng, X.; Ma, T.; Yang, T.; Zhang, J.; Huo, M.; Liu, W.; Yang, Y.; Yuan, B.; Yu, H.; et al. RUNX2 recruits the NuRD(MTA1)/CRL4B complex to promote breast cancer progression and bone metastasis. Cell Death Differ. 2022, 29, 2203–2217. [Google Scholar] [CrossRef]
- Shi, Y.; Hon, M.; Evans, R.M. The peroxisome proliferator-activated receptor delta, an integrator of transcriptional repression and nuclear receptor signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 2613–2618. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Endo, N.; Rutledge, S.J.; Vogel, R.; Shinar, D.; Rodan, G.A. Identification of a new member of the steroid hormone receptor superfamily that is activated by a peroxisome proliferator and fatty acids. Mol. Endocrinol. 1992, 6, 1634–1641. [Google Scholar] [CrossRef] [PubMed]
- Amri, E.Z.; Bonino, F.; Ailhaud, G.; Abumrad, N.A.; Grimaldi, P.A. Cloning of a protein that mediates transcriptional effects of fatty acids in preadipocytes. Homology to peroxisome proliferator-activated receptors. J. Biol. Chem. 1995, 270, 2367–2371. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Barish, G.D.; Wang, Y.-X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef]
- Braissant, O.; Foufelle, F.; Scotto, C.; Dauça, M.; Wahli, W. Differential expression of peroxisome proliferator-activated receptors (PPARs): Tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology 1996, 137, 354–366. [Google Scholar] [CrossRef]
- Wang, Y.-X.; Lee, C.-H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef]
- Genini, D.; Catapano, C.V. Block of nuclear receptor ubiquitination. A mechanism of ligand-dependent control of peroxisome proliferator-activated receptor delta activity. J. Biol. Chem. 2007, 282, 11776–11785. [Google Scholar] [CrossRef]
- Wadosky, K.M.; Willis, M.S. The story so far: Post-translational regulation of peroxisome proliferator-activated receptors by ubiquitination and SUMOylation. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H515–H526. [Google Scholar] [CrossRef]
- Rieck, M.; Wedeken, L.; Müller-Brüsselbach, S.; Meissner, W.; Müller, R. Expression level and agonist-binding affect the turnover, ubiquitination and complex formation of peroxisome proliferator activated receptor beta. FEBS J. 2007, 274, 5068–5076. [Google Scholar] [CrossRef] [PubMed]
- Sprecher, D.L.; Massien, C.; Pearce, G.; Billin, A.N.; Perlstein, I.; Willson, T.M.; Hassall, D.G.; Ancellin, N.; Patterson, S.D.; Lobe, D.C.; et al. Triglyceride:high-density lipoprotein cholesterol effects in healthy subjects administered a peroxisome proliferator activated receptor delta agonist. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 359–365. [Google Scholar] [CrossRef]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef] [PubMed]
- de la Monte, S.M.; Wands, J.R. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef]
- de la Monte, S.M.; Tong, M.; Lester-Coll, N.; Plater, M.J.; Wands, J.R. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: Relevance to Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 89–109. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.; Hennuyer, N.; Bouchaert, E.; Rommens, C.; Grillot, D.; Mezdour, H.; Staels, B. Generation and characterization of a humanized PPARδ mouse model. Br. J. Pharmacol. 2011, 164, 192–208. [Google Scholar] [CrossRef][Green Version]
- Liu, S.; Hatano, B.; Zhao, M.; Yen, C.-C.; Kang, K.; Reilly, S.M.; Gangl, M.R.; Gorgun, C.; Balschi, J.A.; Ntambi, J.M.; et al. Role of peroxisome proliferator-activated receptor {delta}/{beta} in hepatic metabolic regulation. J. Biol. Chem. 2011, 286, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Nicol, C.J.; Ito, S.; Bility, M.T.; Kennett, M.J.; Ward, J.M.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor-beta/delta protects against chemically induced liver toxicity in mice. Hepatology 2008, 47, 225–235. [Google Scholar] [CrossRef]
- Peters, J.M.; Gonzalez, F.J. Sorting out the functional role(s) of peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) in cell proliferation and cancer. Biochim. Biophys. Acta 2009, 1796, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Hollingshead, H.E.; Gonzalez, F.J. Role of peroxisome-proliferator-activated receptor beta/delta (PPARbeta/delta) in gastrointestinal tract function and disease. Clin. Sci. 2008, 115, 107–127. [Google Scholar] [CrossRef]
- Tan, N.S.; Michalik, L.; Noy, N.; Yasmin, R.; Pacot, C.; Heim, M.; Flühmann, B.; Desvergne, B.; Wahli, W. Critical roles of PPAR beta/delta in keratinocyte response to inflammation. Genes Dev. 2001, 15, 3263–3277. [Google Scholar] [CrossRef]
