
Evaluation of Proteasome Inhibitors in the Treatment of Idiopathic Pulmonary Fibrosis

 ,
, 
Abstract
1. Introduction
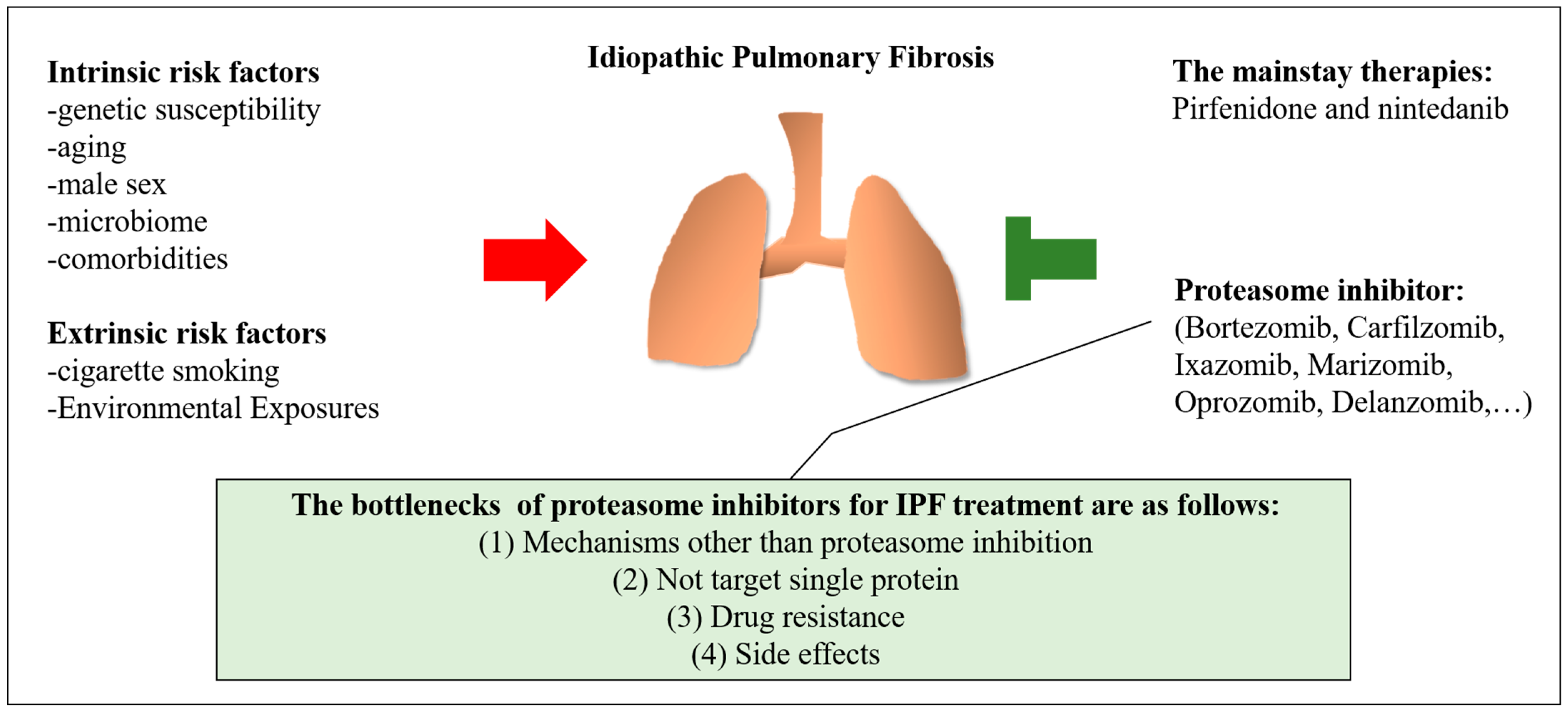
2. Risk Factors for Idiopathic Pulmonary Fibrosis
2.1. Intrinsic Risk Factors
2.2. Extrinsic Risk Factors
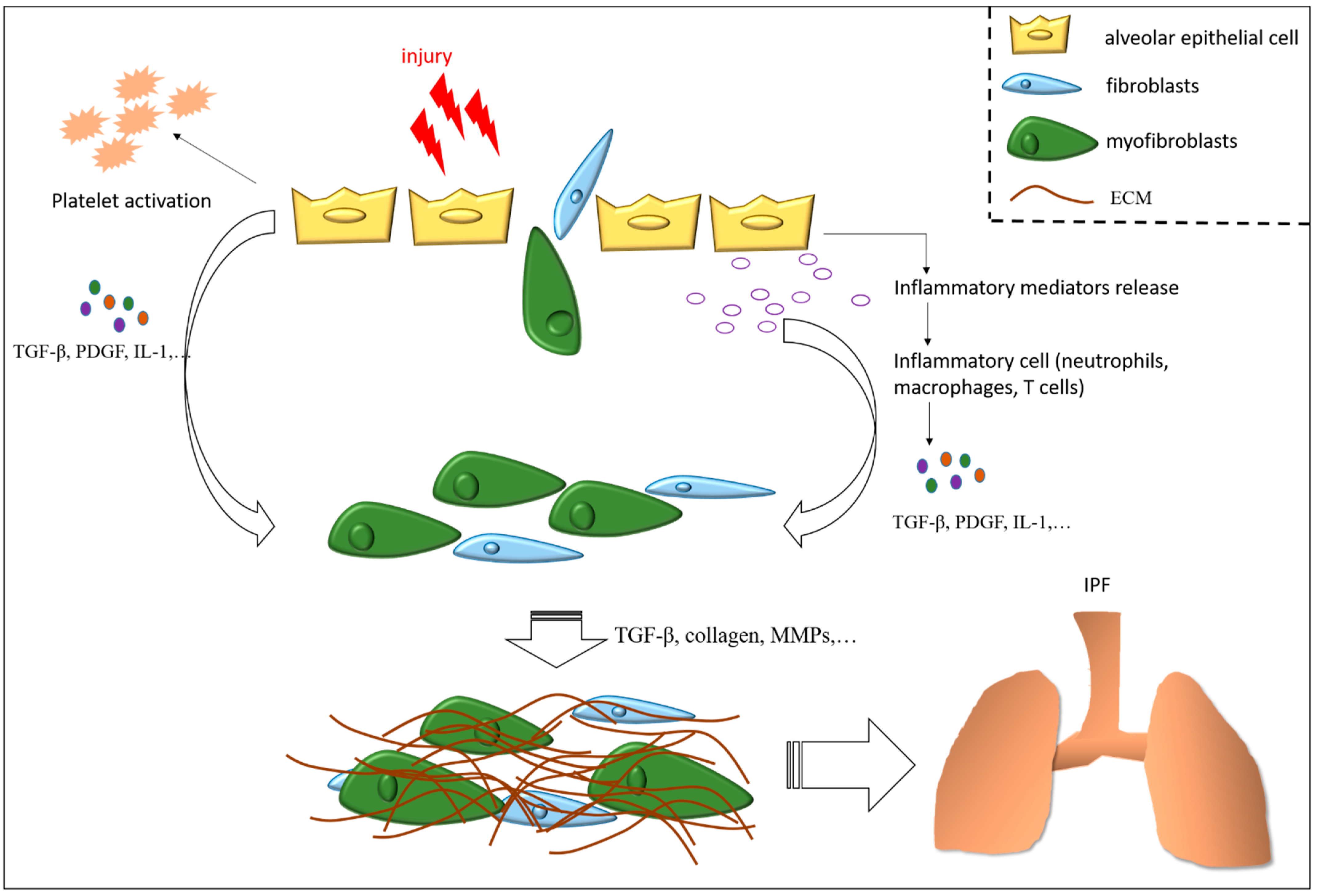
3. Mechanisms of Pulmonary Fibrosis
3.1. Apoptosis Resistance of (Myo)Fibroblasts
3.2. Dysfunction of Pulmonary Vessels
3.3. Mitochondrial Dysfunction
3.4. Autophagy Dysfunction
3.5. Aberrant Epithelia Hyperplasia and Dysfunction
3.6. Lipid Metabolism Disorder
3.7. Transforming Growth Factor-Beta in Idiopathic Pulmonary Fibrosis
3.8. Inflammation
4. The Mainstay of Medication and The Potential of Proteasome Inhibitors for IPF
4.1. Pirfenidone and Nintedanib
4.2. Overview of Proteasome Inhibitors and the Effects of Proteasome Inhibitors in Patients with Pulmonary Fibrosis
4.2.1. MG-132
4.2.2. Bortezomib
4.2.3. Carfilzomib
4.2.4. Oprozomib
4.2.5. Ixazomib
4.2.6. Delanzomib
4.2.7. Marizomib
5. Challenges in the Treatment of Pulmonary Fibrosis with Proteasome Inhibitors
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- García-Sancho, C.; Buendía-Roldán, I.; Fernández-Plata, M.R.; Navarro, C.; Pérez-Padilla, R.; Vargas, M.H.; Loyd, J.E.; Selman, M. Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respir. Med. 2011, 105, 1902–1907. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Chapman, K.; Ullah, S. Idiopathic Pulmonary Fibrosis; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Phan, T.H.G.; Paliogiannis, P.; Nasrallah, G.K.; Giordo, R.; Eid, A.H.; Fois, A.G.; Zinellu, A.; Mangoni, A.A.; Pintus, G. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell. Mol. LifeSci. 2021, 78, 2031–2057. [Google Scholar] [CrossRef] [PubMed]
- Bitterman, P. Fibroblast-Matrix Cross-Talk in Idiopathic Pulmonary Fibrosis: Cross-Links at the Crossroads. Am. J. Respir. Cell Mol. Biol. 2018, 58, 547–548. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef]
- Weiss, C.H.; Budinger, G.R.; Mutlu, G.M.; Jain, M. Proteasomal regulation of pulmonary fibrosis. Proc. Am. Thorac. Soc. 2010, 7, 77–83. [Google Scholar] [CrossRef]
- Fineschi, S.; Bongiovanni, M.; Donati, Y.; Djaafar, S.; Naso, F.; Goffin, L.; Argiroffo, C.B.; Pache, J.C.; Dayer, J.M.; Ferrari-Lacraz, S.; et al. In vivo investigations on anti-fibrotic potential of proteasome inhibition in lung and skin fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 458–465. [Google Scholar] [CrossRef]
- Zaman, T.; Lee, J.S. Risk factors for the development of idiopathic pulmonary fibrosis: A review. Curr. Pulmonol. Rep. 2018, 7, 118–125. [Google Scholar] [CrossRef]
- Kaur, A.; Mathai, S.K.; Schwartz, D.A. Genetics in Idiopathic Pulmonary Fibrosis Pathogenesis, Prognosis, and Treatment. Front. Med. 2017, 4, 154. [Google Scholar] [CrossRef]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef]
- Noth, I.; Zhang, Y.; Ma, S.F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef]
- Leuschner, G.; Klotsche, J.; Kreuter, M.; Prasse, A.; Wirtz, H.; Pittrow, D.; Frankenberger, M.; Behr, J.; Kneidinger, N. Idiopathic Pulmonary Fibrosis in Elderly Patients: Analysis of the INSIGHTS-IPF Observational Study. Front. Med. 2020, 7, 601279. [Google Scholar] [CrossRef] [PubMed]
- Stuart, B.D.; Lee, J.S.; Kozlitina, J.; Noth, I.; Devine, M.S.; Glazer, C.S.; Torres, F.; Kaza, V.; Girod, C.E.; Jones, K.D.; et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: An observational cohort study with independent validation. Lancet Respir. Med. 2014, 2, 557–565. [Google Scholar] [CrossRef]
- Snetselaar, R.; van Batenburg, A.A.; van Oosterhout, M.F.M.; Kazemier, K.M.; Roothaan, S.M.; Peeters, T.; van der Vis, J.J.; Goldschmeding, R.; Grutters, J.C.; van Moorsel, C.H.M. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS ONE 2017, 12, e0189467. [Google Scholar] [CrossRef] [PubMed]
- Voltz, J.W.; Card, J.W.; Carey, M.A.; Degraff, L.M.; Ferguson, C.D.; Flake, G.P.; Bonner, J.C.; Korach, K.S.; Zeldin, D.C. Male sex hormones exacerbate lung function impairment after bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 39, 45–52. [Google Scholar] [CrossRef]
- Lekgabe, E.D.; Royce, S.G.; Hewitson, T.D.; Tang, M.L.; Zhao, C.; Moore, X.L.; Tregear, G.W.; Bathgate, R.A.; Du, X.J.; Samuel, C.S. The effects of relaxin and estrogen deficiency on collagen deposition and hypertrophy of nonreproductive organs. Endocrinol. 2006, 147, 5575–5583. [Google Scholar] [CrossRef]
