
RhoA Signaling in Neurodegenerative Diseases



Abstract
1. Introduction
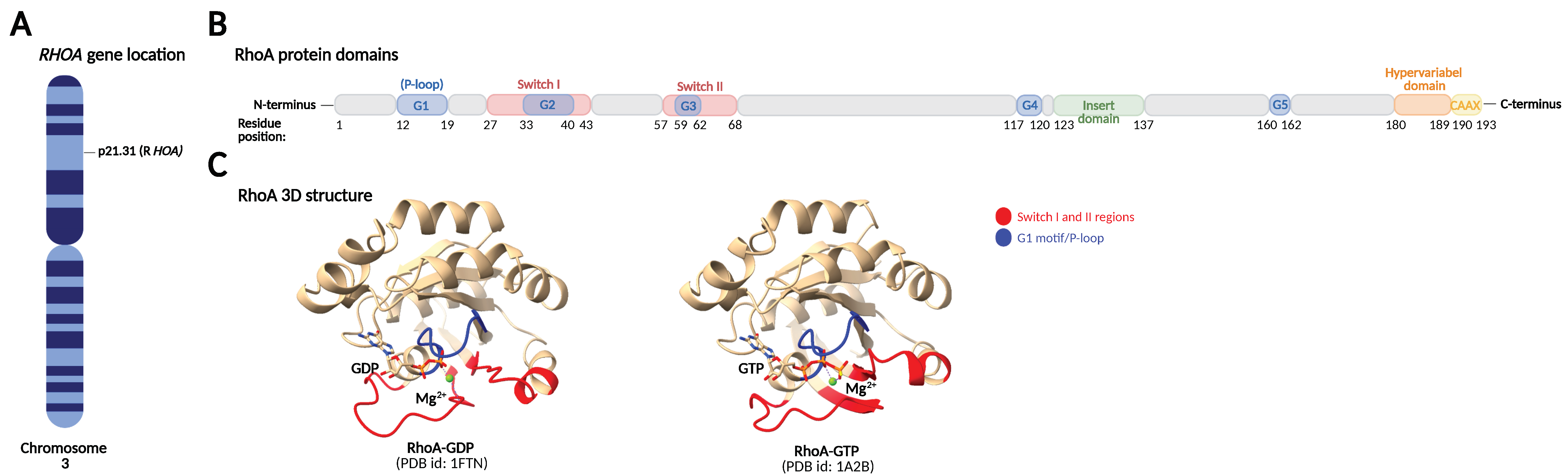
2. RhoA Structure and Regulation of RhoA Activity and Expression
2.1. GDP/GTP Cycling
2.2. Post-Translational Modifications of RhoA
2.2.1. Prenylation
2.2.2. Phosphorylations
2.2.3. Ubiquitinations
2.2.4. Additional PTMs
2.3. Transcriptional Regulation of RhoA
2.4. Post-Transcriptional Regulation of RhoA by miRNAs
3. Downstream Targets of RhoA
3.1. Cytoskeletal Dynamics
3.2. Cell Death
3.3. Mitochondria
3.4. Autophagy
3.5. Neuroinflammation
3.6. Gene Transcription
4. RhoA Signaling in Neurodegenerative Diseases
4.1. Parkinson’s Disease
4.2. Alzheimer’s Disease
4.3. Huntington’s Disease
4.4. Amyotrophic Lateral Sclerosis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stankiewicz, T.R.; Linseman, D.A. Rho family GTPases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell. Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.L.; von Delft, F.; Brennan, P.E. Targeting the Small GTPase Superfamily through Their Regulatory Proteins. Angew. Chem. Int. Ed. Engl. 2020, 59, 6342–6366. [Google Scholar] [CrossRef]
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef] [PubMed]
- DeGeer, J.; Lamarche-Vane, N. Rho GTPases in neurodegeneration diseases. Exp. Cell Res. 2013, 319, 2384–2394. [Google Scholar] [CrossRef] [PubMed]
- Arrazola Sastre, A.; Luque Montoro, M.; Galvez-Martin, P.; Lacerda, H.M.; Lucia, A.M.; Llavero, F.; Zugaza, J.L. Small GTPases of the Ras and Rho Families Switch on/off Signaling Pathways in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6312. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Horikoshi, Y.; Kuriyagawa, C.; Niiyama, Y. Rho/ROCK Pathway and Noncoding RNAs: Implications in Ischemic Stroke and Spinal Cord Injury. Int. J. Mol. Sci. 2021, 22, 11573. [Google Scholar] [CrossRef]
- Mulherkar, S.; Tolias, K.F. RhoA-ROCK Signaling as a Therapeutic Target in Traumatic Brain Injury. Cells 2020, 9, 245. [Google Scholar] [CrossRef]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Zamboni, V.; Jones, R.; Umbach, A.; Ammoni, A.; Passafaro, M.; Hirsch, E.; Merlo, G.R. Rho GTPases in Intellectual Disability: From Genetics to Therapeutic Opportunities. Int. J. Mol. Sci. 2018, 19, 1821. [Google Scholar] [CrossRef]
- Schaefer, A.; Reinhard, N.R.; Hordijk, P.L. Toward understanding RhoGTPase specificity: Structure, function and local activation. Small GTPases 2014, 5, 6. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Bi, F.; Zhou, X.; Zheng, Y. Rho GTPase regulation by miRNAs and covalent modifications. Trends Cell Biol. 2012, 22, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.R.; Olson, M.F. Transcriptional regulation of Rho GTPase signaling. Transcription 2011, 2, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Toma-Fukai, S.; Shimizu, T. Structural Insights into the Regulation Mechanism of Small GTPases by GEFs. Molecules 2019, 24, 3308. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mata, R.; Boulter, E.; Burridge, K. The ‘invisible hand’: Regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 2011, 12, 493–504. [Google Scholar] [CrossRef]
- Choi, E.K.; Kim, J.G.; Kim, H.J.; Cho, J.Y.; Jeong, H.; Park, Y.; Islam, R.; Cap, C.K.; Park, J.B. Regulation of RhoA GTPase and novel target proteins for ROCK. Small GTPases 2020, 11, 95–102. [Google Scholar] [CrossRef]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef]
- Bellanger, J.M.; Lazaro, J.B.; Diriong, S.; Fernandez, A.; Lamb, N.; Debant, A. The two guanine nucleotide exchange factor domains of Trio link the Rac1 and the RhoA pathways in vivo. Oncogene 1998, 16, 147–152. [Google Scholar] [CrossRef]
- Jaiswal, M.; Gremer, L.; Dvorsky, R.; Haeusler, L.C.; Cirstea, I.C.; Uhlenbrock, K.; Ahmadian, M.R. Mechanistic insights into specificity, activity, and regulatory elements of the regulator of G-protein signaling (RGS)-containing Rho-specific guanine nucleotide exchange factors (GEFs) p115, PDZ-RhoGEF (PRG), and leukemia-associated RhoGEF (LARG). J. Biol. Chem. 2011, 286, 18202–18212. [Google Scholar] [CrossRef]
- Arthur, W.T.; Ellerbroek, S.M.; Der, C.J.; Burridge, K.; Wennerberg, K. XPLN, a guanine nucleotide exchange factor for RhoA and RhoB, but not RhoC. J. Biol. Chem. 2002, 277, 42964–42972. [Google Scholar] [CrossRef]
- Sloan, C.M.; Quinn, C.V.; Peters, J.P.; Farley, J.; Goetzinger, C.; Wernli, M.; DeMali, K.A.; Ellerbroek, S.M. Divergence of Rho residue 43 impacts GEF activity. Small GTPases 2012, 3, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Hamel, B.; Monaghan-Benson, E.; Rojas, R.J.; Temple, B.R.; Marston, D.J.; Burridge, K.; Sondek, J. SmgGDS is a guanine nucleotide exchange factor that specifically activates RhoA and RhoC. J. Biol. Chem. 2011, 286, 12141–12148. [Google Scholar] [CrossRef]
- Rossman, K.L.; Der, C.J.; Sondek, J. GEF means go: Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 2005, 6, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.H.; Freuler, F.; Guerini, D.; Siehler, S. Reversible translocation of p115-RhoGEF by G(12/13)-coupled receptors. J. Cell. Biochem. 2008, 104, 1660–1670. [Google Scholar] [CrossRef] [PubMed]
- Song, E.H.; Oh, W.; Ulu, A.; Carr, H.S.; Zuo, Y.; Frost, J.A. Acetylation of the RhoA GEF Net1A controls its subcellular localization and activity. J. Cell Sci. 2015, 128, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Choi, K.C.; Hong, C.W.; Park, H.S.; Choi, E.K.; Kim, Y.S.; Park, J.B. Tyr42 phosphorylation of RhoA GTPase promotes tumorigenesis through nuclear factor (NF)-κB. Free Radic. Biol. Med. 2017, 112, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Self, A.J.; Kasmi, F.; Paterson, H.F.; Hall, A.; Marshall, C.J.; Ellis, C. rho family GTPase activating proteins p190, bcr and rhoGAP show distinct specificities in vitro and in vivo. EMBO J. 1993, 12, 5151–5160. [Google Scholar] [CrossRef]
- Thomas, S.; Overdevest, J.B.; Nitz, M.D.; Williams, P.D.; Owens, C.R.; Sanchez-Carbayo, M.; Frierson, H.F.; Schwartz, M.A.; Theodorescu, D. Src and caveolin-1 reciprocally regulate metastasis via a common downstream signaling pathway in bladder cancer. Cancer Res. 2011, 71, 832–841. [Google Scholar] [CrossRef]
- Lazarini, M.; Traina, F.; Machado-Neto, J.A.; Barcellos, K.S.; Moreira, Y.B.; Brandao, M.M.; Verjovski-Almeida, S.; Ridley, A.J.; Saad, S.T. ARHGAP21 is a RhoGAP for RhoA and RhoC with a role in proliferation and migration of prostate adenocarcinoma cells. Biochim. Biophys. Acta. 2013, 1832, 365–374. [Google Scholar] [CrossRef]
- Hildebrand, J.D.; Taylor, J.M.; Parsons, J.T. An SH3 domain-containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol. Cell. Biol. 1996, 16, 3169–3178. [Google Scholar] [CrossRef]
- Ito, H.; Morishita, R.; Nagata, K.I. Functions of Rhotekin, an Effector of Rho GTPase, and Its Binding Partners in Mammals. Int. J. Mol. Sci. 2018, 19, 2121. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, Y.; Kaibuchi, K.; Hori, Y.; Fujioka, H.; Araki, S.; Ueda, T.; Kikuchi, A.; Takai, Y. Molecular cloning and characterization of a novel type of regulatory protein (GDI) for the rho proteins, ras p21-like small GTP-binding proteins. Oncogene 1990, 5, 1321–1328. [Google Scholar] [PubMed]
- Leonard, D.; Hart, M.J.; Platko, J.V.; Eva, A.; Henzel, W.; Evans, T.; Cerione, R.A. The identification and characterization of a GDP-dissociation inhibitor (GDI) for the CDC42Hs protein. J. Biol. Chem. 1992, 267, 22860–22868. [Google Scholar] [CrossRef]
- Faure, J.; Dagher, M.C. Interactions between Rho GTPases and Rho GDP dissociation inhibitor (Rho-GDI). Biochimie 2001, 83, 409–414. [Google Scholar] [CrossRef]
- Adra, C.N.; Ko, J.; Leonard, D.; Wirth, L.J.; Cerione, R.A.; Lim, B. Identification of a novel protein with GDP dissociation inhibitor activity for the ras-like proteins CDC42Hs and rac I. Genes Chromosomes Cancer 1993, 8, 253–261. [Google Scholar] [CrossRef]
- Zalcman, G.; Closson, V.; Camonis, J.; Honore, N.; Rousseau-Merck, M.F.; Tavitian, A.; Olofsson, B. RhoGDI-3 is a new GDP dissociation inhibitor (GDI). Identification of a non-cytosolic GDI protein interacting with the small GTP-binding proteins RhoB and RhoG. J. Biol. Chem. 1996, 271, 30366–30374. [Google Scholar] [CrossRef]
- Yamashita, T.; Tohyama, M. The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat. Neurosci. 2003, 6, 461–467. [Google Scholar] [CrossRef]