- Schmuth, M.; Haqq, C.M.; Cairns, W.J.; Holder, J.C.; Dorsam, S.; Chang, S.; Lau, P.; Fowler, A.J.; Chuang, G.; Moser, A.H.; et al. Peroxisome proliferator-activated receptor (PPAR)-beta/delta stimulates differentiation and lipid accumulation in keratinocytes. J. Investig. Dermatol. 2004, 122, 971–983. [Google Scholar] [CrossRef]
- Kim, D.J.; Bility, M.T.; Billin, A.N.; Willson, T.M.; Gonzalez, F.J.; Peters, J.M. PPARbeta/delta selectively induces differentiation and inhibits cell proliferation. Cell Death Differ. 2006, 13, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Romanowska, M.; Reilly, L.; Palmer, C.N.A.; Gustafsson, M.C.U.; Foerster, J. Activation of PPARbeta/delta causes a psoriasis-like skin disease in vivo. PLoS ONE 2010, 5, e9701. [Google Scholar] [CrossRef] [PubMed]
- Montagner, A.; Delgado, M.B.; Tallichet-Blanc, C.; Chan, J.S.K.; Sng, M.K.; Mottaz, H.; Degueurce, G.; Lippi, Y.; Moret, C.; Baruchet, M.; et al. Src is activated by the nuclear receptor peroxisome proliferator-activated receptor β/δ in ultraviolet radiation-induced skin cancer. EMBO Mol. Med. 2014, 6, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.C.; Tan, M.J.; Philippe, V.; Tan, S.H.; Tan, C.K.; Ku, C.W.; Goh, Y.Y.; Wahli, W.; Michalik, L.; Tan, N.S. Regulation of epithelial-mesenchymal IL-1 signaling by PPARbeta/delta is essential for skin homeostasis and wound healing. J. Cell Biol. 2009, 184, 817–831. [Google Scholar] [CrossRef]
- Yang, L.; Olsson, B.; Pfeifer, D.; Jönsson, J.-I.; Zhou, Z.-G.; Jiang, X.; Fredriksson, B.-A.; Zhang, H.; Sun, X.-F. Knockdown of peroxisome proliferator-activated receptor-beta induces less differentiation and enhances cell-fibronectin adhesion of colon cancer cells. Oncogene 2010, 29, 516–526. [Google Scholar] [CrossRef]
- Marin, H.E.; Peraza, M.A.; Billin, A.N.; Willson, T.M.; Ward, J.M.; Kennett, M.J.; Gonzalez, F.J.; Peters, J.M. Ligand activation of peroxisome proliferator-activated receptor beta inhibits colon carcinogenesis. Cancer Res. 2006, 66, 4394–4401. [Google Scholar] [CrossRef]
- Santos, G.C.; Zielenska, M.; Prasad, M.; Squire, J.A. Chromosome 6p amplification and cancer progression. J. Clin. Pathol. 2007, 60, 1–7. [Google Scholar] [CrossRef]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef]
- Berglund, L.; Björling, E.; Oksvold, P.; Fagerberg, L.; Asplund, A.; Szigyarto, C.A.-K.; Persson, A.; Ottosson, J.; Wernérus, H.; Nilsson, P.; et al. A genecentric Human Protein Atlas for expression profiles based on antibodies. Mol. Cell. Proteomics 2008, 7, 2019–2027. [Google Scholar] [CrossRef]
- Uhlén, M.; Björling, E.; Agaton, C.; Szigyarto, C.A.-K.; Amini, B.; Andersen, E.; Andersson, A.-C.; Angelidou, P.; Asplund, A.; Asplund, C.; et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteomics 2005, 4, 1920–1932. [Google Scholar] [CrossRef]
- Foreman, J.E.; Sharma, A.K.; Amin, S.; Gonzalez, F.J.; Peters, J.M. Ligand activation of peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) inhibits cell growth in a mouse mammary gland cancer cell line. Cancer Lett. 2010, 288, 219–225. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kittler, R.; Zhou, J.; Hua, S.; Ma, L.; Liu, Y.; Pendleton, E.; Cheng, C.; Gerstein, M.; White, K.P. A comprehensive nuclear receptor network for breast cancer cells. Cell Rep. 2013, 3, 538–551. [Google Scholar] [CrossRef][Green Version]
- Stephen, R.L.; Gustafsson, M.C.U.; Jarvis, M.; Tatoud, R.; Marshall, B.R.; Knight, D.; Ehrenborg, E.; Harris, A.L.; Wolf, C.R.; Palmer, C.N.A. Activation of peroxisome proliferator-activated receptor delta stimulates the proliferation of human breast and prostate cancer cell lines. Cancer Res. 2004, 64, 3162–3170. [Google Scholar] [CrossRef] [PubMed]
- Girroir, E.E.; Hollingshead, H.E.; Billin, A.N.; Willson, T.M.; Robertson, G.P.; Sharma, A.K.; Amin, S.; Gonzalez, F.J.; Peters, J.M. Peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) ligands inhibit growth of UACC903 and MCF7 human cancer cell lines. Toxicology 2008, 243, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.-L.; Morales, J.L.; Zhu, B.; Kang, B.-H.; Gonzalez, F.J.; Peters, J.M. Activation of peroxisome proliferator-activated receptor-β/δ (PPAR-β/δ) inhibits human breast cancer cell line tumorigenicity. Mol. Cancer Ther. 2014, 13, 1008–1017. [Google Scholar] [CrossRef]