- Fang, C.; Huang, H.; Zhang, Q.; Wang, N.; Jing, X.; Guo, J.; Ferianc, M.; Xu, Z. Relation between sex hormones and leucocyte telomere length in men with idiopathic pulmonary fibrosis. Respirol. 2020, 25, 1265–1273. [Google Scholar] [CrossRef]
- Smith, L.C.; Moreno, S.; Robertson, L.; Robinson, S.; Gant, K.; Bryant, A.J.; Sabo-Attwood, T. Transforming growth factor beta1 targets estrogen receptor signaling in bronchial epithelial cells. Respir. Res. 2018, 19, 160. [Google Scholar] [CrossRef]
- Ntolios, P.; Tzilas, V.; Bouros, E.; Avdoula, E.; Karakasiliotis, I.; Bouros, D.; Steiropoulos, P. The Role of Microbiome and Virome in Idiopathic Pulmonary Fibrosis. Biomed. 2021, 9, 442. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Cox, M.J.; Willis-Owen, S.A.; Mallia, P.; Russell, K.E.; Russell, A.M.; Murphy, E.; Johnston, S.L.; Schwartz, D.A.; Wells, A.U.; et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 906–913. [Google Scholar] [CrossRef]
- Fastrès, A.; Felice, F.; Roles, E.; Moermans, C.; Corhay, J.L.; Bureau, F.; Louis, R.; Clercx, C.; Guiot, J. The Lung Microbiome in Idiopathic Pulmonary Fibrosis: A Promising Approach for Targeted Therapies. Int. J. Mol. Sci. 2017, 18, 2735. [Google Scholar] [CrossRef]
- Chioma, O.S.; Hesse, L.E.; Chapman, A.; Drake, W.P. Role of the Microbiome in Interstitial Lung Diseases. Front. Med. 2021, 8, 595522. [Google Scholar] [CrossRef] [PubMed]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. New Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Noth, I.; Zangan, S.M.; Soares, R.V.; Forsythe, A.; Demchuk, C.; Takahashi, S.M.; Patel, S.B.; Strek, M.E.; Krishnan, J.A.; Patti, M.G.; et al. Prevalence of hiatal hernia by blinded multidetector CT in patients with idiopathic pulmonary fibrosis. Eur. Respir. J. 2012, 39, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Collard, H.R.; Anstrom, K.J.; Martinez, F.J.; Noth, I.; Roberts, R.S.; Yow, E.; Raghu, G. Anti-acid treatment and disease progression in idiopathic pulmonary fibrosis: An analysis of data from three randomised controlled trials. Lancet Respir. Med. 2013, 1, 369–376. [Google Scholar] [CrossRef]
- Kreuter, M.; Wuyts, W.; Renzoni, E.; Koschel, D.; Maher, T.M.; Kolb, M.; Weycker, D.; Spagnolo, P.; Kirchgaessler, K.U.; Herth, F.J.; et al. Antacid therapy and disease outcomes in idiopathic pulmonary fibrosis: A pooled analysis. Lancet Respir. Med. 2016, 4, 381–389. [Google Scholar] [CrossRef]
- Raghu, G.; Pellegrini, C.A.; Yow, E.; Flaherty, K.R.; Meyer, K.; Noth, I.; Scholand, M.B.; Cello, J.; Ho, L.A.; Pipavath, S.; et al. Laparoscopic anti-reflux surgery for the treatment of idiopathic pulmonary fibrosis (WRAP-IPF): A multicentre, randomised, controlled phase 2 trial. Lancet Respir. Med. 2018, 6, 707–714. [Google Scholar] [CrossRef]
- Gille, T.; Didier, M.; Boubaya, M.; Moya, L.; Sutton, A.; Carton, Z.; Baran-Marszak, F.; Sadoun-Danino, D.; Israël-Biet, D.; Cottin, V.; et al. Obstructive sleep apnoea and related comorbidities in incident idiopathic pulmonary fibrosis. Eur. Respir. J. 2017, 49, 1601934. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Yang, J.; Xue, Q.; Miao, L.; Cai, L. Pulmonary fibrosis: A possible diabetic complication. Diabetes/Metab. Res. Rev. 2011, 27, 311–317. [Google Scholar] [CrossRef]
- Sato, N.; Takasaka, N.; Yoshida, M.; Tsubouchi, K.; Minagawa, S.; Araya, J.; Saito, N.; Fujita, Y.; Kurita, Y.; Kobayashi, K.; et al. Metformin attenuates lung fibrosis development via NOX4 suppression. Respir. Res. 2016, 17, 107. [Google Scholar] [CrossRef]
- Rangarajan, S.; Bone, N.B.; Zmijewska, A.A.; Jiang, S.; Park, D.W.; Bernard, K.; Locy, M.L.; Ravi, S.; Deshane, J.; Mannon, R.B.; et al. Metformin reverses established lung fibrosis in a bleomycin model. Nat. Med. 2018, 24, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.W.; Johnson, J.E.; Browning, P.J.; Cruz-Gervis, R.A.; Davis, A.; Graham, B.S.; Brigham, K.L.; Oates, J.A., Jr.; Loyd, J.E.; Stecenko, A.A. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J. Clin. Microbiol. 2003, 41, 2633–2640. [Google Scholar] [CrossRef] [PubMed]
- Sides, M.D.; Klingsberg, R.C.; Shan, B.; Gordon, K.A.; Nguyen, H.T.; Lin, Z.; Takahashi, T.; Flemington, E.K.; Lasky, J.A. The Epstein-Barr virus latent membrane protein 1 and transforming growth factor-β1 synergistically induce epithelial-mesenchymal transition in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 2011, 44, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Malizia, A.P.; Keating, D.T.; Smith, S.M.; Walls, D.; Doran, P.P.; Egan, J.J. Alveolar epithelial cell injury with Epstein-Barr virus upregulates TGFbeta1 expression. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L451–L460. [Google Scholar] [CrossRef]