- Takahashi, K.; Sasaki, T.; Mammoto, A.; Takaishi, K.; Kameyama, T.; Tsukita, S.; Takai, Y. Direct interaction of the Rho GDP dissociation inhibitor with ezrin/radixin/moesin initiates the activation of the Rho small G protein. J. Biol. Chem. 1997, 272, 23371–23375. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, J.G.; Moon, M.Y.; Park, S.H.; Park, J.B. IκB kinase γ nuclear factor-κB-essential modulator (IKKγ/NEMO) facilitates RhoA GTPase activation, which, in turn, activates Rho-associated KINASE (ROCK) to phosphorylate IKKbeta in response to transforming growth factor (TGF)- β1. J. Biol. Chem. 2014, 289, 1429–1440. [Google Scholar] [CrossRef]
- Kuhlmann, N.; Wroblowski, S.; Scislowski, L.; Lammers, M. RhoGDIalpha Acetylation at K127 and K141 Affects Binding toward Nonprenylated RhoA. Biochemistry 2016, 55, 304–312. [Google Scholar] [CrossRef]
- Kim, J.G.; Kwon, H.J.; Wu, G.; Park, Y.; Lee, J.Y.; Kim, J.; Kim, S.C.; Choe, M.; Kang, S.G.; Seo, G.Y.; et al. RhoA GTPase oxidation stimulates cell proliferation via nuclear factor-κB activation. Free Radic. Biol. Med. 2017, 103, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Eva, A.; Vecchio, G.; Rao, C.D.; Tronick, S.R.; Aaronson, S.A. The predicted DBL oncogene product defines a distinct class of transforming proteins. Proc. Natl. Acad. Sci. USA 1988, 85, 2061–2065. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.J.; Eva, A.; Evans, T.; Aaronson, S.A.; Cerione, R.A. Catalysis of guanine nucleotide exchange on the CDC42Hs protein by the dbl oncogene product. Nature 1991, 354, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Rossman, K.L.; Liu, B.; Ritola, K.D.; Chiang, D.; Campbell, S.L.; Burridge, K.; Der, C.J. Vav2 is an activator of Cdc42, Rac1, and RhoA. J. Biol. Chem. 2000, 275, 10141–10149. [Google Scholar] [CrossRef]
- Mosteller, R.; Han, J.; Das, B.; Broek, D. Biochemical analysis of regulation of Vav, a guanine-nucleotide exchange factor for Rho family of GTPases. Methods Enzymol. 2000, 325, 38–51. [Google Scholar] [CrossRef]
- Reddy, J.M.; Raut, N.G.R.; Seifert, J.L.; Hynds, D.L. Regulation of Small GTPase Prenylation in the Nervous System. Mol. Neurobiol. 2020, 57, 2220–2231. [Google Scholar] [CrossRef]
- Roberts, P.J.; Mitin, N.; Keller, P.J.; Chenette, E.J.; Madigan, J.P.; Currin, R.O.; Cox, A.D.; Wilson, O.; Kirschmeier, P.; Der, C.J. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 2008, 283, 25150–25163. [Google Scholar] [CrossRef]
- Winter-Vann, A.M.; Casey, P.J. Post-prenylation-processing enzymes as new targets in oncogenesis. Nat. Rev. Cancer 2005, 5, 405–412. [Google Scholar] [CrossRef]
- Lang, P.; Gesbert, F.; Delespine-Carmagnat, M.; Stancou, R.; Pouchelet, M.; Bertoglio, J. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 1996, 15, 510–519. [Google Scholar] [CrossRef]
- Ellerbroek, S.M.; Wennerberg, K.; Burridge, K. Serine phosphorylation negatively regulates RhoA in vivo. J. Biol. Chem. 2003, 278, 19023–19031. [Google Scholar] [CrossRef]
- Tkachenko, E.; Sabouri-Ghomi, M.; Pertz, O.; Kim, C.; Gutierrez, E.; Machacek, M.; Groisman, A.; Danuser, G.; Ginsberg, M.H. Protein kinase A governs a RhoA-RhoGDI protrusion-retraction pacemaker in migrating cells. Nat. Cell Biol. 2011, 13, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Rolli-Derkinderen, M.; Sauzeau, V.; Boyer, L.; Lemichez, E.; Baron, C.; Henrion, D.; Loirand, G.; Pacaud, P. Phosphorylation of serine 188 protects RhoA from ubiquitin/proteasome-mediated degradation in vascular smooth muscle cells. Circ. Res. 2005, 96, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, K.; Ishihara, S.; Mizutani, T.; Kawabata, K.; Haga, H. Compressive stress induces dephosphorylation of the myosin regulatory light chain via RhoA phosphorylation by the adenylyl cyclase/protein kinase A signaling pathway. PLoS ONE 2015, 10, e0117937. [Google Scholar] [CrossRef]
- Nusser, N.; Gosmanova, E.; Makarova, N.; Fujiwara, Y.; Yang, L.; Guo, F.; Luo, Y.; Zheng, Y.; Tigyi, G. Serine phosphorylation differentially affects RhoA binding to effectors: Implications to NGF-induced neurite outgrowth. Cell. Signal. 2006, 18, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Sawada, N.; Itoh, H.; Yamashita, J.; Doi, K.; Inoue, M.; Masatsugu, K.; Fukunaga, Y.; Sakaguchi, S.; Sone, M.; Yamahara, K.; et al. cGMP-dependent protein kinase phosphorylates and inactivates RhoA. Biochem. Biophys. Res. Commun. 2001, 280, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Sauzeau, V.; Le Jeune, H.; Cario-Toumaniantz, C.; Smolenski, A.; Lohmann, S.M.; Bertoglio, J.; Chardin, P.; Pacaud, P.; Loirand, G. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J. Biol. Chem. 2000, 275, 21722–21729. [Google Scholar] [CrossRef]
- Gayard, M.; Guilluy, C.; Rousselle, A.; Viollet, B.; Henrion, D.; Pacaud, P.; Loirand, G.; Rolli-Derkinderen, M. AMPK alpha 1-induced RhoA phosphorylation mediates vasoprotective effect of estradiol. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2634–2642. [Google Scholar] [CrossRef]
- Guilluy, C.; Rolli-Derkinderen, M.; Loufrani, L.; Bourge, A.; Henrion, D.; Sabourin, L.; Loirand, G.; Pacaud, P. Ste20-related kinase SLK phosphorylates Ser188 of RhoA to induce vasodilation in response to angiotensin II Type 2 receptor activation. Circ. Res. 2008, 102, 1265–1274. [Google Scholar] [CrossRef]
- Su, T.; Straight, S.; Bao, L.; Xie, X.; Lehner, C.L.; Cavey, G.S.; Teknos, T.N.; Pan, Q. PKC epsilon Phosphorylates and Mediates the Cell Membrane Localization of RhoA. ISRN Oncol. 2013, 2013, 329063. [Google Scholar] [CrossRef]
- Tang, J.; Ip, J.P.; Ye, T.; Ng, Y.P.; Yung, W.H.; Wu, Z.; Fang, W.; Fu, A.K.; Ip, N.Y. Cdk5-dependent Mst3 phosphorylation and activity regulate neuronal migration through RhoA inhibition. J. Neurosci. 2014, 34, 7425–7436. [Google Scholar] [CrossRef]
- Tong, J.; Li, L.; Ballermann, B.; Wang, Z. Phosphorylation and Activation of RhoA by ERK in Response to Epidermal Growth Factor Stimulation. PLoS ONE 2016, 11, e0147103. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Mialki, R.K.; Dong, S.; Khoo, A.; Mallampalli, R.K.; Zhao, Y.; Zhao, J. A new mechanism of RhoA ubiquitination and degradation: Roles of SCF(FBXL19) E3 ligase and Erk2. Biochim. Biophys. Acta. 2013, 1833, 2757–2764. [Google Scholar] [CrossRef] [PubMed]
- Uezu, A.; Okada, H.; Murakoshi, H.; del Vescovo, C.D.; Yasuda, R.; Diviani, D.; Soderling, S.H. Modified SH2 domain to phototrap and identify phosphotyrosine proteins from subcellular sites within cells. Proc. Natl. Acad. Sci. USA 2012, 109, E2929–E2938. [Google Scholar] [CrossRef]
- Kim, J.G.; Mahmud, S.; Min, J.K.; Lee, Y.B.; Kim, H.; Kang, D.C.; Park, H.S.; Seong, J.; Park, J.B. RhoA GTPase phosphorylated at tyrosine 42 by src kinase binds to β-catenin and contributes transcriptional regulation of vimentin upon Wnt3A. Redox Biol. 2021, 40, 101842. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, S.; Chen, S.; Chen, S.; Geng, Q.; Xu, D. c-Met-dependent phosphorylation of RhoA plays a key role in gastric cancer tumorigenesis. J. Pathol. 2019, 249, 126–136. [Google Scholar] [CrossRef]
- Wang, H.R.; Zhang, Y.; Ozdamar, B.; Ogunjimi, A.A.; Alexandrova, E.; Thomsen, G.H.; Wrana, J.L. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science 2003, 302, 1775–1779. [Google Scholar] [CrossRef] [PubMed]
- Ozdamar, B.; Bose, R.; Barrios-Rodiles, M.; Wang, H.R.; Zhang, Y.; Wrana, J.L. Regulation of the polarity protein Par6 by TGFβ receptors controls epithelial cell plasticity. Science 2005, 307, 1603–1609. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, Z.; Meng, M.; Zhao, Y.; Dong, N.; Yan, H.; Liu, L.; Ding, M.; Peng, H.B.; Shao, F. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol. Cell 2009, 35, 841–855. [Google Scholar] [CrossRef]
- Ibeawuchi, S.R.; Agbor, L.N.; Quelle, F.W.; Sigmund, C.D. Hypertension-causing Mutations in Cullin3 Protein Impair RhoA Protein Ubiquitination and Augment the Association with Substrate Adaptors. J. Biol. Chem. 2015, 290, 19208–19217. [Google Scholar] [CrossRef]
- Heo, J.; Raines, K.W.; Mocanu, V.; Campbell, S.L. Redox regulation of RhoA. Biochemistry 2006, 45, 14481–14489. [Google Scholar] [CrossRef]
- Aghajanian, A.; Wittchen, E.S.; Campbell, S.L.; Burridge, K. Direct activation of RhoA by reactive oxygen species requires a redox-sensitive motif. PLoS ONE 2009, 4, e8045. [Google Scholar] [CrossRef] [PubMed]