- Ghosh, M.; Ai, Y.; Narko, K.; Wang, Z.; Peters, J.M.; Hla, T. PPARdelta is pro-tumorigenic in a mouse model of COX-2-induced mammary cancer. Prostaglandins Other Lipid Mediat. 2009, 88, 97–100. [Google Scholar] [CrossRef][Green Version]
- Yin, Y.; Russell, R.G.; Dettin, L.E.; Bai, R.; Wei, Z.-L.; Kozikowski, A.P.; Kopelovich, L.; Glazer, R.I. Peroxisome proliferator-activated receptor delta and gamma agonists differentially alter tumor differentiation and progression during mammary carcinogenesis. Cancer Res. 2005, 65, 3950–3957. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef]
- Pollock, C.B.; Yin, Y.; Yuan, H.; Zeng, X.; King, S.; Li, X.; Kopelovich, L.; Albanese, C.; Glazer, R.I. PPARδ activation acts cooperatively with 3-phosphoinositide-dependent protein kinase-1 to enhance mammary tumorigenesis. PLoS ONE 2011, 6, e16215. [Google Scholar] [CrossRef]
- Yuan, H.; Lu, J.; Xiao, J.; Upadhyay, G.; Umans, R.; Kallakury, B.; Yin, Y.; Fant, M.E.; Kopelovich, L.; Glazer, R.I. PPARδ induces estrogen receptor-positive mammary neoplasia through an inflammatory and metabolic phenotype linked to mTOR activation. Cancer Res. 2013, 73, 4349–4361. [Google Scholar] [CrossRef]
- Shearer, B.G.; Hoekstra, W.J. Recent advances in peroxisome proliferator-activated receptor science. Curr. Med. Chem. 2003, 10, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Berry, D.C.; Toshkov, I.A.; Cheng, L.; Nikitin, A.Y.; Noy, N. Overcoming retinoic acid-resistance of mammary carcinomas by diverting retinoic acid from PPARbeta/delta to RAR. Proc. Natl. Acad. Sci. USA 2008, 105, 7546–7551. [Google Scholar] [CrossRef] [PubMed]
- Kannan-Thulasiraman, P.; Seachrist, D.D.; Mahabeleshwar, G.H.; Jain, M.K.; Noy, N. Fatty acid-binding protein 5 and PPARbeta/delta are critical mediators of epidermal growth factor receptor-induced carcinoma cell growth. J. Biol. Chem. 2010, 285, 19106–19115. [Google Scholar] [CrossRef]
- Borland, M.G.; Khozoie, C.; Albrecht, P.P.; Zhu, B.; Lee, C.; Lahoti, T.S.; Gonzalez, F.J.; Peters, J.M. Stable over-expression of PPARβ/δ and PPARγ to examine receptor signaling in human HaCaT keratinocytes. Cell Signal. 2011, 23, 2039–2050. [Google Scholar] [CrossRef] [PubMed]
- Borland, M.G.; Foreman, J.E.; Girroir, E.E.; Zolfaghari, R.; Sharma, A.K.; Amin, S.; Gonzalez, F.J.; Ross, A.C.; Peters, J.M. Ligand activation of peroxisome proliferator-activated receptor-beta/delta inhibits cell proliferation in human HaCaT keratinocytes. Mol. Pharmacol. 2008, 74, 1429–1442. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, G.; Shi, Y.; Sun, L.; Gorczynski, R.; Li, Y.-J.; Xu, Z.; Spaner, D.E. PPAR-delta promotes survival of breast cancer cells in harsh metabolic conditions. Oncogenesis 2016, 5, e232. [Google Scholar] [CrossRef]
- Burdick, A.D.; Bility, M.T.; Girroir, E.E.; Billin, A.N.; Willson, T.M.; Gonzalez, F.J.; Peters, J.M. Ligand activation of peroxisome proliferator-activated receptor-beta/delta(PPARbeta/delta) inhibits cell growth of human N/TERT-1 keratinocytes. Cell Signal. 2007, 19, 1163–1171. [Google Scholar] [CrossRef][Green Version]
- Adhikary, T.; Brandt, D.T.; Kaddatz, K.; Stockert, J.; Naruhn, S.; Meissner, W.; Finkernagel, F.; Obert, J.; Lieber, S.; Scharfe, M.; et al. Inverse PPARβ/δ agonists suppress oncogenic signaling to the ANGPTL4 gene and inhibit cancer cell invasion. Oncogene 2013, 32, 5241–5252. [Google Scholar] [CrossRef]
- Gimble, J.M.; Pighetti, G.M.; Lerner, M.R.; Wu, X.; Lightfoot, S.A.; Brackett, D.J.; Darcy, K.; Hollingsworth, A.B. Expression of peroxisome proliferator activated receptor mRNA in normal and tumorigenic rodent mammary glands. Biochem. Biophys. Res. Commun. 1998, 253, 813–817. [Google Scholar] [CrossRef]
- Fajas, L.; Auboeuf, D.; Raspé, E.; Schoonjans, K.; Lefebvre, A.M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.C.; Deeb, S.; et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J. Biol. Chem. 1997, 272, 18779–18789. [Google Scholar] [CrossRef]
- Fang, S.; Livergood, M.C.; Nakagawa, P.; Wu, J.; Sigmund, C.D. Role of the Peroxisome Proliferator Activated Receptors in Hypertension. Circ. Res. 2021, 128, 1021–1039. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.; Szabo, G.; Abdelmalek, M.F.; Byrne, C.D.; Cusi, K.; Dufour, J.-F.; Roden, M.; Sacks, F.; Tacke, F. Nonalcoholic steatohepatitis: The role of peroxisome proliferator-activated receptors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Azhar, S. Peroxisome proliferator-activated receptors, metabolic syndrome and cardiovascular disease. Future Cardiol. 2010, 6, 657–691. [Google Scholar] [CrossRef] [PubMed]