- Milara, J.; Peiró, T.; Serrano, A.; Guijarro, R.; Zaragozá, C.; Tenor, H.; Cortijo, J. Roflumilast N-oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulm. Pharmacol. Ther. 2014, 28, 138–148. [Google Scholar] [CrossRef]
- Huzen, J.; Wong, L.S.; van Veldhuisen, D.J.; Samani, N.J.; Zwinderman, A.H.; Codd, V.; Cawthon, R.M.; Benus, G.F.; van der Horst, I.C.; Navis, G.; et al. Telomere length loss due to smoking and metabolic traits. J. Intern. Med. 2014, 275, 155–163. [Google Scholar] [CrossRef]
- Tanjore, H.; Blackwell, T.S.; Lawson, W.E. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L721–L729. [Google Scholar] [CrossRef]
- Jensen, K.; Nizamutdinov, D.; Guerrier, M.; Afroze, S.; Dostal, D.; Glaser, S. General mechanisms of nicotine-induced fibrogenesis. FASEBJ: Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 4778–4787. [Google Scholar] [CrossRef]
- Hubbard, R.; Cooper, M.; Antoniak, M.; Venn, A.; Khan, S.; Johnston, I.; Lewis, S.; Britton, J. Risk of cryptogenic fibrosing alveolitis in metal workers. Lancet 2000, 355, 466–467. [Google Scholar] [CrossRef]
- Iwai, K.; Mori, T.; Yamada, N.; Yamaguchi, M.; Hosoda, Y. Idiopathic pulmonary fibrosis. Epidemiologic approaches to occupational exposure. Am. J. Respir. Crit. Care Med. 1994, 150, 670–675. [Google Scholar] [CrossRef]
- Kolahian, S.; Fernandez, I.E.; Eickelberg, O.; Hartl, D. Immune Mechanisms in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Chioma, O.S.; Drake, W.P. Role of Microbial Agents in Pulmonary Fibrosis. Yale J. Biol. Med. 2017, 90, 219–227. [Google Scholar] [PubMed]
- Tashiro, J.; Rubio, G.A.; Limper, A.H.; Williams, K.; Elliot, S.J.; Ninou, I.; Aidinis, V.; Tzouvelekis, A.; Glassberg, M.K. Exploring Animal Models That Resemble Idiopathic Pulmonary Fibrosis. Front. Med. 2017, 4, 118. [Google Scholar] [CrossRef] [PubMed]
- Degryse, A.L.; Lawson, W.E. Progress toward improving animal models for idiopathic pulmonary fibrosis. Am. J. Med. Sci. 2011, 341, 444–449. [Google Scholar] [CrossRef] [PubMed]
- Redente, E.F.; Black, B.P.; Backos, D.S.; Bahadur, A.N.; Humphries, S.M.; Lynch, D.A.; Tuder, R.M.; Zemans, R.L.; Riches, D.W.H. Persistent, Progressive Pulmonary Fibrosis and Epithelial Remodeling in Mice. Am. J. Respir. Cell Mol. Biol. 2021, 64, 669–676. [Google Scholar] [CrossRef]
- Degryse, A.L.; Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Jones, B.R.; McMahon, F.B.; Gleaves, L.A.; Blackwell, T.S.; Lawson, W.E. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L442–L452. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.Y.; Tang, X.X. Irreversibility of Pulmonary Fibrosis. Aging Dis. 2022, 13, 73–86. [Google Scholar] [CrossRef]
- Shu, D.Y.; Lovicu, F.J. Myofibroblast transdifferentiation: The dark force in ocular wound healing and fibrosis. Prog. Retin. Eye Res. 2017, 60, 44–65. [Google Scholar] [CrossRef]
- Bamberg, A.; Redente, E.F.; Groshong, S.D.; Tuder, R.M.; Cool, C.D.; Keith, R.C.; Edelman, B.L.; Black, B.P.; Cosgrove, G.P.; Wynes, M.W.; et al. Protein Tyrosine Phosphatase-N13 Promotes Myofibroblast Resistance to Apoptosis in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 198, 914–927. [Google Scholar] [CrossRef]
- Pendyala, S.; Moitra, J.; Kalari, S.; Kleeberger, S.R.; Zhao, Y.; Reddy, S.P.; Garcia, J.G.; Natarajan, V. Nrf2 regulates hyperoxia-induced Nox4 expression in human lung endothelium: Identification of functional antioxidant response elements on the Nox4 promoter. Free. Radic. Biol. Med. 2011, 50, 1749–1759. [Google Scholar] [CrossRef]
- Redente, E.F.; Chakraborty, S.; Sajuthi, S.; Black, B.P.; Edelman, B.L.; Seibold, M.A.; Riches, D.W. Loss of Fas signaling in fibroblasts impairs homeostatic fibrosis resolution and promotes persistent pulmonary fibrosis. JCI Insight 2020, 6, e141618. [Google Scholar] [CrossRef] [PubMed]
- Probst, C.K.; Montesi, S.B.; Medoff, B.D.; Shea, B.S.; Knipe, R.S. Vascular permeability in the fibrotic lung. Eur. Respir. J. 2020, 56, 1900100. [Google Scholar] [CrossRef] [PubMed]
