
MicroRNA-145 Impairs Classical Non-Homologous End-Joining in Response to Ionizing Radiation-Induced DNA Double-Strand Breaks via Targeting DNA-PKcs

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Cell Transfection
2.3. Antibodies
2.4. Western-Blot Analysis
2.5. Reverse Transcription-Quantitative Real-Time PCR (RT-qPCR)
2.6. MicroRNA Luciferase Reporter Assay
2.7. Immunofluorescence Cell Staining
2.8. NHEJ Assay
2.9. HR Assay
2.10. In Vitro NHEJ Assay
2.11. Single-Cell Gel-Electrophoresis (Comet Assay)
2.12. Clonal Survival Assay
2.13. Statistical Analyses
3. Results
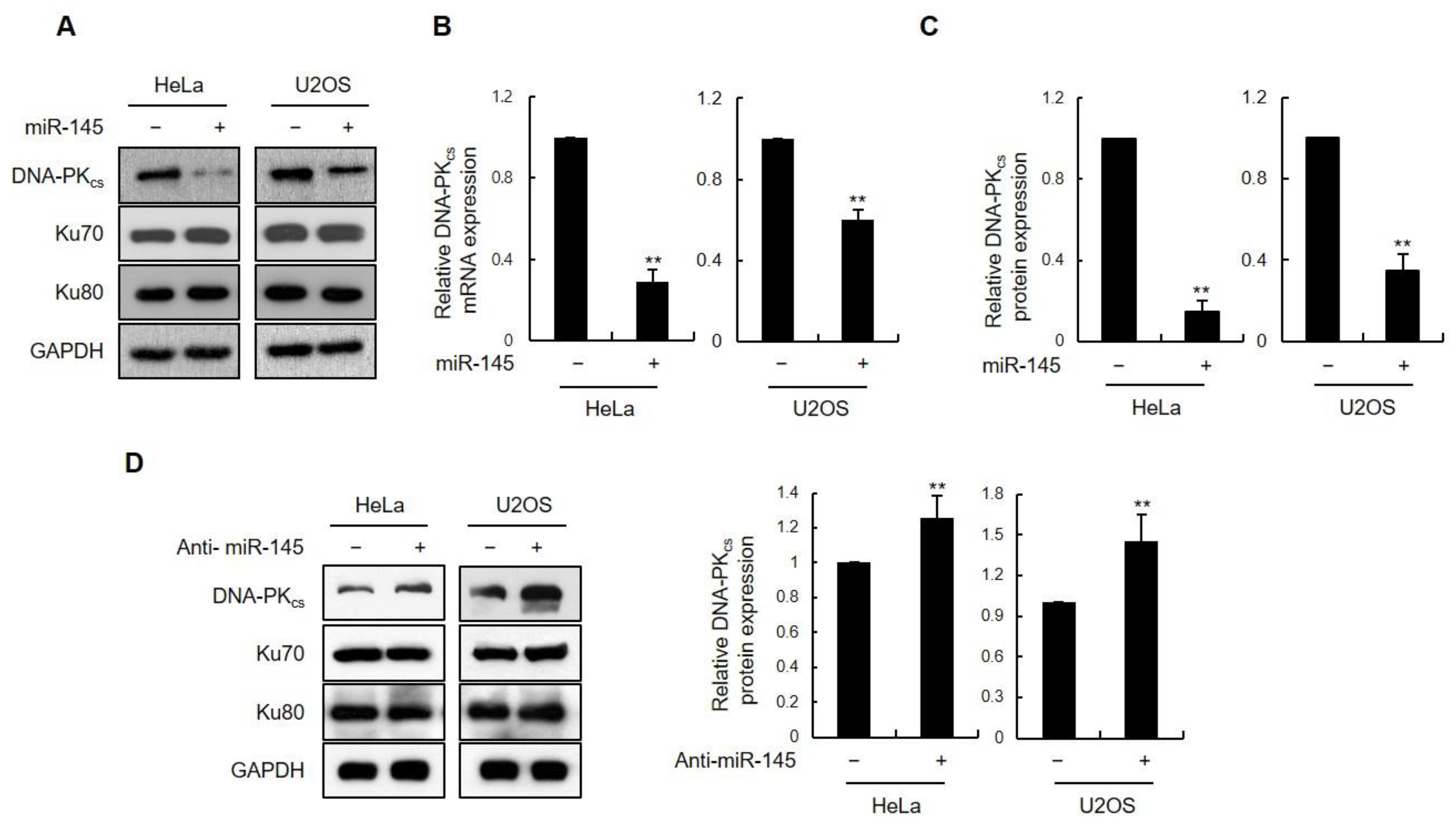
3.1. miR-145 as a DNA-PKcs-Regulating miRNA
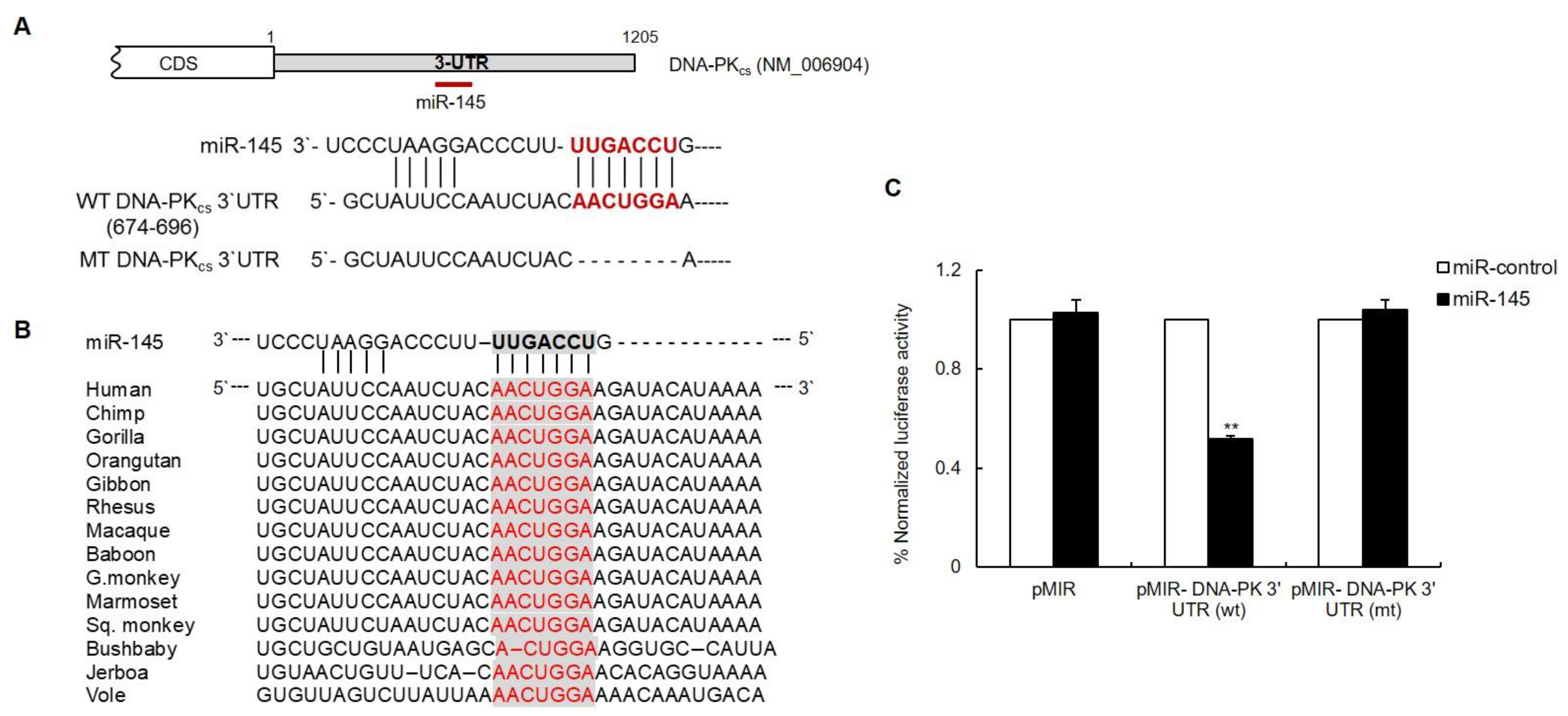
3.2. The DNA-PKcs 3′UTR Is Directly Targeted by miR-145
3.3. miR-145 Causes an Accumulation of Unrepaired DSBs after IR and Affects Cell Survival
3.4. miR-145 Reduces the DSB Repair by Downregulating DNA-PKcs
3.5. miR-145 Overexpression Interferes with Non-Homologous End Joining Repair
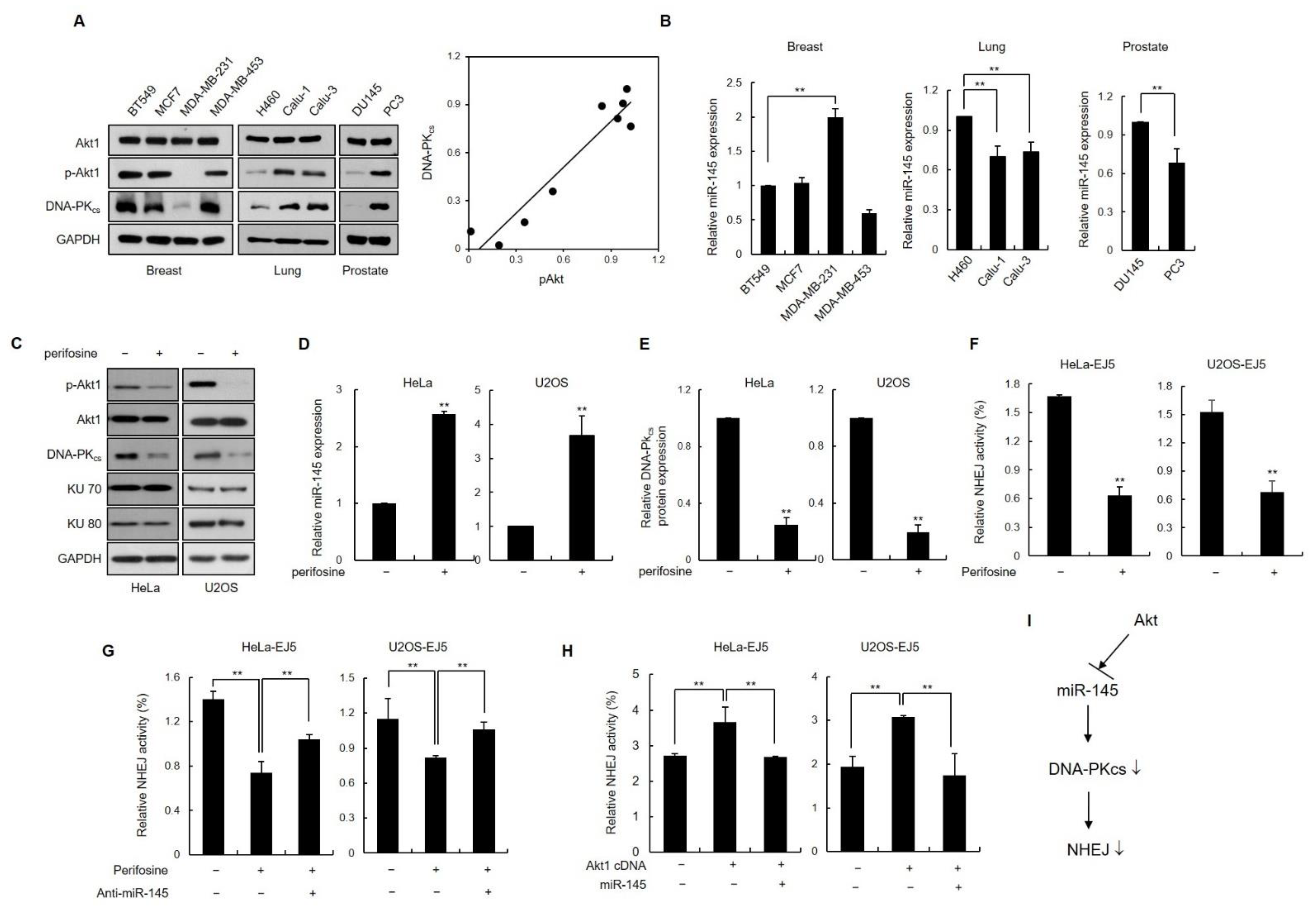
3.6. Inhibition of Akt1 Downregulates DNA-PKcs Expression via Induction of Endogenous miR-145 Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Bassing, C.H.; Alt, F.W. The cellular response to general and programmed DNA double strand breaks. DNA Repair 2004, 3, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Boboila, C.; Alt, F.W.; Schwer, B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 2012, 116, 1–49. [Google Scholar]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef]
- Mahaney, B.L.; Meek, K.; Lees-Miller, S.P. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem. J. 2009, 417, 639–650. [Google Scholar] [CrossRef]
- Van der Burg, M.; Ijspeert, H.; Verkaik, N.S.; Turul, T.; Wiegant, W.W.; Morotomi-Yano, K.; Mari, P.O.; Tezcan, I.; Chen, D.J.; Zdzienicka, M.Z.; et al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J. Clin. Investig. 2009, 119, 91–98. [Google Scholar] [CrossRef]