- Takeyama, K.; Kodera, Y.; Suzawa, M.; Kato, S. [Peroxisome proliferator-activated receptor(PPAR)--structure, function, tissue distribution, gene expression]. Nihon Rinsho. 2000, 58, 357–363. [Google Scholar]
- Janani, C.; Ranjitha Kumari, B.D. PPAR gamma gene--a review. Diabetes Metab. Syndr. 2015, 9, 46–50. [Google Scholar] [CrossRef]
- Hamblin, M.; Chang, L.; Fan, Y.; Zhang, J.; Chen, Y.E. PPARs and the cardiovascular system. Antioxid. Redox Signal. 2009, 11, 1415–1452. [Google Scholar] [CrossRef]
- Li, Y.; Qi, Y.; Huang, T.H.W.; Yamahara, J.; Roufogalis, B.D. Pomegranate flower: A unique traditional antidiabetic medicine with dual PPAR-alpha/-gamma activator properties. Diabetes. Obes. Metab. 2008, 10, 10–17. [Google Scholar] [CrossRef]
- Blaschke, F.; Caglayan, E.; Hsueh, W.A. Peroxisome proliferator-activated receptor gamma agonists: Their role as vasoprotective agents in diabetes. Endocrinol. Metab. Clin. N. Am. 2006, 35, 561–574. [Google Scholar] [CrossRef]
- Rizos, C.V.; Kei, A.; Elisaf, M.S. The current role of thiazolidinediones in diabetes management. Arch. Toxicol. 2016, 90, 1861–1881. [Google Scholar] [CrossRef]
- Kahn, C.R.; Chen, L.; Cohen, S.E. Unraveling the mechanism of action of thiazolidinediones. J. Clin. Investig. 2000, 106, 1305–1307. [Google Scholar] [CrossRef]
- Hauner, H. The mode of action of thiazolidinediones. Diabetes. Metab. Res. Rev. 2002, 18 (Suppl. S2), S10–S15. [Google Scholar] [CrossRef] [PubMed]
- Farshbaf, M.J.; Ghaedi, K.; Shirani, M.; Nasr-Esfahani, M.H. Peroxisome proliferator activated receptor gamma (PPARγ) as a therapeutic target for improvement of cognitive performance in Fragile-X. Med. Hypotheses 2014, 82, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhao, L.-H.; Huang, B.; Wang, R.-Y.; Yuan, S.-X.; Tao, Q.-F.; Xu, Y.; Sun, H.-Y.; Lin, C.; Zhou, W.-P. Pioglitazone, a PPARγ agonist, inhibits growth and invasion of human hepatocellular carcinoma via blockade of the rage signaling. Mol. Carcinog. 2015, 54, 1584–1595. [Google Scholar] [CrossRef]
- Wang, Y.; Tan, H.; Xu, D.; Ma, A.; Zhang, L.; Sun, J.; Yang, Z.; Liu, Y.; Shi, G. The combinatory effects of PPAR-γ agonist and survivin inhibition on the cancer stem-like phenotype and cell proliferation in bladder cancer cells. Int. J. Mol. Med. 2014, 34, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, B.K.; Yang, H.-H.; Sung, P.-J.; Weng, C.-F. Excavatolide B inhibits nonsmall cell lung cancer proliferation by altering peroxisome proliferator activated receptor gamma expression and PTEN/AKT/NF-Kβ expression. Environ. Toxicol. 2017, 32, 290–301. [Google Scholar] [CrossRef]
- Srivastava, N.; Kollipara, R.K.; Singh, D.K.; Sudderth, J.; Hu, Z.; Nguyen, H.; Wang, S.; Humphries, C.G.; Carstens, R.; Huffman, K.E.; et al. Inhibition of cancer cell proliferation by PPARγ is mediated by a metabolic switch that increases reactive oxygen species levels. Cell Metab. 2014, 20, 650–661. [Google Scholar] [CrossRef]
- Pestereva, E.; Kanakasabai, S.; Bright, J.J. PPARγ agonists regulate the expression of stemness and differentiation genes in brain tumour stem cells. Br. J. Cancer 2012, 106, 1702–1712. [Google Scholar] [CrossRef]
- Trindade-da-Silva, C.A.; Reis, C.F.; Vecchi, L.; Napimoga, M.H.; Sperandio, M.; Matias Colombo, B.F.; Alves, P.T.; Ward, L.S.; Ueira-Vieira, C.; Goulart, L.R. 15-Deoxy-Δ(12,14)-prostaglandin J2 Induces Apoptosis and Upregulates SOCS3 in Human Thyroid Cancer Cells. PPAR Res. 2016, 2016, 4106297. [Google Scholar] [CrossRef]
- Wu, K.; Yang, Y.; Liu, D.; Qi, Y.; Zhang, C.; Zhao, J.; Zhao, S. Activation of PPARγ suppresses proliferation and induces apoptosis of esophageal cancer cells by inhibiting TLR4-dependent MAPK pathway. Oncotarget 2016, 7, 44572–44582. [Google Scholar] [CrossRef]
- Zurlo, D.; Ziccardi, P.; Votino, C.; Colangelo, T.; Cerchia, C.; Dal Piaz, F.; Dallavalle, S.; Moricca, S.; Novellino, E.; Lavecchia, A.; et al. The antiproliferative and proapoptotic effects of cladosporols A and B are related to their different binding mode as PPARγ ligands. Biochem. Pharmacol. 2016, 108, 22–35. [Google Scholar] [CrossRef]