- Caporarello, N.; Meridew, J.A.; Aravamudhan, A.; Jones, D.L.; Austin, S.A.; Pham, T.X.; Haak, A.J.; Moo Choi, K.; Tan, Q.; Haresi, A.; et al. Vascular dysfunction in aged mice contributes to persistent lung fibrosis. Aging Cell 2020, 19, e13196. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Zhou, J. CXCR7 attenuates the TGF-β-induced endothelial-to-mesenchymal transition and pulmonary fibrosis. Mol. Biosyst. 2017, 13, 2116–2124. [Google Scholar] [CrossRef] [PubMed]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxidative Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [PubMed]
- Caporarello, N.; Meridew, J.A.; Jones, D.L.; Tan, Q.; Haak, A.J.; Choi, K.M.; Manlove, L.J.; Prakash, Y.S.; Tschumperlin, D.J.; Ligresti, G. PGC1α repression in IPF fibroblasts drives a pathologic metabolic, secretory and fibrogenic state. Thorax 2019, 74, 749–760. [Google Scholar] [CrossRef]
- Wang, N.; Zhu, P.; Huang, R.; Wang, C.; Sun, L.; Lan, B.; He, Y.; Zhao, H.; Gao, Y. PINK1: The guard of mitochondria. Life Sci. 2020, 259, 118247. [Google Scholar] [CrossRef]
- Singh, K.K.; Lovren, F.; Pan, Y.; Quan, A.; Ramadan, A.; Matkar, P.N.; Ehsan, M.; Sandhu, P.; Mantella, L.E.; Gupta, N.; et al. The essential autophagy gene ATG7 modulates organ fibrosis via regulation of endothelial-to-mesenchymal transition. J. Biol. Chem. 2015, 290, 2547–2559. [Google Scholar] [CrossRef]
- Liao, S.X.; Sun, P.P.; Gu, Y.H.; Rao, X.M.; Zhang, L.Y.; Ou-Yang, Y. Autophagy and pulmonary disease. Ther. Adv. Respir. Dis. 2019, 13, 1753466619890538. [Google Scholar] [CrossRef]
- Aspal, M.; Zemans, R.L. Mechanisms of ATII-to-ATI Cell Differentiation during Lung Regeneration. Int. J. Mol. Sci. 2020, 21, 3188. [Google Scholar] [CrossRef]
- Ding, L.; Tang, S.; Tang, W.; Mosley, D.D.; Yu, A.; Sil, D.; Romanova, S.; Bailey, K.L.; Knoell, D.L.; Wyatt, T.A.; et al. Perfluorocarbon Nanoemulsions Enhance Therapeutic siRNA Delivery in the Treatment of Pulmonary Fibrosis. Adv. Sci. 2022, 9, e2103676. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Li, Y.; Yu, J.; Dong, L.; Husain, A.N.; Shen, L.; Weber, C.R. Idiopathic pulmonary fibrosis is associated with tight junction protein alterations. Biochim. Et Biophys. Acta Biomembr. 2020, 1862, 183205. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2020, 180, 107–121.e17. [Google Scholar] [CrossRef] [PubMed]
- Shannon, J.M.; Hyatt, B.A. Epithelial-mesenchymal interactions in the developing lung. Annu. Rev. Physiol. 2004, 66, 625–645. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Role of epithelial cells in idiopathic pulmonary fibrosis: From innocent targets to serial killers. Proc. Am. Thorac. Soc. 2006, 3, 364–372. [Google Scholar] [CrossRef]
- Hadjicharalambous, M.R.; Lindsay, M.A. Idiopathic Pulmonary Fibrosis: Pathogenesis and the Emerging Role of Long Non-Coding RNAs. Int. J. Mol. Sci. 2020, 21, 524. [Google Scholar] [CrossRef]
- Kasper, M.; Barth, K. Potential contribution of alveolar epithelial type I cells to pulmonary fibrosis. Biosci. Rep. 2017, 37, BSR20171301. [Google Scholar] [CrossRef]
- Camelo, A.; Dunmore, R.; Sleeman, M.A.; Clarke, D.L. The epithelium in idiopathic pulmonary fibrosis: Breaking the barrier. Front. Pharmacol. 2014, 4, 173. [Google Scholar] [CrossRef]
- Vockeroth, D.; Gunasekara, L.; Amrein, M.; Possmayer, F.; Lewis, J.F.; Veldhuizen, R.A. Role of cholesterol in the biophysical dysfunction of surfactant in ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L117–L125. [Google Scholar] [CrossRef]
- Sunaga, H.; Matsui, H.; Ueno, M.; Maeno, T.; Iso, T.; Syamsunarno, M.R.; Anjo, S.; Matsuzaka, T.; Shimano, H.; Yokoyama, T.; et al. Deranged fatty acid composition causes pulmonary fibrosis in Elovl6-deficient mice. Nat. Commun. 2013, 4, 2563. [Google Scholar] [CrossRef]
- Romero, F.; Hong, X.; Shah, D.; Kallen, C.B.; Rosas, I.; Guo, Z.; Schriner, D.; Barta, J.; Shaghaghi, H.; Hoek, J.B.; et al. Lipid Synthesis Is Required to Resolve Endoplasmic Reticulum Stress and Limit Fibrotic Responses in the Lung. Am. J. Respir. Cell Mol. Biol. 2018, 59, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef] [PubMed]
- Lv, T.; Jiang, K.; Wang, J.; Tang, N.; Dai, H.; Wang, C. Single-cell RNA sequencing profiling of the effects of aging on alveolar stem cells. Sci. China Life Sci. 2019, 62, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef]
- Kim, K.K.; Wei, Y.; Szekeres, C.; Kugler, M.C.; Wolters, P.J.; Hill, M.L.; Frank, J.A.; Brumwell, A.N.; Wheeler, S.E.; Kreidberg, J.A.; et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J. Clin. Investig. 2009, 119, 213–224. [Google Scholar]