- Jhappan, C.; Morse, H.C., 3rd; Fleischmann, R.D.; Gottesman, M.M.; Merlino, G. DNA-PKcs: A T-cell tumour suppressor encoded at the mouse scid locus. Nat. Genet. 1997, 17, 483–486. [Google Scholar] [CrossRef]
- Okayasu, R.; Suetomi, K.; Yu, Y.; Silver, A.; Bedford, J.S.; Cox, R.; Ullrich, R.L. A deficiency in DNA repair and DNA-PKcs expression in the radiosensitive BALB/c mouse. Cancer Res. 2000, 60, 4342–4345. [Google Scholar]
- Pierce, A.J.; Hu, P.; Han, M.; Ellis, N.; Jasin, M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001, 15, 3237–3242. [Google Scholar] [CrossRef]
- Allen, C.; Kurimasa, A.; Brenneman, M.A.; Chen, D.J.; Nickoloff, J.A. DNA-dependent protein kinase suppresses double-strand break-induced and spontaneous homologous recombination. Proc. Natl. Acad. Sci. USA 2002, 99, 3758–3763. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Du, L.; Nagabayashi, G.; Seeger, R.C.; Gatti, R.A. ATM is down-regulated by N-Myc-regulated microRNA-421. Proc. Natl. Acad. Sci. USA 2010, 107, 1506–1511. [Google Scholar] [CrossRef] [PubMed]
- Moskwa, P.; Buffa, F.M.; Pan, Y.; Panchakshari, R.; Gottipati, P.; Muschel, R.J.; Beech, J.; Kulshrestha, R.; Abdelmohsen, K.; Weinstock, D.M.; et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol. Cell 2011, 41, 210–220. [Google Scholar] [CrossRef]
- Garcia, A.I.; Buisson, M.; Bertrand, P.; Rimokh, R.; Rouleau, E.; Lopez, B.S.; Lidereau, R.; Mikaelian, I.; Mazoyer, S. Down-regulation of BRCA1 expression by miR-146a and miR-146b-5p in triple negative sporadic breast cancers. EMBO Mol. Med. 2011, 3, 279–290. [Google Scholar] [CrossRef]
- Chowdhury, D.; Choi, Y.E.; Brault, M.E. Charity begins at home: Non-coding RNA functions in DNA repair. Nat. Rev. Mol. Cell Biol. 2013, 14, 181–189. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, S.J.; Jeong, S.Y.; Kim, M.J.; Jun, S.; Lee, H.S.; Chang, I.Y.; Lim, S.C.; Yoon, S.P.; Yong, J.; et al. MicroRNA-22 Suppresses DNA Repair and Promotes Genomic Instability through Targeting of MDC1. Cancer Res. 2015, 75, 1298–1310. [Google Scholar] [CrossRef]