- Weidner, C.; Rousseau, M.; Micikas, R.J.; Fischer, C.; Plauth, A.; Wowro, S.J.; Siems, K.; Hetterling, G.; Kliem, M.; Schroeder, F.C.; et al. Amorfrutin C Induces Apoptosis and Inhibits Proliferation in Colon Cancer Cells through Targeting Mitochondria. J. Nat. Prod. 2016, 79, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Kohno, H.; Yasui, Y.; Suzuki, R.; Hosokawa, M.; Miyashita, K.; Tanaka, T. Dietary seed oil rich in conjugated linolenic acid from bitter melon inhibits azoxymethane-induced rat colon carcinogenesis through elevation of colonic PPARgamma expression and alteration of lipid composition. Int. J. Cancer 2004, 110, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Yasui, Y.; Hosokawa, M.; Sahara, T.; Suzuki, R.; Ohgiya, S.; Kohno, H.; Tanaka, T.; Miyashita, K. Bitter gourd seed fatty acid rich in 9c,11t,13t-conjugated linolenic acid induces apoptosis and up-regulates the GADD45, p53 and PPARgamma in human colon cancer Caco-2 cells. Prostaglandins. Leukot. Essent. Fatty Acids 2005, 73, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Mueller, E.; Sarraf, P.; Tontonoz, P.; Evans, R.M.; Martin, K.J.; Zhang, M.; Fletcher, C.; Singer, S.; Spiegelman, B.M. Terminal differentiation of human breast cancer through PPAR gamma. Mol. Cell 1998, 1, 465–470. [Google Scholar] [CrossRef]
- Moon, H.-S.; Guo, D.-D.; Lee, H.-G.; Choi, Y.-J.; Kang, J.-S.; Jo, K.; Eom, J.-M.; Yun, C.-H.; Cho, C.-S. Alpha-eleostearic acid suppresses proliferation of MCF-7 breast cancer cells via activation of PPARgamma and inhibition of ERK 1/2. Cancer Sci. 2010, 101, 396–402. [Google Scholar] [CrossRef]
- Bonofiglio, D.; Gabriele, S.; Aquila, S.; Catalano, S.; Gentile, M.; Middea, E.; Giordano, F.; Andò, S. Estrogen receptor alpha binds to peroxisome proliferator-activated receptor response element and negatively interferes with peroxisome proliferator-activated receptor gamma signaling in breast cancer cells. Clin. Cancer Res. 2005, 11, 6139–6147. [Google Scholar] [CrossRef]
- Bonofiglio, D.; Aquila, S.; Catalano, S.; Gabriele, S.; Belmonte, M.; Middea, E.; Qi, H.; Morelli, C.; Gentile, M.; Maggiolini, M.; et al. Peroxisome proliferator-activated receptor-gamma activates p53 gene promoter binding to the nuclear factor-kappaB sequence in human MCF7 breast cancer cells. Mol. Endocrinol. 2006, 20, 3083–3092. [Google Scholar] [CrossRef]
- Bonofiglio, D.; Gabriele, S.; Aquila, S.; Qi, H.; Belmonte, M.; Catalano, S.; Andò, S. Peroxisome proliferator-activated receptor gamma activates fas ligand gene promoter inducing apoptosis in human breast cancer cells. Breast Cancer Res. Treat. 2009, 113, 423–434. [Google Scholar] [CrossRef]
- Chinnaiyan, A.M.; Dixit, V.M. Portrait of an executioner: The molecular mechanism of FAS/APO-1-induced apoptosis. Semin. Immunol. 1997, 9, 69–76. [Google Scholar] [CrossRef]
- Debatin, K.-M. Apoptosis pathways in cancer and cancer therapy. Cancer Immunol. Immunother. 2004, 53, 153–159. [Google Scholar] [CrossRef]
- Pinkoski, M.J.; Green, D.R. Fas ligand, death gene. Cell Death Differ. 1999, 6, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Catalano, S.; Mauro, L.; Bonofiglio, D.; Pellegrino, M.; Qi, H.; Rizza, P.; Vizza, D.; Bossi, G.; Andò, S. In vivo and in vitro evidence that PPARγ ligands are antagonists of leptin signaling in breast cancer. Am. J. Pathol. 2011, 179, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Demetri, G.D.; Mueller, E.; Sarraf, P.; Spiegelman, B.M.; Winer, E.P. Use of the peroxisome proliferator-activated receptor (PPAR) gamma ligand troglitazone as treatment for refractory breast cancer: A phase II study. Breast Cancer Res. Treat. 2003, 79, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Gale, E.A. Lessons from the glitazones: A story of drug development. Lancet 2001, 357, 1870–1875. [Google Scholar] [CrossRef]
- Krentz, A.J.; Bailey, C.J. Oral antidiabetic agents: Current role in type 2 diabetes mellitus. Drugs 2005, 65, 385–411. [Google Scholar] [CrossRef]
- Garcia-Vallvé, S.; Guasch, L.; Tomas-Hernández, S.; del Bas, J.M.; Ollendorff, V.; Arola, L.; Pujadas, G.; Mulero, M. Peroxisome Proliferator-Activated Receptor γ (PPARγ) and Ligand Choreography: Newcomers Take the Stage. J. Med. Chem. 2015, 58, 5381–5394. [Google Scholar] [CrossRef]
- Yee, L.D.; Williams, N.; Wen, P.; Young, D.C.; Lester, J.; Johnson, M.V.; Farrar, W.B.; Walker, M.J.; Povoski, S.P.; Suster, S.; et al. Pilot study of rosiglitazone therapy in women with breast cancer: Effects of short-term therapy on tumor tissue and serum markers. Clin Cancer Res. 2007, 13, 246–252. [Google Scholar] [CrossRef]
- Elstner, E.; Müller, C.; Koshizuka, K.; Williamson, E.A.; Park, D.; Asou, H.; Shintaku, P.; Said, J.W.; Heber, D.; Koeffler, H.P. Ligands for peroxisome proliferator-activated receptorgamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc. Natl. Acad. Sci. USA 1998, 95, 8806–8811. [Google Scholar] [CrossRef]