- Velagacherla, V.; Mehta, C.H.; Nayak, Y.; Nayak, U.Y. Molecular pathways and role of epigenetics in the idiopathic pulmonary fibrosis. Life Sci. 2022, 291, 120283. [Google Scholar] [CrossRef]
- Hernandez, D.M.; Kang, J.H.; Choudhury, M.; Andrianifahanana, M.; Yin, X.; Limper, A.H.; Leof, E.B. IPF pathogenesis is dependent upon TGFβ induction of IGF-1. FASEB J: Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 5363–5388. [Google Scholar] [CrossRef]
- Kolb, M.; Margetts, P.J.; Anthony, D.C.; Pitossi, F.; Gauldie, J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Investig. 2001, 107, 1529–1536. [Google Scholar] [CrossRef]
- Lappalainen, U.; Whitsett, J.A.; Wert, S.E.; Tichelaar, J.W.; Bry, K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am. J. Respir. Cell Mol. Biol. 2005, 32, 311–318. [Google Scholar] [CrossRef]
- Wilson, M.S.; Madala, S.K.; Ramalingam, T.R.; Gochuico, B.R.; Rosas, I.O.; Cheever, A.W.; Wynn, T.A. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J. Exp. Med. 2010, 207, 535–552. [Google Scholar] [CrossRef]
- Kinder, B.W.; Brown, K.K.; Schwarz, M.I.; Ix, J.H.; Kervitsky, A.; King, T.E., Jr. Baseline BAL neutrophilia predicts early mortality in idiopathic pulmonary fibrosis. Chest 2008, 133, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Baroni, G.S.; D’Ambrosio, L.; Curto, P.; Casini, A.; Mancini, R.; Jezequel, A.M.; Benedetti, A. Interferon gamma decreases hepatic stellate cell activation and extracellular matrix deposition in rat liver fibrosis. Hepatol. 1996, 23, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Chiaramonte, M.G.; Donaldson, D.D.; Cheever, A.W.; Wynn, T.A. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J. Clin. Investig. 1999, 104, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Heukels, P.; Moor, C.C.; von der Thüsen, J.H.; Wijsenbeek, M.S.; Kool, M. Inflammation and immunity in IPF pathogenesis and treatment. Respir. Med. 2019, 147, 79–91. [Google Scholar] [CrossRef]
- Caminati, A.; Cassandro, R.; Torre, O.; Harari, S. Severe idiopathic pulmonary fibrosis: What can be done? Eur. Respir.Rev.: Off. J. Eur. Respir. Soc. 2017, 26, 170047. [Google Scholar] [CrossRef]
- Miles, T.; Hoyne, G.F.; Knight, D.A.; Fear, M.W.; Mutsaers, S.E.; Prêle, C.M. The contribution of animal models to understanding the role of the immune system in human idiopathic pulmonary fibrosis. Clin. Transl. Immunol. 2020, 9, e1153. [Google Scholar] [CrossRef]
- Kolb, M.; Bonniaud, P.; Galt, T.; Sime, P.J.; Kelly, M.M.; Margetts, P.J.; Gauldie, J. Differences in the fibrogenic response after transfer of active transforming growth factor-beta1 gene to lungs of “fibrosis-prone” and “fibrosis-resistant” mouse strains. Am. J. Respir. Cell Mol. Biol. 2002, 27, 141–150. [Google Scholar] [CrossRef]
- Kolb, M.; Bonella, F.; Wollin, L. Therapeutic targets in idiopathic pulmonary fibrosis. Respir. Med. 2017, 131, 49–57. [Google Scholar] [CrossRef]
- Saito, S.; Alkhatib, A.; Kolls, J.K.; Kondoh, Y.; Lasky, J.A. Pharmacotherapy and adjunctive treatment for idiopathic pulmonary fibrosis (IPF). J. Thorac. Dis. 2019, 11, S1740–S1754. [Google Scholar] [CrossRef]
- Lancaster, L.H.; de Andrade, J.A.; Zibrak, J.D.; Padilla, M.L.; Albera, C.; Nathan, S.D.; Wijsenbeek, M.S.; Stauffer, J.L.; Kirchgaessler, K.U.; Costabel, U. Pirfenidone safety and adverse event management in idiopathic pulmonary fibrosis. Eur. Respir.Rev.: Off. J. Eur. Respir. Soc. 2017, 26, 170057. [Google Scholar] [CrossRef]
- Cottin, V.; Maher, T. Long-term clinical and real-world experience with pirfenidone in the treatment of idiopathic pulmonary fibrosis. Eur. Respir.Rev.: Off. J. Eur. Respir. Soc. 2015, 24, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Corte, T.; Bonella, F.; Crestani, B.; Demedts, M.G.; Richeldi, L.; Coeck, C.; Pelling, K.; Quaresma, M.; Lasky, J.A. Safety, tolerability and appropriate use of nintedanib in idiopathic pulmonary fibrosis. Respir. Res. 2015, 16, 116. [Google Scholar] [CrossRef] [PubMed]
- Wollin, L.; Wex, E.; Pautsch, A.; Schnapp, G.; Hostettler, K.E.; Stowasser, S.; Kolb, M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1434–1445. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. New Engl. J. Med. 2018, 379, 797–798. [Google Scholar] [CrossRef]
- Kato, M.; Sasaki, S.; Nakamura, T.; Kurokawa, K.; Yamada, T.; Ochi, Y.; Ihara, H.; Takahashi, F.; Takahashi, K. Gastrointestinal adverse effects of nintedanib and the associated risk factors in patients with idiopathic pulmonary fibrosis. Sci. Rep. 2019, 9, 12062. [Google Scholar] [CrossRef]