- Rafikov, R.; Dimitropoulou, C.; Aggarwal, S.; Kangath, A.; Gross, C.; Pardo, D.; Sharma, S.; Jezierska-Drutel, A.; Patel, V.; Snead, C.; et al. Lipopolysaccharide-induced lung injury involves the nitration-mediated activation of RhoA. J. Biol. Chem. 2014, 289, 4710–4722. [Google Scholar] [CrossRef] [PubMed]
- Worby, C.A.; Mattoo, S.; Kruger, R.P.; Corbeil, L.B.; Koller, A.; Mendez, J.C.; Zekarias, B.; Lazar, C.; Dixon, J.E. The fic domain: Regulation of cell signaling by adenylylation. Mol. Cell 2009, 34, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, G.; Sehr, P.; Wilm, M.; Selzer, J.; Mann, M.; Aktories, K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 1997, 387, 725–729. [Google Scholar] [CrossRef]
- Singh, U.S.; Kunar, M.T.; Kao, Y.L.; Baker, K.M. Role of transglutaminase II in retinoic acid-induced activation of RhoA-associated kinase-2. EMBO J. 2001, 20, 2413–2423. [Google Scholar] [CrossRef]
- Walther, D.J.; Peter, J.U.; Winter, S.; Holtje, M.; Paulmann, N.; Grohmann, M.; Vowinckel, J.; Alamo-Bethencourt, V.; Wilhelm, C.S.; Ahnert-Hilger, G.; et al. Serotonylation of small GTPases is a signal transduction pathway that triggers platelet α-granule release. Cell 2003, 115, 851–862. [Google Scholar] [CrossRef]
- Ihara, K.; Muraguchi, S.; Kato, M.; Shimizu, T.; Shirakawa, M.; Kuroda, S.; Kaibuchi, K.; Hakoshima, T. Crystal structure of human RhoA in a dominantly active form complexed with a GTP analogue. J. Biol. Chem. 1998, 273, 9656–9666. [Google Scholar] [CrossRef]
- Kristelly, R.; Gao, G.; Tesmer, J.J. Structural determinants of RhoA binding and nucleotide exchange in leukemia-associated Rho guanine-nucleotide exchange factor. J. Biol. Chem. 2004, 279, 47352–47362. [Google Scholar] [CrossRef]
- Li, R.; Zheng, Y. Residues of the Rho family GTPases Rho and Cdc42 that specify sensitivity to Dbl-like guanine nucleotide exchange factors. J. Biol. Chem. 1997, 272, 4671–4679. [Google Scholar] [CrossRef]
- De la Vega, M.; Burrows, J.F.; Johnston, J.A. Ubiquitination: Added complexity in Ras and Rho family GTPase function. Small GTPases 2011, 2, 192–201. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Islam, R.; Cho, J.Y.; Jeong, H.; Cap, K.C.; Park, Y.; Hossain, A.J.; Park, J.B. Regulation of RhoA GTPase and various transcription factors in the RhoA pathway. J. Cell. Physiol. 2018, 233, 6381–6392. [Google Scholar] [CrossRef] [PubMed]
- Nomikou, E.; Livitsanou, M.; Stournaras, C.; Kardassis, D. Transcriptional and post-transcriptional regulation of the genes encoding the small GTPases RhoA, RhoB, and RhoC: Implications for the pathogenesis of human diseases. Cell. Mol. Life Sci. 2018, 75, 2111–2124. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.H.; Lee, S.W.; Li, C.F.; Wang, J.; Yang, W.L.; Wu, C.Y.; Wu, J.; Nakayama, K.I.; Kang, H.Y.; Huang, H.Y.; et al. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat. Cell Biol. 2010, 12, 457–467. [Google Scholar] [CrossRef]
- Dopeso, H.; Rodrigues, P.; Bilic, J.; Bazzocco, S.; Carton-Garcia, F.; Macaya, I.; de Marcondes, P.G.; Anguita, E.; Masanas, M.; Jimenez-Flores, L.M.; et al. Mechanisms of inactivation of the tumour suppressor gene RHOA in colorectal cancer. Br. J. Cancer 2018, 118, 106–116. [Google Scholar] [CrossRef]
- Asensio-Juan, E.; Gallego, C.; Martinez-Balbas, M.A. The histone demethylase PHF8 is essential for cytoskeleton dynamics. Nucleic Acids Res. 2012, 40, 9429–9440. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Xiang, L.; Lee, S.J.; Chaturvedi, P.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Hypoxia-inducible factors mediate coordinated RhoA-ROCK1 expression and signaling in breast cancer cells. Proc. Natl. Acad. Sci. USA 2014, 111, E384–E393. [Google Scholar] [CrossRef]
- Gonzalez-Rodriguez, P.; Falcon, D.; Castro, M.J.; Urena, J.; Lopez-Barneo, J.; Castellano, A. Hypoxic induction of T-type Ca2+ channels in rat cardiac myocytes: Role of HIF-1α and RhoA/ROCK signalling. J. Physiol. 2015, 593, 4729–4745. [Google Scholar] [CrossRef]
- Yin, C.P.; Guan, S.H.; Zhang, B.; Wang, X.X.; Yue, S.W. Upregulation of HIF-1α protects neuroblastoma cells from hypoxia-induced apoptosis in a RhoA-dependent manner. Mol. Med. Rep. 2015, 12, 7123–7131. [Google Scholar] [CrossRef]
- Vertelov, G.; Kharazi, L.; Muralidhar, M.G.; Sanati, G.; Tankovich, T.; Kharazi, A. High targeted migration of human mesenchymal stem cells grown in hypoxia is associated with enhanced activation of RhoA. Stem Cell Res. Ther. 2013, 4, 5. [Google Scholar] [CrossRef]
- Ozturk, E.; Hobiger, S.; Despot-Slade, E.; Pichler, M.; Zenobi-Wong, M. Hypoxia regulates RhoA and Wnt/β-catenin signaling in a context-dependent way to control re-differentiation of chondrocytes. Sci. Rep. 2017, 7, 9032. [Google Scholar] [CrossRef] [PubMed]
- Goto, K.; Chiba, Y.; Matsusue, K.; Hattori, Y.; Maitani, Y.; Sakai, H.; Kimura, S.; Misawa, M. The proximal STAT6 and NF-κB sites are responsible for IL-13- and TNF-α-induced RhoA transcriptions in human bronchial smooth muscle cells. Pharmacol. Res. 2010, 61, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Sauzeau, V.; Rolli-Derkinderen, M.; Marionneau, C.; Loirand, G.; Pacaud, P. RhoA expression is controlled by nitric oxide through cGMP-dependent protein kinase activation. J. Biol. Chem. 2003, 278, 9472–9480. [Google Scholar] [CrossRef] [PubMed]
- Nomikou, E.; Stournaras, C.; Kardassis, D. Functional analysis of the promoters of the small GTPases RhoA and RhoB in embryonic stem cells. Biochem. Biophys. Res. Commun. 2017, 491, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Kong, W.; Yang, H.; He, L.; Zhao, J.J.; Coppola, D.; Dalton, W.S.; Cheng, J.Q. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol. Cell. Biol. 2008, 28, 6773–6784. [Google Scholar] [CrossRef]
- Korourian, A.; Roudi, R.; Shariftabrizi, A.; Madjd, Z. MicroRNA-31 inhibits RhoA-mediated tumor invasion and chemotherapy resistance in MKN-45 gastric adenocarcinoma cells. Exp. Biol. Med. 2017, 242, 1842–1847. [Google Scholar] [CrossRef]
- Ge, F.; Wang, C.; Wang, W.; Liu, W.; Wu, B. MicroRNA-31 inhibits tumor invasion and metastasis by targeting RhoA in human gastric cancer. Oncol. Rep. 2017, 38, 1133–1139. [Google Scholar] [CrossRef]
- Wang, S.C.; Lin, X.L.; Li, J.; Zhang, T.T.; Wang, H.Y.; Shi, J.W.; Yang, S.; Zhao, W.T.; Xie, R.Y.; Wei, F.; et al. MicroRNA-122 triggers mesenchymal-epithelial transition and suppresses hepatocellular carcinoma cell motility and invasion by targeting RhoA. PLoS ONE 2014, 9, e101330. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, W.; Yang, X.; Zhao, D.; Li, F.; Wang, H. MicroRNA-146a inhibits cell migration and invasion by targeting RhoA in breast cancer. Oncol. Rep. 2016, 36, 189–196. [Google Scholar] [CrossRef]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Xu, R.; Wang, J.; Hou, B.; Xie, A. MiR-133b ameliorates axon degeneration induced by MPP+ via targeting RhoA. Neuroscience 2016, 325, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.C.; Zheng, J.Y.; Tang, L.J.; Huang, B.S.; Li, K.; Tao, Y.; Yu, W.; Zhu, R.L.; Li, S.; Li, L.X. MiR-133b Promotes neurite outgrowth by targeting RhoA expression. Cell. Physiol. Biochem. 2015, 35, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.K.; Wang, X.F.; Lu, Q.B.; Xu, X.M. Altered microRNA expression following traumatic spinal cord injury. Exp. Neurol. 2009, 219, 424–429. [Google Scholar] [CrossRef]
- Yu, Y.M.; Gibbs, K.M.; Davila, J.; Campbell, N.; Sung, S.; Todorova, T.I.; Otsuka, S.; Sabaawy, H.E.; Hart, R.P.; Schachner, M. MicroRNA miR-133b is essential for functional recovery after spinal cord injury in adult zebrafish. Eur. J. Neurosci. 2011, 33, 1587–1597. [Google Scholar] [CrossRef]
- Xin, H.; Li, Y.; Liu, Z.; Wang, X.; Shang, X.; Cui, Y.; Zhang, Z.G.; Chopp, M. MiR-133b promotes neural plasticity and functional recovery after treatment of stroke with multipotent mesenchymal stromal cells in rats via transfer of exosome-enriched extracellular particles. Stem Cells 2013, 31, 2737–2746. [Google Scholar] [CrossRef]
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; Mills, A.A.; Karayiorgou, M.; et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 2008, 40, 751–760. [Google Scholar] [CrossRef]
- Xu, B.; Hsu, P.K.; Stark, K.L.; Karayiorgou, M.; Gogos, J.A. Derepression of a neuronal inhibitor due to miRNA dysregulation in a schizophrenia-related microdeletion. Cell 2013, 152, 262–275. [Google Scholar] [CrossRef]