- Zhang, J.; Jing, L.; Tan, S.; Zeng, E.M.; Lin, Y.; He, L.; Hu, Z.; Liu, J.; Guo, Z. Inhibition of miR-1193 leads to synthetic lethality in glioblastoma multiforme cells deficient of DNA-PKcs. Cell Death Dis. 2020, 11, 602. [Google Scholar] [CrossRef]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, S.J.; Hariharasudhan, G.; Kim, M.J.; Jung, S.M.; Jeong, S.Y.; Chang, I.Y.; Kim, C.; Kim, E.; Yu, J.; et al. ID3 regulates the MDC1-mediated DNA damage response in order to maintain genome stability. Nat. Commun. 2017, 8, 903. [Google Scholar] [CrossRef]
- Pierce, A.J.; Johnson, R.D.; Thompson, L.H.; Jasin, M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999, 13, 2633–2638. [Google Scholar] [CrossRef]
- Shahi, A.; Lee, J.H.; Kang, Y.; Lee, S.H.; Hyun, J.W.; Chang, I.Y.; Jun, J.Y.; You, H.J. Mismatch-repair protein MSH6 is associated with Ku70 and regulates DNA double-strand break repair. Nucleic Acids Res. 2011, 39, 2130–2143. [Google Scholar] [CrossRef] [PubMed]
- Sundaravinayagam, D.; Kim, H.R.; Wu, T.; Kim, H.H.; Lee, H.S.; Jun, S.; Cha, J.H.; Kee, Y.; You, H.J.; Lee, J.H. miR146a-mediated targeting of FANCM during inflammation compromises genome integrity. Oncotarget 2016, 7, 45976. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Garcia, D.M.; Baek, D.; Shin, C.; Bell, G.W.; Grimson, A.; Bartel, D.P. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat. Struct. Mol. Biol. 2011, 18, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. GammaH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef]
- Seluanov, A.; Mittelman, D.; Pereira-Smith, O.M.; Wilson, J.H.; Gorbunova, V. DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc. Natl. Acad. Sci. USA 2004, 101, 7624–7629. [Google Scholar] [CrossRef]
- Luo, J.; Manning, B.D.; Cantley, L.C. Targeting the PI3K-Akt pathway in human cancer: Rationale and promise. Cancer Cell 2003, 4, 257–262. [Google Scholar] [CrossRef]