- Luedtke, D.; Marzin, K.; Jungnik, A.; von Wangenheim, U.; Dallinger, C. Effects of Ketoconazole and Rifampicin on the Pharmacokinetics of Nintedanib in Healthy Subjects. Eur. J. Drug Metab. Pharmacokinet. 2018, 43, 533–541. [Google Scholar] [CrossRef]
- Richeldi, L.; Cottin, V.; du Bois, R.M.; Selman, M.; Kimura, T.; Bailes, Z.; Schlenker-Herceg, R.; Stowasser, S.; Brown, K.K. Nintedanib in patients with idiopathic pulmonary fibrosis: Combined evidence from the TOMORROW and INPULSIS(®) trials. Respir. Med. 2016, 113, 74–79. [Google Scholar] [CrossRef]
- Nathan, S.D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glaspole, I.; Glassberg, M.K.; Kardatzke, D.R.; Daigl, M.; Kirchgaessler, K.U.; Lancaster, L.H.; et al. Effect of pirfenidone on mortality: Pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir. Med. 2017, 5, 33–41. [Google Scholar] [CrossRef]
- Grześk, G.; Woźniak-Wiśniewska, A.; Błażejewski, J.; Górny, B.; Wołowiec, Ł.; Rogowicz, D.; Nowaczyk, A. The Interactions of Nintedanib and Oral Anticoagulants-Molecular Mechanisms and Clinical Implications. Int. J. Mol. Sci. 2020, 22, 282. [Google Scholar] [CrossRef]
- Richeldi, L.; Fletcher, S.; Adamali, H.; Chaudhuri, N.; Wiebe, S.; Wind, S.; Hohl, K.; Baker, A.; Schlenker-Herceg, R.; Stowasser, S.; et al. No relevant pharmacokinetic drug-drug interaction between nintedanib and pirfenidone. Eur. Respir. J. 2019, 53, 1801060. [Google Scholar] [CrossRef]
- Ogura, T.; Taniguchi, H.; Azuma, A.; Inoue, Y.; Kondoh, Y.; Hasegawa, Y.; Bando, M.; Abe, S.; Mochizuki, Y.; Chida, K.; et al. Safety and pharmacokinetics of nintedanib and pirfenidone in idiopathic pulmonary fibrosis. Eur. Respir. J. 2015, 45, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Landis-Piwowar, K.R.; Milacic, V.; Chen, D.; Yang, H.; Zhao, Y.; Chan, T.H.; Yan, B.; Dou, Q.P. The proteasome as a potential target for novel anticancer drugs and chemosensitizers. Drug Resist. Updates: Rev. Comment. Antimicrob. Anticancer. Chemother. 2006, 9, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Tomaszewski, J.E. Proteasome inhibitors. Biochem. Pharmacol. 2015, 96, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Semren, N.; Habel-Ungewitter, N.C.; Fernandez, I.E.; Königshoff, M.; Eickelberg, O.; Stöger, T.; Meiners, S. Validation of the 2nd Generation Proteasome Inhibitor Oprozomib for Local Therapy of Pulmonary Fibrosis. PLoS ONE 2015, 10, e0136188. [Google Scholar] [CrossRef]
- Fricker LD: Proteasome Inhibitor Drugs. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 457–476. [CrossRef]
- Juvekar, A.; Manna, S.; Ramaswami, S.; Chang, T.P.; Vu, H.Y.; Ghosh, C.C.; Celiker, M.Y.; Vancurova, I. Bortezomib induces nuclear translocation of IκBα resulting in gene-specific suppression of NF-κB--dependent transcription and induction of apoptosis in CTCL. Mol. Cancer Res. 2011, 9, 183–194. [Google Scholar] [CrossRef]
- Wu, X.; Chen, Z.; Yang, Y.; Dong, Y.; Liu, H.; Kuang, S.; Luo, K. Impact of proteasome inhibitor MG-132 on expression of NF-κB, IL-1β and histological remodeling after myocardial infarction. Exp. Ther. Med. 2018, 16, 1365–1372. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, C.; Zhou, F.; Chen, X. PDK1 inhibitor GSK2334470 synergizes with proteasome inhibitor MG-132 in multiple myeloma cells by inhibiting full AKT activity and increasing nuclear accumulation of the PTEN protein. Oncol. Rep. 2018, 39, 2951–2959. [Google Scholar] [CrossRef]
- Meiners, S.; Hocher, B.; Weller, A.; Laule, M.; Stangl, V.; Guenther, C.; Godes, M.; Mrozikiewicz, A.; Baumann, G.; Stangl, K. Downregulation of matrix metalloproteinases and collagens and suppression of cardiac fibrosis by inhibition of the proteasome. Hypertens. 2004, 44, 471–477. [Google Scholar] [CrossRef][Green Version]
- Han, L.; Zhu, B.; Chen, H.; Jin, Y.; Liu, J.; Wang, W. Proteasome inhibitor MG132 inhibits the process of renal interstitial fibrosis. Exp. Ther. Med. 2019, 17, 2953–2962. [Google Scholar] [CrossRef]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ Are Mechanoregulators of TGF-β-Smad Signaling and Renal Fibrogenesis. J. Am. Soc.Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef] [PubMed]