- Liu, M.; Lang, N.; Chen, X.; Tang, Q.; Liu, S.; Huang, J.; Zheng, Y.; Bi, F. miR-185 targets RhoA and Cdc42 expression and inhibits the proliferation potential of human colorectal cells. Cancer Lett. 2011, 301, 151–160. [Google Scholar] [CrossRef]
- Hill, J.J.; Hashimoto, T.; Lewis, D.A. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 2006, 11, 557–566. [Google Scholar] [CrossRef]
- Ide, M.; Lewis, D.A. Altered cortical CDC42 signaling pathways in schizophrenia: Implications for dendritic spine deficits. Biol. Psychiatry 2010, 68, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Forstner, A.J.; Degenhardt, F.; Schratt, G.; Nothen, M.M. MicroRNAs as the cause of schizophrenia in 22q11.2 deletion carriers, and possible implications for idiopathic disease: A mini-review. Front. Mol. Neurosci. 2013, 6, 47. [Google Scholar] [CrossRef] [PubMed]
- Jian, Q.; An, Q.; Zhu, D.; Hui, K.; Liu, Y.; Chi, S.; Li, C. MicroRNA 340 is involved in UVB-induced dendrite formation through the regulation of RhoA expression in melanocytes. Mol. Cell. Biol. 2014, 34, 3407–3420. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, Z.F.; Wu, H.; Wang, W. miR-142-5p Improves Neural Differentiation and Proliferation of Adipose-Derived Stem Cells. Cell. Physiol. Biochem. 2018, 50, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, D.; Guo, D. MiR-124 Promote Neurogenic Transdifferentiation of Adipose Derived Mesenchymal Stromal Cells Partly through RhoA/ROCK1, but Not ROCK2 Signaling Pathway. PLoS ONE 2016, 11, e0146646. [Google Scholar] [CrossRef] [PubMed]
- Amin, E.; Dubey, B.N.; Zhang, S.C.; Gremer, L.; Dvorsky, R.; Moll, J.M.; Taha, M.S.; Nagel-Steger, L.; Piekorz, R.P.; Somlyo, A.V.; et al. Rho-kinase: Regulation, (dys)function, and inhibition. Biol. Chem. 2013, 394, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Guiler, W.; Koehler, A.; Boykin, C.; Lu, Q. Pharmacological Modulators of Small GTPases of Rho Family in Neurodegenerative Diseases. Front. Cell. Neurosci. 2021, 15, 661612. [Google Scholar] [CrossRef]
- Koch, J.C.; Tatenhorst, L.; Roser, A.E.; Saal, K.A.; Tonges, L.; Lingor, P. ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharmacol. Ther. 2018, 189, 1–21. [Google Scholar] [CrossRef]
- Arimura, N.; Menager, C.; Fukata, Y.; Kaibuchi, K. Role of CRMP-2 in neuronal polarity. J. Neurobiol. 2004, 58, 34–47. [Google Scholar] [CrossRef]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef]
- Watanabe, N.; Madaule, P.; Reid, T.; Ishizaki, T.; Watanabe, G.; Kakizuka, A.; Saito, Y.; Nakao, K.; Jockusch, B.M.; Narumiya, S. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 1997, 16, 3044–3056. [Google Scholar] [CrossRef] [PubMed]
- Narumiya, S.; Tanji, M.; Ishizaki, T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 2009, 28, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Piekny, A.; Werner, M.; Glotzer, M. Cytokinesis: Welcome to the Rho zone. Trends Cell Biol. 2005, 15, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, F.T.; Gai, M.; Berto, G.E.; Di Cunto, F. Of rings and spines: The multiple facets of Citron proteins in neural development. Small GTPases 2020, 11, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Naydenov, N.G.; Koblinski, J.E.; Ivanov, A.I. Anillin is an emerging regulator of tumorigenesis, acting as a cortical cytoskeletal scaffold and a nuclear modulator of cancer cell differentiation. Cell. Mol. Life Sci. 2021, 78, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Camera, P.; da Silva, J.S.; Griffiths, G.; Giuffrida, M.G.; Ferrara, L.; Schubert, V.; Imarisio, S.; Silengo, L.; Dotti, C.G.; Di Cunto, F. Citron-N is a neuronal Rho-associated protein involved in Golgi organization through actin cytoskeleton regulation. Nat. Cell Biol. 2003, 5, 1071–1078. [Google Scholar] [CrossRef]
- Camera, P.; Schubert, V.; Pellegrino, M.; Berto, G.; Vercelli, A.; Muzzi, P.; Hirsch, E.; Altruda, F.; Dotti, C.G.; Di Cunto, F. The RhoA-associated protein Citron-N controls dendritic spine maintenance by interacting with spine-associated Golgi compartments. EMBO Rep. 2008, 9, 384–392. [Google Scholar] [CrossRef]
- Mardakheh, F.K.; Self, A.; Marshall, C.J. RHO binding to FAM65A regulates Golgi reorientation during cell migration. J. Cell Sci. 2016, 129, 4466–4479. [Google Scholar] [CrossRef]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobio.l 2014, 115, 157–188. [Google Scholar] [CrossRef]
- Shi, J.; Wei, L. Rho kinase in the regulation of cell death and survival. Arch. Immunol. Ther. Exp. 2007, 55, 61–75. [Google Scholar] [CrossRef]
- Bertholet, A.M.; Delerue, T.; Millet, A.M.; Moulis, M.F.; David, C.; Daloyau, M.; Arnaune-Pelloquin, L.; Davezac, N.; Mils, V.; Miquel, M.C.; et al. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol. Dis. 2016, 90, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hu, C.; Huang, J.; Liu, W.; Lai, W.; Leng, F.; Tang, Q.; Liu, Y.; Wang, Q.; Zhou, M.; et al. ROCK1 induces dopaminergic nerve cell apoptosis via the activation of Drp1-mediated aberrant mitochondrial fission in Parkinson’s disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, Y.; Long, J.; Wang, J.; Haudek, S.B.; Overbeek, P.; Chang, B.H.; Schumacker, P.T.; Danesh, F.R. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012, 15, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Brand, C.S.; Tan, V.P.; Brown, J.H.; Miyamoto, S. RhoA regulates Drp1 mediated mitochondrial fission through ROCK to protect cardiomyocytes. Cell. Signal. 2018, 50, 48–57. [Google Scholar] [CrossRef]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef]
- Soudani, N.; Ghantous, C.M.; Farhat, Z.; Shebaby, W.N.; Zibara, K.; Zeidan, A. Calcineurin/NFAT Activation-Dependence of Leptin Synthesis and Vascular Growth in Response to Mechanical Stretch. Front. Physiol. 2016, 7, 433. [Google Scholar] [CrossRef]
- Schwarz, T.L. Mitochondrial trafficking in neurons. Cold Spring Harb. Perspect. Biol. 2013, 5, a011304. [Google Scholar] [CrossRef]
- Minin, A.A.; Kulik, A.V.; Gyoeva, F.K.; Li, Y.; Goshima, G.; Gelfand, V.I. Regulation of mitochondria distribution by RhoA and formins. J. Cell Sci. 2006, 119, 659–670. [Google Scholar] [CrossRef]
- Moskal, N.; Riccio, V.; Bashkurov, M.; Taddese, R.; Datti, A.; Lewis, P.N.; Angus McQuibban, G. ROCK inhibitors upregulate the neuroprotective Parkin-mediated mitophagy pathway. Nat. Commun. 2020, 11, 88. [Google Scholar] [CrossRef]
- McCoy, M.K.; Kaganovich, A.; Rudenko, I.N.; Ding, J.; Cookson, M.R. Hexokinase activity is required for recruitment of parkin to depolarized mitochondria. Hum. Mol. Genet. 2014, 23, 145–156. [Google Scholar] [CrossRef]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.O.; Wong, H.K.; Oyama, F.; Goswami, A.; Okuno, M.; Kino, Y.; Miyazaki, H.; Nukina, N. Inhibition of Rho kinases enhances the degradation of mutant huntingtin. J. Biol. Chem. 2009, 284, 13153–13164. [Google Scholar] [CrossRef] [PubMed]
- Mleczak, A.; Millar, S.; Tooze, S.A.; Olson, M.F.; Chan, E.Y. Regulation of autophagosome formation by Rho kinase. Cell. Signal. 2013, 25, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Koch, J.C.; Tonges, L.; Barski, E.; Michel, U.; Bahr, M.; Lingor, P. ROCK2 is a major regulator of axonal degeneration, neuronal death and axonal regeneration in the CNS. Cell Death Dis. 2014, 5, e1225. [Google Scholar] [CrossRef]
- Liu, F.T.; Yang, Y.J.; Wu, J.J.; Li, S.; Tang, Y.L.; Zhao, J.; Liu, Z.Y.; Xiao, B.G.; Zuo, J.; Liu, W.; et al. Fasudil, a Rho kinase inhibitor, promotes the autophagic degradation of A53T α-synuclein by activating the JNK 1/Bcl-2/beclin 1 pathway. Brain Res. 2016, 1632, 9–18. [Google Scholar] [CrossRef]
- Hamano, T.; Shirafuji, N.; Yen, S.H.; Yoshida, H.; Kanaan, N.M.; Hayashi, K.; Ikawa, M.; Yamamura, O.; Fujita, Y.; Kuriyama, M.; et al. Rho-kinase ROCK inhibitors reduce oligomeric tau protein. Neurobiol. Aging 2020, 89, 41–54. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Perez-Rojas, J.M.; Hernandez-Damian, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Kast, D.J.; Dominguez, R. The Cytoskeleton-Autophagy Connection. Curr. Biol. 2017, 27, R318–R326. [Google Scholar] [CrossRef]
- Nishimura, Y.; Itoh, K.; Yoshioka, K.; Ikeda, K.; Himeno, M. A role for small GTPase RhoA in regulating intracellular membrane traffic of lysosomes in invasive rat hepatoma cells. Histochem. J. 2002, 34, 189–213. [Google Scholar] [CrossRef]