- Li, H.F.; Kim, J.S.; Waldman, T. Radiation-induced Akt activation modulates radioresistance in human glioblastoma cells. Radiat. Oncol. 2009, 4, 43. [Google Scholar] [CrossRef]
- Kim, I.A.; Bae, S.S.; Fernandes, A.; Wu, J.; Muschel, R.J.; McKenna, W.G.; Birnbaum, M.J.; Bernhard, E.J. Selective inhibition of Ras, phosphoinositide 3 kinase, and Akt isoforms increases the radiosensitivity of human carcinoma cell lines. Cancer Res. 2005, 65, 7902–7910. [Google Scholar] [CrossRef]
- Toulany, M.; Minjgee, M.; Saki, M.; Holler, M.; Meier, F.; Eicheler, W.; Rodemann, H.P. ERK2-dependent reactivation of Akt mediates the limited response of tumor cells with constitutive K-RAS activity to PI3K inhibition. Cancer Biol. Ther. 2014, 15, 317–328. [Google Scholar] [CrossRef]
- Brognard, J.; Clark, A.S.; Ni, Y.; Dennis, P.A. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001, 61, 3986–3997. [Google Scholar] [PubMed]
- Sachdeva, M.; Zhu, S.; Wu, F.; Wu, H.; Walia, V.; Kumar, S.; Elble, R.; Watabe, K.; Mo, Y.Y. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc. Natl. Acad. Sci. USA 2009, 106, 3207–3212. [Google Scholar] [CrossRef] [PubMed]
- Kondapaka, S.B.; Singh, S.S.; Dasmahapatra, G.P.; Sausville, E.A.; Roy, K.K. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol. Cancer Ther. 2003, 2, 1093–1103. [Google Scholar] [PubMed]
- Goodwin, J.F.; Knudsen, K.E. Beyond DNA repair: DNA-PK function in cancer. Cancer Discov. 2014, 4, 1126–1139. [Google Scholar] [CrossRef]
- Kienker, L.J.; Shin, E.K.; Meek, K. Both V(D)J recombination and radioresistance require DNA-PK kinase activity, though minimal levels suffice for V(D)J recombination. Nucleic Acids Res. 2000, 28, 2752–2761. [Google Scholar] [CrossRef]
- Kurimasa, A.; Kumano, S.; Boubnov, N.V.; Story, M.D.; Tung, C.S.; Peterson, S.R.; Chen, D.J. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol. Cell. Biol. 1999, 19, 3877–3884. [Google Scholar] [CrossRef]