- Aravamudhan, A.; Haak, A.J.; Choi, K.M.; Meridew, J.A.; Caporarello, N.; Jones, D.L.; Tan, Q.; Ligresti, G.; Tschumperlin, D.J. TBK1 regulates YAP/TAZ and fibrogenic fibroblast activation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L852–L863. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.F.; Kane, R.; Farrell, A.T.; Abraham, S.; Benson, K.; Brower, M.E.; Bradley, S.; Gobburu, J.V.; Goheer, A.; Lee, S.L.; et al. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin. Cancer Res. 2004, 10, 3954–3964. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z.; Stinchcombe, T.E.; Mitchell, B.S.; Shea, T.C.; Baldwin, A.S.; Stahl, S.; Adams, J.; Esseltine, D.L.; Elliott, P.J.; Pien, C.S.; et al. Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J. Clin.Oncol. 2002, 20, 4420–4427. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauhan, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Hatake, K.; Mishima, Y.; Terui, Y. Proteasome inhibitors. Cancer Chemother. 2004, 31, 999–1002. [Google Scholar]
- Jares, P.; Colomer, D.; Campo, E. Molecular pathogenesis of mantle cell lymphoma. J. Clin. Investig. 2012, 122, 3416–3423. [Google Scholar] [CrossRef]
- Uziel, O.; Cohen, O.; Beery, E.; Nordenberg, J.; Lahav, M. The effect of Bortezomib and Rapamycin on Telomerase Activity in Mantle Cell Lymphoma. Transl. Oncol. 2014, 7, 741–751. [Google Scholar] [CrossRef]
- Pan, X.; Hussain, F.N.; Iqbal, J.; Feuerman, M.H.; Hussain, M.M. Inhibiting proteasomal degradation of microsomal triglyceride transfer protein prevents CCl4-induced steatosis. J. Biol. Chem. 2007, 282, 17078–17089. [Google Scholar] [CrossRef]
- Wagner-Ballon, O.; Pisani, D.F.; Gastinne, T.; Tulliez, M.; Chaligné, R.; Lacout, C.; Auradé, F.; Villeval, J.L.; Gonin, P.; Vainchenker, W.; et al. Proteasome inhibitor bortezomib impairs both myelofibrosis and osteosclerosis induced by high thrombopoietin levels in mice. Blood 2007, 110, 345–353. [Google Scholar] [CrossRef]
- Mutlu, G.M.; Budinger, G.R.; Wu, M.; Lam, A.P.; Zirk, A.; Rivera, S.; Urich, D.; Chiarella, S.E.; Go, L.H.; Ghosh, A.K.; et al. Proteasomal inhibition after injury prevents fibrosis by modulating TGF-β(1) signalling. Thorax 2012, 67, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Penke, L.R.K.; Speth, J.; Wettlaufer, S.; Draijer, C.; Peters-Golden, M. Bortezomib Inhibits Lung Fibrosis and Fibroblast Activation without Proteasome Inhibition. Am. J. Respir. Cell Mol. Biol. 2022, 66, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Alonci, A.; Gerace, D.; Russo, S.; Innao, V.; Calabrò, L.; Musolino, C. New orally active proteasome inhibitors in multiple myeloma. Leuk. Res. 2014, 38, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, Y.; Gu, H.; Dong, M.; Cai, Z. Emerging agents and regimens for multiple myeloma. J. Hematol. Oncol. 2020, 13, 150. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fang, Y.; Fan, R.A.; Kirk, C.J. Proteasome Inhibitors and Their Pharmacokinetics, Pharmacodynamics, and Metabolism. Int. J. Mol. Sci. 2021, 22, 11595. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.V.; Hertlein, E.; Lu, Y.; Sass, E.J.; Lapalombella, R.; Chen, T.L.; Davis, M.E.; Woyach, J.A.; Lehman, A.; Jarjoura, D.; et al. The proteasome inhibitor carfilzomib functions independently of p53 to induce cytotoxicity and an atypical NF-κB response in chronic lymphocytic leukemia cells. Clin. Cancer Res. 2013, 19, 2406–2419. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Singh, A.V.; Aujay, M.; Kirk, C.J.; Bandi, M.; Ciccarelli, B.; Raje, N.; Richardson, P.; Anderson, K.C. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma. Blood 2010, 116, 4906–4915. [Google Scholar] [CrossRef] [PubMed]
- Kupperman, E.; Lee, E.C.; Cao, Y.; Bannerman, B.; Fitzgerald, M.; Berger, A.; Yu, J.; Yang, Y.; Hales, P.; Bruzzese, F.; et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010, 70, 1970–1980. [Google Scholar] [CrossRef]
- Dorsey, B.D.; Iqbal, M.; Chatterjee, S.; Menta, E.; Bernardini, R.; Bernareggi, A.; Cassarà, P.G.; D’Arasmo, G.; Ferretti, E.; De Munari, S.; et al. Discovery of a potent, selective, and orally active proteasome inhibitor for the treatment of cancer. J. Med. Chem. 2008, 51, 1068–1072. [Google Scholar] [CrossRef]
- Raninga, P.V.; Lee, A.; Sinha, D.; Dong, L.F.; Datta, K.K.; Lu, X.; Kalita-de Croft, P.; Dutt, M.; Hill, M.; Pouliot, N.; et al. Marizomib suppresses triple-negative breast cancer via proteasome and oxidative phosphorylation inhibition. Theranostics 2020, 10, 5259–5275. [Google Scholar] [CrossRef]