- Nishimura, Y.; Itoh, K.; Yoshioka, K.; Uehata, M.; Himeno, M. Small guanosine triphosphatase Rho/Rho-associated kinase as a novel regulator of intracellular redistribution of lysosomes in invasive tumor cells. Cell Tissue Res. 2000, 301, 341–351. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Scheiblich, H.; Bicker, G. Regulation of Microglial Phagocytosis by RhoA/ROCK-Inhibiting Drugs. Cell. Mol. Neurobiol. 2017, 37, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ye, P.; Wang, D.; Liu, Y.; Cao, L.; Wang, Y.; Xu, Y.; Zhu, C. Involvement of RhoA/ROCK Signaling in Aβ-Induced Chemotaxis, Cytotoxicity and Inflammatory Response of Microglial BV2 Cells. Cell. Mol. Neurobiol. 2019, 39, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Roser, A.E.; Tonges, L.; Lingor, P. Modulation of Microglial Activity by Rho-Kinase (ROCK) Inhibition as Therapeutic Strategy in Parkinson’s Disease and Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2017, 9, 94. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Yu, J.; Guo, M.; Meng, J.; Liu, C.; Xie, Y.; Feng, L.; Xiao, B.; Ma, C. Rho kinase inhibitor fasudil regulates microglia polarization and function. Neuroimmunomodulation 2013, 20, 313–322. [Google Scholar] [CrossRef]
- Ding, J.; Li, Q.Y.; Wang, X.; Sun, C.H.; Lu, C.Z.; Xiao, B.G. Fasudil protects hippocampal neurons against hypoxia-reoxygenation injury by suppressing microglial inflammatory responses in mice. J. Neurochem. 2010, 114, 1619–1629. [Google Scholar] [CrossRef]
- He, Q.; Li, Y.H.; Guo, S.S.; Wang, Y.; Lin, W.; Zhang, Q.; Wang, J.; Ma, C.G.; Xiao, B.G. Inhibition of Rho-kinase by Fasudil protects dopamine neurons and attenuates inflammatory response in an intranasal lipopolysaccharide-mediated Parkinson’s model. Eur. J. Neurosci. 2016, 43, 41–52. [Google Scholar] [CrossRef]
- Moon, M.Y.; Kim, H.J.; Li, Y.; Kim, J.G.; Jeon, Y.J.; Won, H.Y.; Kim, J.S.; Kwon, H.Y.; Choi, I.G.; Ro, E.; et al. Involvement of small GTPase RhoA in the regulation of superoxide production in BV2 cells in response to fibrillar Abeta peptides. Cell. Signal. 2013, 25, 1861–1869. [Google Scholar] [CrossRef]
- Cap, K.C.; Kim, J.G.; Hamza, A.; Park, J.B. P-Tyr42 RhoA GTPase amplifies superoxide formation through p47phox, phosphorylated by ROCK. Biochem. Biophys. Res. Commun. 2020, 523, 972–978. [Google Scholar] [CrossRef]
- Labandeira-Garcia, J.L.; Rodriguez-Perez, A.I.; Villar-Cheda, B.; Borrajo, A.; Dominguez-Meijide, A.; Guerra, M.J. Rho Kinase and Dopaminergic Degeneration: A Promising Therapeutic Target for Parkinson’s Disease. Neuroscientist 2015, 21, 616–629. [Google Scholar] [CrossRef]
- Holtje, M.; Hoffmann, A.; Hofmann, F.; Mucke, C.; Grosse, G.; Van Rooijen, N.; Kettenmann, H.; Just, I.; Ahnert-Hilger, G. Role of Rho GTPase in astrocyte morphology and migratory response during in vitro wound healing. J. Neurochem. 2005, 95, 1237–1248. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.L.; Perreau, V.M.; Chen, M.J.; Cate, H.S.; Merlo, D.; Cheung, N.S.; O’Shea, R.D.; Beart, P.M. Transcriptomic profiling of astrocytes treated with the Rho kinase inhibitor fasudil reveals cytoskeletal and pro-survival responses. J. Cell. Physiol. 2012, 227, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Tonges, L.; Gunther, R.; Suhr, M.; Jansen, J.; Balck, A.; Saal, K.A.; Barski, E.; Nientied, T.; Gotz, A.A.; Koch, J.C.; et al. Rho kinase inhibition modulates microglia activation and improves survival in a model of amyotrophic lateral sclerosis. Glia 2014, 62, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Rajasekharan, S.; Bin, J.M.; Antel, J.P.; Kennedy, T.E. A central role for RhoA during oligodendroglial maturation in the switch from netrin-1-mediated chemorepulsion to process elaboration. J. Neurochem. 2010, 113, 1589–1597. [Google Scholar] [CrossRef]
- Harboe, M.; Torvund-Jensen, J.; Kjaer-Sorensen, K.; Laursen, L.S. Ephrin-A1-EphA4 signaling negatively regulates myelination in the central nervous system. Glia 2018, 66, 934–950. [Google Scholar] [CrossRef]
- Paintlia, A.S.; Paintlia, M.K.; Singh, A.K.; Singh, I. Modulation of Rho-Rock signaling pathway protects oligodendrocytes against cytokine toxicity via PPAR-α-dependent mechanism. Glia 2013, 61, 1500–1517. [Google Scholar] [CrossRef]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Yue, J.; Lopez, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef]
- Sanchez, M.; Gastaldi, L.; Remedi, M.; Caceres, A.; Landa, C. Rotenone-induced toxicity is mediated by Rho-GTPases in hippocampal neurons. Toxicol. Sci. 2008, 104, 352–361. [Google Scholar] [CrossRef]
- Zhou, Z.; Kim, J.; Insolera, R.; Peng, X.; Fink, D.J.; Mata, M. Rho GTPase regulation of alpha-synuclein and VMAT2: Implications for pathogenesis of Parkinson’s disease. Mol. Cell. Neurosci. 2011, 48, 29–37. [Google Scholar] [CrossRef]
- Barcia, C.; Ros, C.M.; Annese, V.; Carrillo-de Sauvage, M.A.; Ros-Bernal, F.; Gomez, A.; Yuste, J.E.; Campuzano, C.M.; de Pablos, V.; Fernandez-Villalba, E.; et al. ROCK/Cdc42-mediated microglial motility and gliapse formation lead to phagocytosis of degenerating dopaminergic neurons in vivo. Sci. Rep. 2012, 2, 809. [Google Scholar] [CrossRef] [PubMed]
- Tonges, L.; Frank, T.; Tatenhorst, L.; Saal, K.A.; Koch, J.C.; Szego, E.M.; Bahr, M.; Weishaupt, J.H.; Lingor, P. Inhibition of rho kinase enhances survival of dopaminergic neurons and attenuates axonal loss in a mouse model of Parkinson’s disease. Brain 2012, 135, 3355–3370. [Google Scholar] [CrossRef] [PubMed]
- Villar-Cheda, B.; Dominguez-Meijide, A.; Joglar, B.; Rodriguez-Perez, A.I.; Guerra, M.J.; Labandeira-Garcia, J.L. Involvement of microglial RhoA/Rho-kinase pathway activation in the dopaminergic neuron death. Role of angiotensin via angiotensin type 1 receptors. Neurobiol. Dis. 2012, 47, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Borrajo, A.; Rodriguez-Perez, A.I.; Villar-Cheda, B.; Guerra, M.J.; Labandeira-Garcia, J.L. Inhibition of the microglial response is essential for the neuroprotective effects of Rho-kinase inhibitors on MPTP-induced dopaminergic cell death. Neuropharmacology 2014, 85, 1–8. [Google Scholar] [CrossRef]
- Tatenhorst, L.; Eckermann, K.; Dambeck, V.; Fonseca-Ornelas, L.; Walle, H.; Lopes da Fonseca, T.; Koch, J.C.; Becker, S.; Tonges, L.; Bahr, M.; et al. Fasudil attenuates aggregation of alpha-synuclein in models of Parkinson’s disease. Acta Neuropathol. Commun. 2016, 4, 39. [Google Scholar] [CrossRef]
- Hou, L.; Bao, X.; Zang, C.; Yang, H.; Sun, F.; Che, Y.; Wu, X.; Li, S.; Zhang, D.; Wang, Q. Integrin CD11b mediates α-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. 2018, 14, 600–608. [Google Scholar] [CrossRef]
- Mattii, L.; Pardini, C.; Ippolito, C.; Bianchi, F.; Sabbatini, A.R.M.; Vaglini, F. Rho-inhibition and neuroprotective effect on rotenone-treated dopaminergic neurons in vitro. Neurotoxicology 2019, 72, 51–60. [Google Scholar] [CrossRef]
- Bogetofte, H.; Jensen, P.; Okarmus, J.; Schmidt, S.I.; Agger, M.; Ryding, M.; Norregaard, P.; Fenger, C.; Zeng, X.; Graakjaer, J.; et al. Perturbations in RhoA signalling cause altered migration and impaired neuritogenesis in human iPSC-derived neural cells with PARK2 mutation. Neurobiol. Dis. 2019, 132, 104581. [Google Scholar] [CrossRef]
- Lopez-Lopez, A.; Labandeira, C.M.; Labandeira-Garcia, J.L.; Munoz, A. Rho kinase inhibitor fasudil reduces l-DOPA-induced dyskinesia in a rat model of Parkinson’s disease. Br. J. Pharmacol. 2020, 177, 5622–5641. [Google Scholar] [CrossRef]
- Zhou, Y.; Su, Y.; Li, B.; Liu, F.; Ryder, J.W.; Wu, X.; Gonzalez-DeWhitt, P.A.; Gelfanova, V.; Hale, J.E.; May, P.C.; et al. Nonsteroidal anti-inflammatory drugs can lower amyloidogenic Aβ42 by inhibiting Rho. Science 2003, 302, 1215–1217. [Google Scholar] [CrossRef]
- Amano, M.; Kaneko, T.; Maeda, A.; Nakayama, M.; Ito, M.; Yamauchi, T.; Goto, H.; Fukata, Y.; Oshiro, N.; Shinohara, A.; et al. Identification of Tau and MAP2 as novel substrates of Rho-kinase and myosin phosphatase. J. Neurochem. 2003, 87, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Pedrini, S.; Carter, T.L.; Prendergast, G.; Petanceska, S.; Ehrlich, M.E.; Gandy, S. Modulation of statin-activated shedding of Alzheimer APP ectodomain by ROCK. PLoS Med. 2005, 2, e18. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.M.; Wilkinson, B.L.; Golde, T.E.; Landreth, G. Statins reduce amyloid-beta production through inhibition of protein isoprenylation. J. Biol. Chem. 2007, 282, 26832–26844. [Google Scholar] [CrossRef] [PubMed]