- Visser, H.; Thomas, A.D. microRNAs and the DNA damage response: How is cell fate determined? DNA Repair 2021, 108, 103245. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Gatti, R.A. MicroRNA: New players in the DNA damage response. J. Mol. Cell Biol. 2011, 3, 151–158. [Google Scholar] [CrossRef]
- Chiyomaru, T.; Enokida, H.; Tatarano, S.; Kawahara, K.; Uchida, Y.; Nishiyama, K.; Fujimura, L.; Kikkawa, N.; Seki, N.; Nakagawa, M. miR-145 and miR-133a function as tumour suppressors and directly regulate FSCN1 expression in bladder cancer. Br. J. Cancer 2010, 102, 883–891. [Google Scholar] [CrossRef]
- Gregersen, L.H.; Jacobsen, A.B.; Frankel, L.B.; Wen, J.; Krogh, A.; Lund, A.H. MicroRNA-145 targets YES and STAT1 in colon cancer cells. PLoS ONE 2010, 5, e8836. [Google Scholar] [CrossRef]
- Wang, S.; Bian, C.; Yang, Z.; Bo, Y.; Li, J.; Zeng, L.; Zhou, H.; Zhao, R.C. miR-145 inhibits breast cancer cell growth through RTKN. Int. J. Oncol. 2009, 34, 1461–1466. [Google Scholar] [PubMed]
- Xu, Q.; Liu, L.Z.; Qian, X.; Chen, Q.; Jiang, Y.; Li, D.; Lai, L.; Jiang, B.H. miR-145 directly targets p70S6K1 in cancer cells to inhibit tumor growth and angiogenesis. Nucleic Acids Res. 2012, 40, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Eades, G.; Wolfson, B.; Zhang, Y.; Li, Q.; Yao, Y.; Zhou, Q. lincRNA-RoR and miR-145 regulate invasion in triple-negative breast cancer via targeting ARF6. Mol. Cancer Res. 2015, 13, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhyay, M.; Banerjee, A.; Sarkar, N.; Panigrahi, R.; Datta, S.; Pal, A.; Singh, S.P.; Biswas, A.; Chakrabarti, S.; Chakravarty, R. Tumor suppressor micro RNA miR-145 and onco micro RNAs miR-21 and miR-222 expressions are differentially modulated by hepatitis B virus X protein in malignant hepatocytes. BMC Cancer 2014, 14, 721. [Google Scholar] [CrossRef]