- Park, J.E.; Miller, Z.; Jun, Y.; Lee, W.; Kim, K.B. Next-generation proteasome inhibitors for cancer therapy. Transl.Res. 2018, 198, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Levin, N.; Spencer, A.; Harrison, S.J.; Chauhan, D.; Burrows, F.J.; Anderson, K.C.; Reich, S.D.; Richardson, P.G.; Trikha, M. Marizomib irreversibly inhibits proteasome to overcome compensatory hyperactivation in multiple myeloma and solid tumour patients. Br. J. Haematol. 2016, 174, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.; et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer Cell 2005, 8, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Shih, Y.H.; Shieh, T.M.; Tseng, Y.H. Proteasome Inhibitors Interrupt the Activation of Non-Canonical NF-κB Signaling Pathway and Induce Cell Apoptosis in Cytarabine-Resistant HL60 Cells. Int. J. Mol. Sci. 2021, 23, 361. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-C.; Liu, Y.-C.; Wu, Y.-H.; Lo, S.-H.; Wang, S.-C.; Li, C.-Y.; Dai, Z.-K.; Hsu, J.-H.; Yeh, C.-Y.; Tseng, Y.-H. Proteasome Inhibitors Decrease the Viability of Pulmonary Arterial Smooth Muscle Cells by Restoring Mitofusin-2 Expression under Hypoxic Conditions. Biomed. 2022, 10, 873. [Google Scholar]
- Visovsky, C. Chemotherapy-induced peripheral neuropathy. Cancer Investig. 2003, 21, 439–451. [Google Scholar] [CrossRef]
- Areti A, Yerra VG, Naidu V, Kumar A: Oxidative stress and nerve damage: Role in chemotherapy induced peripheral neuropathy. Redox Biol. 2014, 2, 289–295. [CrossRef]
- Kaplan, G.S.; Torcun, C.C.; Grune, T.; Ozer, N.K.; Karademir, B. Proteasome inhibitors in cancer therapy: Treatment regimen and peripheral neuropathy as a side effect. Free. Radic. Biol. Med. 2017, 103, 1–13. [Google Scholar] [CrossRef]
- Alé, A.; Bruna, J.; Navarro, X.; Udina, E. Neurotoxicity induced by antineoplastic proteasome inhibitors. Neurotoxicology 2014, 43, 28–35. [Google Scholar] [CrossRef]
- Meiners, S.; Ludwig, A.; Stangl, V.; Stangl, K. Proteasome inhibitors: Poisons and remedies. Med. Res. Rev. 2008, 28, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Spencer, A.; Harrison, S.; Zonder, J.; Badros, A.; Laubach, J.; Bergin, K.; Khot, A.; Zimmerman, T.; Chauhan, D.; Levin, N.; et al. A phase 1 clinical trial evaluating marizomib, pomalidomide and low-dose dexamethasone in relapsed and refractory multiple myeloma (NPI-0052-107): Final study results. Br. J. Haematol. 2018, 180, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Sitaraman, S.; Na, C.L.; Yang, L.; Filuta, A.; Bridges, J.P.; Weaver, T.E. Proteasome dysfunction in alveolar type 2 epithelial cells is associated with acute respiratory distress syndrome. Sci. Rep. 2019, 9, 12509. [Google Scholar] [CrossRef] [PubMed]
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Investig. 2020, 130, 5088–5099. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Compound | FDA Approval | Class | Effect | Activity | Administration |
|---|---|---|---|---|---|
| MG-132 | just used in laboratories | peptide aldehydes | reversible | T-L, CT-L | N/A |
| Bortezomib | FDA approval in 2003 | boronic acid | reversible | CT-L | IV, SC |
| Carfilzomib | FDA approval in 2012 | epoxyketones | irreversible | CT-L | IV |
| Oprozomib | currently in clinical trials | epoxyketones | irreversible | CT-L | Oral |
| Ixazomib | FDA approval in 2015 | boronic acid | reversible | CT-L | IV, Oral |
| Delanzomib | currently in clinical trials | boronic acid | reversible | CT-L | IV, Oral |
| Marizomib | currently in clinical trials | salinosporamide | irreversible | T-L, CT-L, C-L | IV, Oral |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, I.-C.; Liu, Y.-C.; Wu, Y.-H.; Lo, S.-H.; Dai, Z.-K.; Hsu, J.-H.; Tseng, Y.-H. Evaluation of Proteasome Inhibitors in the Treatment of Idiopathic Pulmonary Fibrosis. Cells 2022, 11, 1543. https://doi.org/10.3390/cells11091543
Chen I-C, Liu Y-C, Wu Y-H, Lo S-H, Dai Z-K, Hsu J-H, Tseng Y-H. Evaluation of Proteasome Inhibitors in the Treatment of Idiopathic Pulmonary Fibrosis. Cells. 2022; 11(9):1543. https://doi.org/10.3390/cells11091543
Chicago/Turabian StyleChen, I-Chen, Yi-Ching Liu, Yen-Hsien Wu, Shih-Hsing Lo, Zen-Kong Dai, Jong-Hau Hsu, and Yu-Hsin Tseng. 2022. "Evaluation of Proteasome Inhibitors in the Treatment of Idiopathic Pulmonary Fibrosis" Cells 11, no. 9: 1543. https://doi.org/10.3390/cells11091543
APA StyleChen, I.-C., Liu, Y.-C., Wu, Y.-H., Lo, S.-H., Dai, Z.-K., Hsu, J.-H., & Tseng, Y.-H. (2022). Evaluation of Proteasome Inhibitors in the Treatment of Idiopathic Pulmonary Fibrosis. Cells, 11(9), 1543. https://doi.org/10.3390/cells11091543