- Mustafa, A.; Kruger, W.D. Suppression of tumor formation by a cyclooxygenase-2 inhibitor and a peroxisome proliferator-activated receptor gamma agonist in an in vivo mouse model of spontaneous breast cancer. Clin. Cancer Res. 2008, 14, 4935–4942. [Google Scholar] [CrossRef]
- Bonofiglio, D.; Cione, E.; Qi, H.; Pingitore, A.; Perri, M.; Catalano, S.; Vizza, D.; Panno, M.L.; Genchi, G.; Fuqua, S.A.W.; et al. Combined low doses of PPARgamma and RXR ligands trigger an intrinsic apoptotic pathway in human breast cancer cells. Am. J. Pathol. 2009, 175, 1270–1280. [Google Scholar] [CrossRef]
- Yamazaki, K.; Shimizu, M.; Okuno, M.; Matsushima-Nishiwaki, R.; Kanemura, N.; Araki, H.; Tsurumi, H.; Kojima, S.; Weinstein, I.B.; Moriwaki, H. Synergistic effects of RXR alpha and PPAR gamma ligands to inhibit growth in human colon cancer cells--phosphorylated RXR alpha is a critical target for colon cancer management. Gut 2007, 56, 1557–1563. [Google Scholar] [CrossRef]
- Jiang, Y.; Huang, Y.; Cheng, C.; Lu, W.; Zhang, Y.; Liu, X.; Zou, L.; Ben, Q.; Shen, A. Combination of thiazolidinedione and hydralazine suppresses proliferation and induces apoptosis by PPARγ up-expression in MDA-MB-231 cells. Exp. Mol. Pathol. 2011, 91, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Girnun, G.D.; Naseri, E.; Vafai, S.B.; Qu, L.; Szwaya, J.D.; Bronson, R.; Alberta, J.A.; Spiegelman, B.M. Synergy between PPARgamma ligands and platinum-based drugs in cancer. Cancer Cell 2007, 11, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Bräutigam, K.; Biernath-Wüpping, J.; Bauerschlag, D.O.; von Kaisenberg, C.S.; Jonat, W.; Maass, N.; Arnold, N.; Meinhold-Heerlein, I. Combined treatment with TRAIL and PPARγ ligands overcomes chemoresistance of ovarian cancer cell lines. J. Cancer Res. Clin. Oncol. 2011, 137, 875–886. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Xin, B.; Shigeto, T.; Mizunuma, H. Combination of ciglitazone, a peroxisome proliferator-activated receptor gamma ligand, and cisplatin enhances the inhibition of growth of human ovarian cancers. J. Cancer Res. Clin. Oncol. 2011, 137, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Cesario, R.M.; Stone, J.; Yen, W.-C.; Bissonnette, R.P.; Lamph, W.W. Differentiation and growth inhibition mediated via the RXR:PPARgamma heterodimer in colon cancer. Cancer Lett. 2006, 240, 225–233. [Google Scholar] [CrossRef]
- Desreumaux, P.; Dubuquoy, L.; Nutten, S.; Peuchmaur, M.; Englaro, W.; Schoonjans, K.; Derijard, B.; Desvergne, B.; Wahli, W.; Chambon, P.; et al. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimer. A basis for new therapeutic strategies. J. Exp. Med. 2001, 193, 827–838. [Google Scholar] [CrossRef]
- Fu, H.; Zhang, J.; Pan, J.; Zhang, Q.; Lu, Y.; Wen, W.; Lubet, R.A.; Szabo, E.; Chen, R.; Wang, Y.; et al. Chemoprevention of lung carcinogenesis by the combination of aerosolized budesonide and oral pioglitazone in A/J mice. Mol. Carcinog. 2011, 50, 913–921. [Google Scholar] [CrossRef]
- Reddy, R.C.; Srirangam, A.; Reddy, K.; Chen, J.; Gangireddy, S.; Kalemkerian, G.P.; Standiford, T.J.; Keshamouni, V.G. Chemotherapeutic drugs induce PPAR-gamma expression and show sequence-specific synergy with PPAR-gamma ligands in inhibition of non-small cell lung cancer. Neoplasia 2008, 10, 597–603. [Google Scholar] [CrossRef]
- Aronica, S.M.; Kraus, W.L.; Katzenellenbogen, B.S. Estrogen action via the cAMP signaling pathway: Stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc. Natl. Acad. Sci. USA 1994, 91, 8517–8521. [Google Scholar] [CrossRef]
- Migliaccio, A.; Di Domenico, M.; Castoria, G.; de Falco, A.; Bontempo, P.; Nola, E.; Auricchio, F. Tyrosine kinase/p21ras/MAP-kinase pathway activation by estradiol-receptor complex in MCF-7 cells. EMBO J. 1996, 15, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Keller, H.; Givel, F.; Perroud, M.; Wahli, W. Signaling cross-talk between peroxisome proliferator-activated receptor/retinoid X receptor and estrogen receptor through estrogen response elements. Mol. Endocrinol. 1995, 9, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Miyoshi, K.; Claudio, E.; Siebenlist, U.K.; Gonzalez, F.J.; Flaws, J.; Wagner, K.-U.; Hennighausen, L. Loss of the peroxisome proliferation-activated receptor gamma (PPARgamma ) does not affect mammary development and propensity for tumor formation but leads to reduced fertility. J. Biol. Chem. 2002, 277, 17830–17835. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Yuan, H.; Zeng, X.; Kopelovich, L.; Glazer, R.I. Inhibition of peroxisome proliferator-activated receptor gamma increases estrogen receptor-dependent tumor specification. Cancer Res. 2009, 69, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Saez, E.; Rosenfeld, J.; Livolsi, A.; Olson, P.; Lombardo, E.; Nelson, M.; Banayo, E.; Cardiff, R.D.; Izpisua-Belmonte, J.C.; Evans, R.M. PPAR gamma signaling exacerbates mammary gland tumor development. Genes Dev. 2004, 18, 528–540. [Google Scholar] [CrossRef]