- Qiu, T.; Zhou, X.; Wang, J.; Du, Y.; Xu, J.; Huang, Z.; Zhu, W.; Shu, Y.; Liu, P. miR-145, miR-133a and miR-133b inhibit proliferation, migration, invasion and cell cycle progression via targeting transcription factor Sp1 in gastric cancer. FEBS Lett. 2014, 588, 1168–1177. [Google Scholar] [CrossRef]
- Lhakhang, T.W.; Chaudhry, M.A. Interactome of Radiation-Induced microRNA-predicted target genes. Comp. Funct. Genom. 2012, 2012, 569731. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L.; Li, B.; Huo, M.; Mu, M.; Liu, J.; Han, J. miR-145 downregulates the expression of cyclin-dependent kinase 6 in human cervical ca rcinoma cells. Exp. Ther. Med. 2014, 8, 591–594. [Google Scholar] [CrossRef]
- Chang, S.; Gao, L.; Yang, Y.; Tong, D.; Guo, B.; Liu, L.; Li, Z.; Song, T.; Huang, C. miR-145 mediates the antiproliferative and gene regulatory effects of vitamin D3 by directly targeting E2F3 in gastric cancer cells. Oncotarget 2015, 6, 7675–7685. [Google Scholar] [CrossRef]
- Zou, C.; Xu, Q.; Mao, F.; Li, D.; Bian, C.; Liu, L.Z.; Jiang, Y.; Chen, X.; Qi, Y.; Zhang, X.; et al. miR-145 inhibits tumor angiogenesis and growth by N-RAS and VEGF. Cell Cycle 2012, 11, 2137–2145. [Google Scholar] [CrossRef]
- Czochor, J.R.; Glazer, P.M. microRNAs in cancer cell response to ionizing radiation. Antioxid. Redox Signal. 2014, 21, 293–312. [Google Scholar] [CrossRef]
- Song, M.; Xie, D.; Gao, S.; Bai, C.J.; Zhu, M.X.; Guan, H.; Zhou, P.K. A biomarker panel of radiation-upregulated miRNA as signature for ionizing radiation exposure. Life 2020, 10, 361. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Q.; Zhang, Y.; Wang, Z.; Ding, J.; Song, Y.; Zhao, W. Cisplatin-mediated down-regulation of miR-145 contributes to up-regulation of PD-L1 via the c-Myc transcription factor in cisplatin-resistant ovarian carcinoma cells. Clin. Exp. Immunol. 2020, 200, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Ng, W.L.; Zhang, X.; Wang, P.; Zhang, Z.; Mo, Y.Y.; Mao, H.; Hao, C.; Olson, J.J.; Curran, W.J.; et al. Targeting DNA-PKcs and ATM with miR-101 sensitizes tumors to radiation. PLoS ONE 2010, 5, e11397. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Jonkers, J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [Google Scholar] [CrossRef]