- Petratos, S.; Li, Q.X.; George, A.J.; Hou, X.; Kerr, M.L.; Unabia, S.E.; Hatzinisiriou, I.; Maksel, D.; Aguilar, M.I.; Small, D.H. The beta-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain 2008, 131, 90–108. [Google Scholar] [CrossRef]
- Huesa, G.; Baltrons, M.A.; Gomez-Ramos, P.; Moran, A.; Garcia, A.; Hidalgo, J.; Frances, S.; Santpere, G.; Ferrer, I.; Galea, E. Altered distribution of RhoA in Alzheimer’s disease and AβPP overexpressing mice. J. Alzheimer’s Dis. 2010, 19, 37–56. [Google Scholar] [CrossRef]
- Chacon, P.J.; Garcia-Mejias, R.; Rodriguez-Tebar, A. Inhibition of RhoA GTPase and the subsequent activation of PTP1B protects cultured hippocampal neurons against amyloid beta toxicity. Mol. Neurodegener. 2011, 6, 14. [Google Scholar] [CrossRef]
- Hamano, T.; Yen, S.H.; Gendron, T.; Ko, L.W.; Kuriyama, M. Pitavastatin decreases tau levels via the inactivation of Rho/ROCK. Neurobiol. Aging 2012, 33, 2306–2320. [Google Scholar] [CrossRef]
- Herskowitz, J.H.; Feng, Y.; Mattheyses, A.L.; Hales, C.M.; Higginbotham, L.A.; Duong, D.M.; Montine, T.J.; Troncoso, J.C.; Thambisetty, M.; Seyfried, N.T.; et al. Pharmacologic inhibition of ROCK2 suppresses amyloid-β production in an Alzheimer’s disease mouse model. J. Neurosci. 2013, 33, 19086–19098. [Google Scholar] [CrossRef]
- Tsushima, H.; Emanuele, M.; Polenghi, A.; Esposito, A.; Vassalli, M.; Barberis, A.; Difato, F.; Chieregatti, E. HDAC6 and RhoA are novel players in Abeta-driven disruption of neuronal polarity. Nat. Commun. 2015, 6, 7781. [Google Scholar] [CrossRef]
- Gentry, E.G.; Henderson, B.W.; Arrant, A.E.; Gearing, M.; Feng, Y.; Riddle, N.C.; Herskowitz, J.H. Rho Kinase Inhibition as a Therapeutic for Progressive Supranuclear Palsy and Corticobasal Degeneration. J. Neurosci. 2016, 36, 1316–1323. [Google Scholar] [CrossRef]
- Henderson, B.W.; Gentry, E.G.; Rush, T.; Troncoso, J.C.; Thambisetty, M.; Montine, T.J.; Herskowitz, J.H. Rho-associated protein kinase 1 (ROCK1) is increased in Alzheimer’s disease and ROCK1 depletion reduces amyloid-β levels in brain. J. Neurochem. 2016, 138, 525–531. [Google Scholar] [CrossRef]
- Gu, Q.F.; Yu, J.Z.; Wu, H.; Li, Y.H.; Liu, C.Y.; Feng, L.; Zhang, G.X.; Xiao, B.G.; Ma, C.G. Therapeutic effect of Rho kinase inhibitor FSD-C10 in a mouse model of Alzheimer’s disease. Exp. Ther. Med. 2018, 16, 3929–3938. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Salazar, S.V.; Cox, T.O.; Strittmatter, S.M. Pyk2 Signaling through Graf1 and RhoA GTPase Is Required for Amyloid-beta Oligomer-Triggered Synapse Loss. J. Neurosci. 2019, 39, 1910–1929. [Google Scholar] [CrossRef]
- Hu, Y.B.; Ren, R.J.; Zhang, Y.F.; Huang, Y.; Cui, H.L.; Ma, C.; Qiu, W.Y.; Wang, H.; Cui, P.J.; Chen, H.Z.; et al. Rho-associated coiled-coil kinase 1 activation mediates amyloid precursor protein site-specific Ser655 phosphorylation and triggers amyloid pathology. Aging Cell 2019, 18, e13001. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Xu, H.; Shen, J.X.; Pan, C.; Yu, Z.H.; Chen, J.J.; Zhu, X.L.; Cai, Y.F.; Lu, Y.P. RhoA/Rock2/Limk1/cofilin1 pathway is involved in attenuation of neuronal dendritic spine loss by paeonol in the frontal cortex of D-galactose and aluminuminduced Alzheimer’s diseaselike rat model. Acta Neurobiol. Exp. 2020, 80, 225–244. [Google Scholar] [CrossRef]
- Patnaik, A.; Zagrebelsky, M.; Korte, M.; Holz, A. Signaling via the p75 neurotrophin receptor facilitates amyloid-β-induced dendritic spine pathology. Sci. Rep. 2020, 10, 13322. [Google Scholar] [CrossRef] [PubMed]
- Pollitt, S.K.; Pallos, J.; Shao, J.; Desai, U.A.; Ma, A.A.; Thompson, L.M.; Marsh, J.L.; Diamond, M.I. A rapid cellular FRET assay of polyglutamine aggregation identifies a novel inhibitor. Neuron 2003, 40, 685–694. [Google Scholar] [CrossRef][Green Version]
- Shao, J.; Welch, W.J.; Diprospero, N.A.; Diamond, M.I. Phosphorylation of profilin by ROCK1 regulates polyglutamine aggregation. Mol. Cell. Biol. 2008, 28, 5196–5208. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Huang, Y.; Ma, A.A.; Lin, E.; Diamond, M.I. Y-27632 improves rotarod performance and reduces huntingtin levels in R6/2 mice. Neurobiol. Dis. 2009, 36, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.O.; Nukina, N. Enhanced degradation of mutant huntingtin by rho kinase inhibition is mediated through activation of proteasome and macroautophagy. Autophagy 2009, 5, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Deyts, C.; Galan-Rodriguez, B.; Martin, E.; Bouveyron, N.; Roze, E.; Charvin, D.; Caboche, J.; Betuing, S. Dopamine D2 receptor stimulation potentiates PolyQ-Huntingtin-induced mouse striatal neuron dysfunctions via Rho/ROCK-II activation. PLoS ONE 2009, 4, e8287. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yasumura, D.; Ma, A.A.; Matthes, M.T.; Yang, H.; Nielson, G.; Huang, Y.; Szoka, F.C.; Lavail, M.M.; Diamond, M.I. Intravitreal administration of HA-1077, a ROCK inhibitor, improves retinal function in a mouse model of huntington disease. PLoS ONE 2013, 8, e56026. [Google Scholar] [CrossRef] [PubMed]
- Brito, V.; Giralt, A.; Enriquez-Barreto, L.; Puigdellivol, M.; Suelves, N.; Zamora-Moratalla, A.; Ballesteros, J.J.; Martin, E.D.; Dominguez-Iturza, N.; Morales, M.; et al. Neurotrophin receptor p75(NTR) mediates Huntington’s disease-associated synaptic and memory dysfunction. J. Clin. Investig. 2014, 124, 4411–4428. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, J.L.; Day, M.; Peterson, J.D.; Xie, Z.; Kress, G.J.; Rafalovich, I.; Kondapalli, J.; Gertler, T.S.; Flajolet, M.; Greengard, P.; et al. Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington’s disease. Neuron 2014, 83, 178–188. [Google Scholar] [CrossRef]
- Miguez, A.; Garcia-Diaz Barriga, G.; Brito, V.; Straccia, M.; Giralt, A.; Gines, S.; Canals, J.M.; Alberch, J. Fingolimod (FTY720) enhances hippocampal synaptic plasticity and memory in Huntington’s disease by preventing p75NTR up-regulation and astrocyte-mediated inflammation. Hum. Mol. Genet. 2015, 24, 4958–4970. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, L.A.; Darwish, H.A.; Abdelsalam, R.M.; Amin, H.A. Role of Rho Kinase Inhibition in the Protective Effect of Fasudil and Simvastatin Against 3-Nitropropionic Acid-Induced Striatal Neurodegeneration and Mitochondrial Dysfunction in Rats. Mol. Neurobiol. 2016, 53, 3927–3938. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, K.L.; Chopra, V.; Rosas, H.D.; Malarick, K.; Hersch, S. Rho Kinase Pathway Alterations in the Brain and Leukocytes in Huntington’s Disease. Mol. Neurobiol. 2016, 53, 2132–2140. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.A.; Belichenko, N.P.; Ford, E.C.; Semaan, S.; Monbureau, M.; Aiyaswamy, S.; Holman, C.M.; Condon, C.; Shamloo, M.; Massa, S.M.; et al. A small molecule p75NTR ligand normalizes signalling and reduces Huntington’s disease phenotypes in R6/2 and BACHD mice. Hum. Mol. Genet. 2016, 25, 4920–4938. [Google Scholar] [CrossRef] [PubMed]
- Galan-Rodriguez, B.; Martin, E.; Brouillet, E.; Deglon, N.; Betuing, S.; Caboche, J. Coupling of D2R Short but not D2R Long receptor isoform to the Rho/ROCK signaling pathway renders striatal neurons vulnerable to mutant huntingtin. Eur. J. Neurosci. 2017, 45, 198–206. [Google Scholar] [CrossRef]
- Hu, J.H.; Chernoff, K.; Pelech, S.; Krieger, C. Protein kinase and protein phosphatase expression in the central nervous system of G93A mSOD over-expressing mice. J. Neurochem. 2003, 85, 422–431. [Google Scholar] [CrossRef]
- Takata, M.; Tanaka, H.; Kimura, M.; Nagahara, Y.; Tanaka, K.; Kawasaki, K.; Seto, M.; Tsuruma, K.; Shimazawa, M.; Hara, H. Fasudil, a rho kinase inhibitor, limits motor neuron loss in experimental models of amyotrophic lateral sclerosis. Br. J. Pharmacol. 2013, 170, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Conti, A.; Riva, N.; Pesca, M.; Iannaccone, S.; Cannistraci, C.V.; Corbo, M.; Previtali, S.C.; Quattrini, A.; Alessio, M. Increased expression of Myosin binding protein H in the skeletal muscle of amyotrophic lateral sclerosis patients. Biochim. Biophys. Acta. 2014, 1842, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Gunther, R.; Balck, A.; Koch, J.C.; Nientiedt, T.; Sereda, M.; Bahr, M.; Lingor, P.; Tonges, L. Rho Kinase Inhibition with Fasudil in the SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis-Symptomatic Treatment Potential after Disease Onset. Front. Pharmacol. 2017, 8, 17. [Google Scholar] [CrossRef]