- Tian, L.; Zhou, J.; Casimiro, M.C.; Liang, B.; Ojeifo, J.O.; Wang, M.; Hyslop, T.; Wang, C.; Pestell, R.G. Activating peroxisome proliferator-activated receptor gamma mutant promotes tumor growth in vivo by enhancing angiogenesis. Cancer Res. 2009, 69, 9236–9244. [Google Scholar] [CrossRef]
- Xu, Y.-Y.; Liu, H.; Su, L.; Xu, N.; Xu, D.-H.; Liu, H.-Y.; Spaner, D.; Bed-David, Y.; Li, Y.-J. PPARγ inhibits breast cancer progression by upregulating PTPRF expression. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 9965–9977. [Google Scholar] [CrossRef]
- Kotta-Loizou, I.; Giaginis, C.; Theocharis, S. The role of peroxisome proliferator-activated receptor-γ in breast cancer. Anticancer. Agents Med. Chem. 2012, 12, 1025–1044. [Google Scholar] [CrossRef]
- Yang, P.-B.; Hou, P.-P.; Liu, F.-Y.; Hong, W.-B.; Chen, H.-Z.; Sun, X.-Y.; Li, P.; Zhang, Y.; Ju, C.-Y.; Luo, L.-J.; et al. Blocking PPARγ interaction facilitates Nur77 interdiction of fatty acid uptake and suppresses breast cancer progression. Proc. Natl. Acad. Sci. USA 2020, 117, 27412–27422. [Google Scholar] [CrossRef]
- Papadaki, I.; Mylona, E.; Giannopoulou, I.; Markaki, S.; Keramopoulos, A.; Nakopoulou, L. PPARgamma expression in breast cancer: Clinical value and correlation with ERbeta. Histopathology 2005, 46, 37–42. [Google Scholar] [CrossRef]
- Kulkarni, S.; Patil, D.B.; Diaz, L.K.; Wiley, E.L.; Morrow, M.; Khan, S.A. COX-2 and PPARgamma expression are potential markers of recurrence risk in mammary duct carcinoma in-situ. BMC Cancer 2008, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, Q.; Zhang, J.; Yang, G.; Shao, Z.; Luo, J.; Fan, M.; Ni, C.; Wu, Z.; Hu, X. Fenofibrate induces apoptosis of triple-negative breast cancer cells via activation of NF-κB pathway. BMC Cancer. 2014, 14, 96. [Google Scholar] [CrossRef] [PubMed]
- Kwong, S.C.; Jamil, A.H.A.; Rhodes, A.; Taib, N.A.; Chung, I. Metabolic role of fatty acid binding protein 7 in mediating triple-negative breast cancer cell death via PPAR-α signaling. J. Lipid Res. 2019, 60, 1807–1817. [Google Scholar] [CrossRef]
- Apaya, M.K.; Hsiao, P.W.; Yang, Y.C.; Shyur, L.F. Deregulating the CYP2C19/Epoxy-Eicosatrienoic Acid-Associated FABP4/FABP5 Signaling Network as a Therapeutic Approach for Metastatic Triple-Negative Breast Cancer. Cancers 2020, 12, 199. [Google Scholar] [CrossRef]
- Volkers, N. Diabetes and cancer: Scientists search for a possible link. J. Natl. Cancer Inst. 2000, 92, 192–194. [Google Scholar] [CrossRef]
- Giovannucci, E.; Harlan, D.M.; Archer, M.C.; Bergenstal, R.M.; Gapstur, S.M.; Habel, L.A.; Pollak, M.; Regensteiner, J.G.; Yee, D. Diabetes and cancer: A consensus report. Diabetes Care 2010, 33, 1674–1685. [Google Scholar] [CrossRef]
- Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Kooperberg, C.; Stefanick, M.L.; Jackson, R.D.; Beresford, S.A.A.; Howard, B.V.; Johnson, K.C.; et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002, 288, 321–333. [Google Scholar] [CrossRef]
- Youssef, J.; Badr, M. Peroxisome proliferator-activated receptors and cancer: Challenges and opportunities. Br. J. Pharmacol. 2011, 164, 68–82. [Google Scholar] [CrossRef]






| The Role in Breast Cancer | Binding Ligand | The Effect on Breast Cancer | |
|---|---|---|---|
| PPARα | cancer-promoting | arachidonic acid | Promoted cell growth and proliferation, especially MCF7 in cells (ER++++) [104] |
| Wy-14,643 | Increased target gene CYP1B1 mRNA and protein levels in MCF7 cells promoted cancer progression [106] | ||
| GW6471 | Reduced cell viability, cell proliferation, and spheroid formation lead to apoptosis and metabolic dysfunction of stem cells [107] | ||
| cancer-suppressing | Wy-14,643 | Inhibited the ability of DMBA to induce tumor formation in rats and induced tumor volume regression [108] | |
| clofibrate | Inhibited cell proliferation and growth, affecting various lipid metabolism pathways [109] | ||
| -- | Inhibited the proliferation and invasion of cells in vitro, inhibited cancer occurrence, bone metastasis, and tumor stemness in vivo [110] |