- Xue, G.; Ren, Z.; Chen, Y.; Zhu, J.; Du, Y.; Pan, D.; Li, X.; Hu, B. A feedback regulation between miR-145 and DNA methyltransferase 3b in prostate cancer cell and their responses to irradiation. Cancer Lett. 2015, 361, 121–127. [Google Scholar] [CrossRef]
- Shi, M.; Du, L.; Liu, D.; Qian, L.; Hu, M.; Yu, M.; Yang, Z.; Zhao, M.; Chen, C.; Guo, L.; et al. Glucocorticoid regulation of a novel HPV-E6-p53-miR-145 pathway modulates invasion and therapy resistance of cervical cancer cells. J. Pathol. 2012, 228, 148–157. [Google Scholar] [CrossRef]
- Lu, H.; He, Y.; Lin, L.; Qi, Z.; Ma, L.; Li, L.; Su, Y. Long non-coding RNA MALAT1 modulates radiosensitivity of HR-HPV+ cervical cancer via sponging miR-145. Tumor Biol. 2016, 37, 1683–1691. [Google Scholar] [CrossRef]
- Liu, R.L.; Dong, Y.; Deng, Y.Z.; Wang, W.J.; Li, W.D. Tumor suppressor miR-145 reverses drug resistance by directly targeting DNA damage-related gene RAD18 in colorectal cancer. Tumor Biol. 2015, 36, 5011–5019. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Xu, N.; Hegarat, N.; Black, E.J.; Scott, M.T.; Hochegger, H.; Gillespie, D.A. Akt/PKB suppresses DNA damage processing and checkpoint activation in late G2. J. Cell Biol. 2010, 190, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Plo, I.; Laulier, C.; Gauthier, L.; Lebrun, F.; Calvo, F.; Lopez, B.S. AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51. Cancer Res. 2008, 68, 9404–9412. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Song, W.; Zhang, F.; Yan, J.; Yang, Q. Akt1 inhibits homologous recombination in Brca1-deficient cells by blocking the Chk1-Rad51 pathway. Oncogene 2013, 32, 1943–1949. [Google Scholar] [CrossRef] [PubMed]