- Joshi, A.R.; Muke, I.; Bobylev, I.; Lehmann, H.C. ROCK inhibition improves axonal regeneration in a preclinical model of amyotrophic lateral sclerosis. J. Comp. Neurol. 2019, 527, 2334–2340. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Corti, O.; Hampe, C.; Darios, F.; Ibanez, P.; Ruberg, M.; Brice, A. Parkinson’s disease: From causes to mechanisms. Comptes Rendus Biol. 2005, 328, 131–142. [Google Scholar] [CrossRef]
- Corti, O.; Lesage, S.; Brice, A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef]
- Yang, L.; Mao, K.; Yu, H.; Chen, J. Neuroinflammatory Responses and Parkinson’ Disease: Pathogenic Mechanisms and Therapeutic Targets. J. Neuroimmune Pharmacol. 2020, 15, 830–837. [Google Scholar] [CrossRef]
- Gcwensa, N.Z.; Russell, D.L.; Cowell, R.M.; Volpicelli-Daley, L.A. Molecular Mechanisms Underlying Synaptic and Axon Degeneration in Parkinson’s Disease. Front. Cell. Neurosci. 2021, 15, 626128. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Wang, Y.; Huang, Z.; Zou, Q.; Pu, Y.; Yu, C.; Cai, Z. Role of RhoA/ROCK signaling in Alzheimer’s disease. Behav. Brain Res. 2021, 414, 113481. [Google Scholar] [CrossRef] [PubMed]
- Boimel, M.; Grigoriadis, N.; Lourbopoulos, A.; Touloumi, O.; Rosenmann, D.; Abramsky, O.; Rosenmann, H. Statins reduce the neurofibrillary tangle burden in a mouse model of tauopathy. J. Neuropathol. Exp. Neurol. 2009, 68, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Forner, S.; Baglietto-Vargas, D.; Martini, A.C.; Trujillo-Estrada, L.; LaFerla, F.M. Synaptic Impairment in Alzheimer’s Disease: A Dysregulated Symphony. Trends Neurosci. 2017, 40, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| GTP/GDP Regulatory Proteins | Factors | References |
|---|---|---|
| GEFs | Dbl | [42,43] |
| Vav1-3 | [44,45] | |
| Trio | [18] | |
| p115-RhoGEF | [19] | |
| p190-RhoGEF | [19] | |
| LARG | [19] | |
| PDZ-RhoGEF | [19] | |
| XPLN | [20,21] | |
| SmgGDS | [22] | |
| Net1 | [25] | |
| GAPs | p190-RhoGAP | [27] |
| ARHGAP21 | [29] | |
| Graf1 | [30] | |
| GDIs | RhoGDI-1 | [32,33] |
| RhoGDI-2 | [34] | |
| GDFs | p75NTR | [37] |
| ERM proteins | [38] | |
| IKKγ/NEMO | [39] |
| PTM | Enzyme/Factor | Sites | Effect | References |
|---|---|---|---|---|
| Prenylation | GGTase-I | Cys190 | Membrane anchoring | [46] |
| Phosphorylations | PKA | Ser188 | Plasma membrane retraction by increasing interaction with GDI | [49,50,51] |
| Protects GTP-bound RhoA from ubiquitin-mediated proteasomal degradation | [52] | |||
| Decreases binding to RhoA effector protein ROCK | [53,54] | |||
| PKG | Ser188 | Translocation to the cytosol by increasing interaction with GDI | [50,55] | |
| Protects GTP-bound RhoA from proteasomal degradation | [52,56] | |||
| AMPKa1 | Ser188 | Inactivation | [57] | |
| SLK | Ser188 | Inactivation | [58] | |
| PKC | Thr127 Ser188 | Translocation to the plasma membrane | [59] | |
| Mst3 | Ser26 | Inactivation by hindering GEF interaction | [60] | |
| ERK | Ser88 Thr100 | Upregulates RhoA activity | [61] | |
| Unknown | Induce ubiquitin-mediated proteasomal degradation | [62] | ||
| Bcr-Abl Src | Tyr34 Tyr66 | Inhibits effector protein binding and GEF interactions | [63] | |
| Src | Tyr42 | Activation by GDI dissociation and GEF interaction | [26,64] | |
| c-Met | Tyr42 | Induce ubiquitin-mediated proteasomal degradation | [65] | |
| Ubiquitinations | SMURF1 | Lys6 Lys7 | Targets active GTP-bound RhoA for proteasomal degradation | [66,67] |
| CUL3BACURD | Unknown | Targets inactive GDP-bound RhoA for proteasomal degradation | [68,69] | |
| SCFFBXL19 | Lys135 | Targets both active and inactive RhoA for proteasomal degradation | [62] | |
| Oxidation | ROS | Cys16 Cys20 | Inactivation by preventing guanine nucleotide binding and GEF association | [70] |
| Activation by GDI dissociation and GEF interaction possible requiring combined P-Tyr42 | [41,71] | |||
| Nitration | NO | Tyr34 | Activation | [72] |
| Adenylation | Fic domain-containing proteins | Tyr34 | Inactivation by steric hindrance of the GDP/GTP binding site in the switch I region | [73] |
| Transglutamination | Transglutaminase | Gln63 | Constitutive activation by abolishing the intrinsic and GAP-stimulated GTPase activity | [74,75,76] |
| Neurodegenerative Disease | Study Model | Finding | Target | Inhibitor(s) | References |
|---|---|---|---|---|---|
| Parkinson’s disease (PD) | Rat primary hippocampal and mesencephalic cultures | Rotenone treatment increases RhoA activity and inhibition of ROCK rescues rotenone-induced inhibition of neurite outgrowth | ROCK | Y27632 | [169] |
| Mouse MN9D dopaminergic cell line | RhoA inhibition leads to neurite extension and reduces α-synuclein expression | RhoA | C3 transferase and db-cAMP | [170] | |
| MPTP mouse model | ROCK inhibition prevents microglia from eliminating dopaminergic neurons in MPTP-treated mice | ROCK | HA-1077 | [171] | |
| MPTP mouse model and rat primary midbrain dopaminergic neurons | ROCK inhibition enhances survival of dopaminergic neurons and attenuates axonal loss | ROCK | Fasudil | [172] | |
| MPTP mouse model and rat primary mesencephalic cultures | Upregulation of RhoA and ROCK in the substantia nigra pars compacta of MPTP-treated mice. ROCK inhibition protects against MPTP-induced dopaminergic cell death both in vivo and in vitro | ROCK | Y27632 | [173] | |
| Rat primary mesencephalic cultures | Inhibition of microglial ROCK is essential to protect against MPTP-induced dopaminergic cell death, but ROCK inhibition also induces a direct effect against axonal retraction in surviving dopaminergic neurons | ROCK | Y27632 | [174] | |
| Transgenic mouse model expressing human A53T α-synuclein | ROCK inhibition decreases midbrain α-synuclein pathology and improves motor and cognitive functions | ROCK | Fasudil | [175] | |
| Rat primary mesencephalic cultures and PC12 cells | MPTP treatment upregulates RhoA expression, and RhoA inhibition attenuates MPTP-induced α-synuclein upregulation and ameliorates axon degeneration | RhoA | miR-133b overexpression | [102] | |
| Human SH-SY5Y cells | Increased ROCK activity in A53T α-synuclein-overexpressing cells. ROCK inhibition induces clearance of A53T α-synuclein by activating autophagy | ROCK | Fasudil | [145] | |
| Murine primary microglial cultures | α-synuclein induces microglial ROS production through CD11b integrin-mediated RhoA/NOX activation | RhoA | siRNA | [176] | |
| Murine primary mesencephalic cultures | Rotenone induces RhoA activation, and RhoA inhibition protects dopaminergic neurons against rotenone-induced neurite damage | RhoA | C3 transferase and simvastatin | [177] | |
| MPTP-treated PC12 cells and MPTP mouse model | ROCK inhibition rescues Drp1-mediated aberrant mitochondrial fission and apoptosis of dopaminergic neurons both in vitro and in vivo | ROCK | Y27632 | [132] | |
| Human-induced pluripotent stem cell-derived neurons with PARK2 mutation | Increased RhoA signaling caused altered migration and impaired neuritogenesis, which could be rescued by RhoA inhibition | RhoA | Rhosin | [178] | |
| Human HEK293 cells, SH-SY5Y cells, and paraquat Drosophila model | A screen of ~3000 compounds identified several ROCK inhibitors that rescued mitochondrial damage by upregulating parkin-mediated mitophagy. The ROCK inhibitor SR3677 was found to be most efficient | ROCK | SR3677 | [139] | |
| 6-OHDA lesioned rats | Increased RhoA and ROCK expression in 6-OHDA lesioned rats with dyskinesia. ROCK inhibition reduces the development of dyskinesia and also inhibits already established dyskinesia | ROCK | Fasudil | [179] | |
| Alzheimer’s disease (AD) | Human SH-SY5Y cells | Aβ production by secretase-dependent cleavage of APP is reduced by RhoA/ROCK inhibition | RhoA and ROCK | NSAIDs, C3 transferase, Y27632 and overexpression of dominant negative RhoA | [180] |
| Tau-transfected COS7 cells | ROCK phosphorylates tau and reduces the activity of tau to promote microtubule assembly | - | - | [181] | |
| Mouse N2a cells | Statins stimulate sAPPα shedding by ROCK inhibition | RhoA | Atorvastatin and simvastatin | [182] | |
| Mouse N2a cells | Statins reduce Aβ production through inhibition of Rho and Rab family proteins | RhoA | Simvastatin and lovastatin | [183] | |
| Human SH-SY5Y cells | Aβ exposure leads to inhibition of neurite outgrowth through increased RhoA activation, which is rescued by ROCK inhibition | ROCK | Y27632 | [184] | |