| The Role in Breast Cancer | Binding Ligand | The Effect on Breast Cancer | |
|---|---|---|---|
| PPARβ/δ | cancer-promoting | GW501,516 | Promoted the proliferation of MCF7 and T47D cells (ER+) instead of MAD-MB-231 and BT-20 cells (ER-), promoted VEGFα and FLT-1 expression [143] |
| GW501,516 | Accelerated adenosquamous carcinoma and mammary squamous cell tumor formation in mice, increased activation of PDK1 [147] | ||
| GW501,516 | Accelerated tumor formation, did not alter PDK1 protein levels [149] | ||
| GW501,516 | Accelerated the oncogenic process and increased tumor diversity, especially ER+/PR+/HER2- tumors [150] | ||
| -- | Promoted tumor growth and the expression of genes, including PDK1 and cell proliferation-related genes [152] | ||
| -- | Promoted the expression of FABP5 and PDK1 in MCF7 cells, promoted cell proliferation, and induced tumorigenesis [153] | ||
| -- | Promoted the survival of MCF7 cells under harsh microenvironmental conditions [156] | ||
| ST247 or DG172 | Inhibited serum and TGFβ-induced invasion of MDA-MB-231 cells [158] | ||
| -- | Increased tumor volume and lung metastasis in mice [156] | ||
| cancer-suppressing | GW0742 or GW501,516 | Inhibited the growth of MCF7 cells [144] | |
| GW0742 or GW501,516 | Inhibited the proliferation and clone formation, and promoted apoptosis in mouse C20 cells [141] | ||
| GW0742 | Inhibited the proliferation of MCF7 cells instead of MDA-MB-231 cells, inhibited the clone formation of MDA-MB-231 cells significantly more than that of MCF7 cells, and inhibited the volume of mouse xenografts [145] | ||
| -- | Inhibited hyperplasia and tumorigenesis in mice and inhibited the expression of epithelial cell proliferation-related genes (e.g., Ki-67, Cyclin D1, etc.) [146] | ||
| no effect | GW0742 or GSK3787 | Had no effect on the proliferation of MCF7 cells, despite both of them influencing the mRNA level of the target gene Angptl4 in vitro and in vivo [30] |
| The Role in Breast Cancer | Binding Ligand | The Effect on Breast Cancer | |
|---|---|---|---|
| PPARγ | cancer-promoting | -- | Promoted Wnt signaling and induced transgenic mice to develop tumors much faster with a higher degree of malignancy and differentiation of the tumors [215] |
| -- | Promoted the growth of tumors and angiogenesis in mice, increasing Angptl4 expression and endothelial stem cells [216] | ||
| -- | Interacted with Nur77, recruited Trim13 to target the ubiquitin proteasomal degradation of Nur77, and promoted cancer progression [219] | ||
| cancer-suppressing | TZD | Induced terminal differentiation of malignant mammary epithelial cells [184] | |
| GW7845 | Inhibited NMU-induced tumor incidence, tumor number, and tumor weight in rats [79]. | ||
| TGZ | Inhibited DMBA-induced tumor progression in rats, reduced malignancy incidence, and induced regression or stasis of total tumor volume [108] | ||
| α-eleostearic acid | Reduced MCF7 cell viability and promoted cell apoptosis [185] | ||
| RGZ | Inhibited PI3K/AKT pathway, inhibited proliferation of MCF7 cells [186] | ||
| RGZ | Promoted the expression of p53 in MCF7, induced caspase 9 cleavage and DNA fragmentation, and promoted cell growth arrest and apoptosis [187] | ||
| RGZ | Promoted target gene FasL expression, activated the cascade of caspases, and induced apoptosis [191] | ||
| TZD | Inhibited MAPK/STAT3/AKT phosphorylation-mediated leptin signaling in MCF7 cells inhibited cell proliferation and promoted cell apoptosis [192] | ||
| BRL49,653 | Inhibited the PI3K/AKT pathway and promoted PTEN expression in MCF7 cells, inhibiting cell growth [186] | ||
| -- | PPARγ silcence promoted Wnt signaling and induced transgenic mice to develope higher tumor diversity, especially ER+ ductal tumors [214] | ||
| -- | Inhibited cell proliferation and migration in vitro, inhibited tumor growth, and distant metastasis in mice [217] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, B.; Xin, Z.; Ren, P.; Wu, H. The Role of PPARs in Breast Cancer. Cells 2023, 12, 130. https://doi.org/10.3390/cells12010130
Zhao B, Xin Z, Ren P, Wu H. The Role of PPARs in Breast Cancer. Cells. 2023; 12(1):130. https://doi.org/10.3390/cells12010130
Chicago/Turabian StyleZhao, Binggong, Zhiqiang Xin, Ping Ren, and Huijian Wu. 2023. "The Role of PPARs in Breast Cancer" Cells 12, no. 1: 130. https://doi.org/10.3390/cells12010130
APA StyleZhao, B., Xin, Z., Ren, P., & Wu, H. (2023). The Role of PPARs in Breast Cancer. Cells, 12(1), 130. https://doi.org/10.3390/cells12010130