- Guirouilh-Barbat, J.K.; Wilhelm, T.; Lopez, B.S. AKT1/BRCA1 in the control of homologous recombination and genetic stability: The missing link between hereditary and sporadic breast cancers. Oncotarget 2010, 1, 691–699. [Google Scholar] [CrossRef]
- Kandel, E.S.; Skeen, J.; Majewski, N.; Di Cristofano, A.; Pandolfi, P.P.; Feliciano, C.S.; Gartel, A.; Hay, N. Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage. Mol. Cell. Biol. 2002, 22, 7831–7841. [Google Scholar] [CrossRef]
- Toulany, M.; Rodemann, H.P. Potential of Akt mediated DNA repair in radioresistance of solid tumors overexpressing erbB-PI3K-Akt pathway. Transl. Cancer Res. 2013, 2, 190–202. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sudhanva, M.S.; Hariharasudhan, G.; Jun, S.; Seo, G.; Kamalakannan, R.; Kim, H.H.; Lee, J.-H. MicroRNA-145 Impairs Classical Non-Homologous End-Joining in Response to Ionizing Radiation-Induced DNA Double-Strand Breaks via Targeting DNA-PKcs. Cells 2022, 11, 1509. https://doi.org/10.3390/cells11091509
Sudhanva MS, Hariharasudhan G, Jun S, Seo G, Kamalakannan R, Kim HH, Lee J-H. MicroRNA-145 Impairs Classical Non-Homologous End-Joining in Response to Ionizing Radiation-Induced DNA Double-Strand Breaks via Targeting DNA-PKcs. Cells. 2022; 11(9):1509. https://doi.org/10.3390/cells11091509
Chicago/Turabian StyleSudhanva, Muddenahalli Srinivasa, Gurusamy Hariharasudhan, Semo Jun, Gwanwoo Seo, Radhakrishnan Kamalakannan, Hyun Hee Kim, and Jung-Hee Lee. 2022. "MicroRNA-145 Impairs Classical Non-Homologous End-Joining in Response to Ionizing Radiation-Induced DNA Double-Strand Breaks via Targeting DNA-PKcs" Cells 11, no. 9: 1509. https://doi.org/10.3390/cells11091509
APA StyleSudhanva, M. S., Hariharasudhan, G., Jun, S., Seo, G., Kamalakannan, R., Kim, H. H., & Lee, J.-H. (2022). MicroRNA-145 Impairs Classical Non-Homologous End-Joining in Response to Ionizing Radiation-Induced DNA Double-Strand Breaks via Targeting DNA-PKcs. Cells, 11(9), 1509. https://doi.org/10.3390/cells11091509