| Postmortem human AD brains and APP-overexpressing mouse model | RhoA is decreased in human AD brains, and remaining RhoA colocalizes with hyperphosphorylated tau. In APP-overexpressing mice, RhoA was decreased within synapses but increased in degenerating neurites | - | - | [185] | |
| Murine primary hippocampal neurons and PC12 cells | Aβ activates RhoA by binding p75NTR. Inhibition of RhoA prevents the deleterious effect of Aβ on cultured hippocampal neurons | RhoA | C3 transferase and overexpression of dominant negative RhoA | [186] | |
| Human M1C cells expressing tau and murine primary cortical neurons | Inhibition of RhoA/ROCK signaling using pitavastatin reduces total tau and phosphorylated tau levels | RhoA | Pitavastatin | [187] | |
| Human SH-SY5Y cells, murine primary cortical neurons, HEK293 cells, and 5XFAD mouse model | Selective ROCK2 inhibition reduces Aβ production by inhibiting BACE1 activity. The mechanism involves altered BACE1 endocytic distribution and APP trafficking to lysosomes | ROCK2 | SR3677 | [188] | |
| Murine primary hippocampal neurons | Soluble Aβ disrupts actin and microtubule dynamics via activation of RhoA and inhibition of histone deacetylase 6, which is rescued by ROCK inhibition | ROCK | Y27632 | [189] | |
| Human SH-SY5Y cells, murine primary cortical neurons, and tau-expressing drosophila | ROCK inhibition diminishes total and phosphorylated tau levels through enhancing autophagy and reducing tau mRNA | ROCK | shRNA, SR3677, and Fasudil | [190] | |
| Postmortem human AD brains, murine primary cortical neurons, and ROCK1−/− mice | ROCK1 protein level is increased in human AD brains, and ROCK1 inhibition reduces Aβ levels | ROCK1 | shRNA | [191] | |
| APP/PS1 mouse model | ROCK inhibition attenuated Aβ burden, tau phosphorylation and BACE expression and increased expression of synapse-associated proteins and neurotrophic factors | ROCK | FSD-C10 | [192] | |
| APP/PS1 mouse model, WT mice injected with fAβ and BV2 microglial cells | Increased RhoA expression in reactive microglia in vivo. RhoA/ROCK signaling is essential for Aβ-induced chemotactic migration, cytotoxicity, and inflammatory responses in microglial BV2 cells. ROCK inhibition suppresses the inflammatory responses | ROCK | Fasudil and Y27632 | [153] | |
| Murine primary hippocampal neurons, HEK293 cells, APP mouse model, and Pyk2−/− mouse model | Aβ induces an increase in actin contractility via Pyk2/RhoGAP Graf1/RhoA-regulated ROCK activation, culminating in dendritic spine retraction. Spine loss is rescued by RhoA and ROCK inhibition | RhoA and ROCK | Y27632 and overexpression of dominant negative RhoA | [193] | |
| APP/PS1 mouse model | APP is a substrate for ROCK, which phosphorylates its Ser655 residue to promote amyloidogenic processing of APP by BACE1. ROCK inhibition rescues Aβ pathology and improves learning and memory in APP/PS1 mice | ROCK | shRNA and Y27632 | [194] | |
| Human M1C cells expressing WT tau, murine primary neurons, and rTG4510 mouse model | ROCK inhibition reduces total tau levels, tau phosphorylation, and oligomerization and upregulates autophagy and proteasome pathways | ROCK | H1152, Y-27632, and Fasudil | [146] | |
| D-galactose and aluminum rat model | Paeonol rescues neuronal dendritic spine loss through inhibition of the RhoA/ROCK/LIMK1/cofilin1 pathway | RhoA | Paeonol | [195] | |
| p75NTR−/− murine primary hippocampal neurons | Aβ activates RhoA through p75NTR. Inhibition of p75NTR-mediated RhoA activation or ROCK protects neurons from Aβ-induced dendritic spine pathology | p75NTR and ROCK | TAT-Pep5 peptide and Y27632 | [196] | |
| Huntington’s disease (HD) | COS7 cells, HEK293 cells, C17-2 cells, and HD drosophila model | Y27632 was identified in a compound screen to reduce polyglutamine toxicity. Verification in cellular and drosophila models of HD shows reduced Htt aggregation and toxicity | ROCK | Y27632 | [197] |
| HEK293 cells and rat primary cortical neurons | ROCK inhibition reduces Htt aggregation | ROCK | Y27632 | [198] | |
| R6/2 mouse model | ROCK inhibition improves rotarod performance and reduces soluble mutant Htt in R6/2 mice, but no effect on Htt aggregation, cellular atrophy in the striatum, or lifespan | ROCK | Y27632 | [199] | |
| Mouse N2a cells and MEFs | ROCK inhibition reduces Htt aggregation via activation of the ubiquitin proteasome system and macroautophagy | ROCK | Y27632 | [200] | |
| Mouse primary striatal neurons | Dopaminergic D2 receptor stimulation act in synergy with mutant Htt to increase aggregates formation and striatal cell death through activation of RhoA/ROCK. This could be rescued by ROCK inhibition | ROCK | siRNA, Y27632 and Fasudil | [201] | |
| R6/2 mouse model | ROCK inhibition improved retinal function in the R6/2 mouse model | ROCK | Fasudil | [202] | |
| R6/1 and HdhQ7/Q111 knock-in mouse models, p75NTR-overexpressing primary hippocampal neurons and postmortem human HD brains | p75NTR levels is increased in HD mouse models and in post-mortem brain tissue from HD patients. Normalizing p75NTR levels prevented memory and synaptic deficits in HD mutant mice. Inhibition of RhoA normalized dendritic spine density in primary hippocampal cultures | p75NTR and RhoA | shRNA and C3 transferase | [203] | |
| Mutant BACHD mice | Plasticity in indirect pathway spiny projection neurons from BACHD mutant mice can be rescued by inhibition of p75NTR/RhoA signaling | p75NTR and ROCK | TAT-Pep5 peptide and Y27632 | [204] | |
| R6/1 mouse model | Inhibition of p75NTR rescues dendritic spine loss through negative regulation of RhoA | p75NTR | FTY720 | [205] | |
| 3-NP rat model | Inhibition of RhoA/ROCK signaling inhibited 3-NP-induced neurotoxicity and mitochondrial dysfunction | RhoA and ROCK | Simvastatin and Fasudil | [206] | |
| HD human blood leukocytes, postmortem HD brain tissue, and R6/2 mouse model | Increased mRNA expression of RhoA, ROCK, and downstream cytoskeletal-related effector proteins in leukocytes and frontal cortex of postmortem brain tissue from HD patients and in striatum of R6/2 mice | - | - | [207] | |
| R6/2 and BACHD mouse models | Normalizing p75NTR signaling reduces key HD neuropathologies, including Htt aggregation and dendritic spine loss, and improves cognition and motor performance in HD mouse models. The effect was mediated by downregulation of ROCK and PTEN | p75NTR | LM11A-31 (ligand) | [208] | |
| HEK293 cells and mouse primary striatal neurons | The dopamine D2 receptor short isoform, but not the long isoform, is coupled to the RhoA/ROCK/cofilin pathway and its involvement in striatal vulnerability to mutant Htt | - | - | [209] | |
| Amyotrophic lateral sclerosis (ALS) | SOD1-G93A mouse model | Increased ROCK expression in the spinal cord of G93A SOD1 mice | - | - | [210] |
| SOD1-G93A mouse model | ROCK inhibition delays disease onset and extends survival in SOD1-G93A mice. ROCK inhibition reduced the phosphorylation of PTEN, resulting in neuronal protection via increasing phosphorylated Akt | ROCK | Fasudil | [211] | |
| Human ALS skeletal muscle biopsies | Increased ROCK expression in skeletal muscles from sporadic ALS patients | - | - | [212] | |
| SOD1-G93A mouse model | ROCK inhibition delays onset and extends survival in SOD1-G93A mice via reduced microgliosis and decreased release of proinflammatory cytokines and chemokines | ROCK | Fasudil | [163] | |
| SOD1-G93A mouse model | ROCK inhibition in a more advanced disease stage in SOD1-G93A mice improved motor function in male mice but did not increase motor neuron survival or reduce microglial infiltration | ROCK | Fasudil | [213] | |
| SOD1-G93A mouse model | ROCK inhibition improved axonal regeneration of injured motor axons after sciatic crush in SOD1-G93A mice | ROCK | Y27632 | [214] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, S.I.; Blaabjerg, M.; Freude, K.; Meyer, M. RhoA Signaling in Neurodegenerative Diseases. Cells 2022, 11, 1520. https://doi.org/10.3390/cells11091520
Schmidt SI, Blaabjerg M, Freude K, Meyer M. RhoA Signaling in Neurodegenerative Diseases. Cells. 2022; 11(9):1520. https://doi.org/10.3390/cells11091520
Chicago/Turabian StyleSchmidt, Sissel Ida, Morten Blaabjerg, Kristine Freude, and Morten Meyer. 2022. "RhoA Signaling in Neurodegenerative Diseases" Cells 11, no. 9: 1520. https://doi.org/10.3390/cells11091520
APA StyleSchmidt, S. I., Blaabjerg, M., Freude, K., & Meyer, M. (2022). RhoA Signaling in Neurodegenerative Diseases. Cells, 11(9), 1520. https://doi.org/10.3390/cells11091520