
Unraveling the Molecular Basis of the Dystrophic Process in Limb-Girdle Muscular Dystrophy LGMD-R12 by Differential Gene Expression Profiles in Diseased and Healthy Muscles

 , and
, and
Abstract
:1. Introduction
2. Patients and Methods
2.1. Patients and Controls
2.2. Muscle Biopsies
2.3. RNA Procedures
2.3.1. RNA Extraction
2.3.2. RNA Quality Control
2.3.3. Library Preparation
2.3.4. Sequencing (RNA-Seq)
2.4. Data Analysis
2.4.1. Preprocessing
2.4.2. Mapping
2.4.3. Counting
2.5. RNA-Seq Analysis
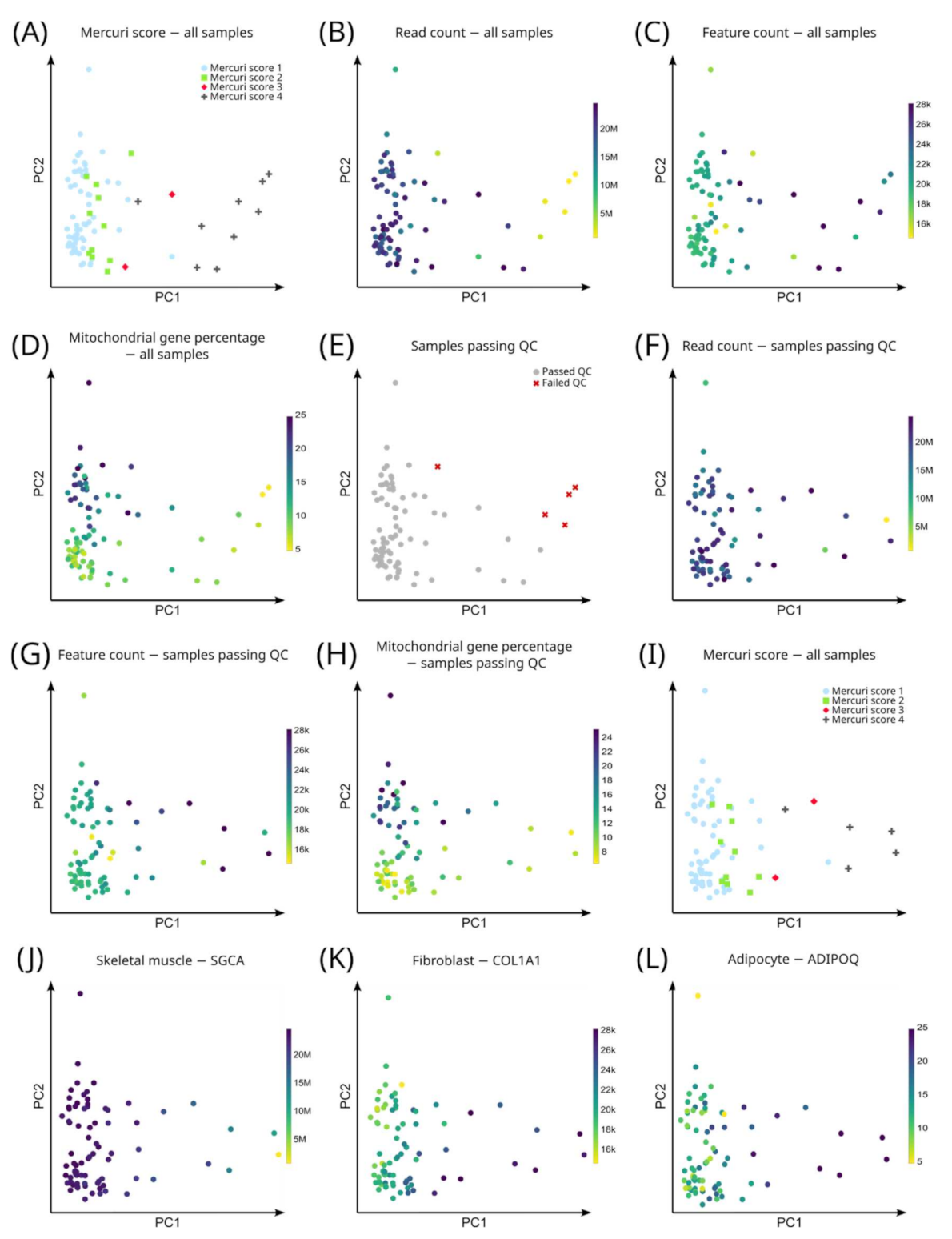
2.5.1. Quality Control and Data Normalization
2.5.2. Principal Component Analysis (PCA)
2.5.3. Pair-Wise Differential Analysis
2.5.4. Gene Set Enrichment Analysis (GSEA)
2.5.5. Heatmap Analysis
2.5.6. Deconvolution Analysis
2.6. Single Cell scRNA-Seq Analysis
2.6.1. Quality Control and Data Normalization
2.6.2. Dimension Reduction
2.6.3. Clustering Analysis and Annotation
2.6.4. Pair-Wise Differential Analysis
2.6.5. Marker Gene Analysis
2.6.6. Heatmap Analysis
3. Results
3.1. Demographic Data
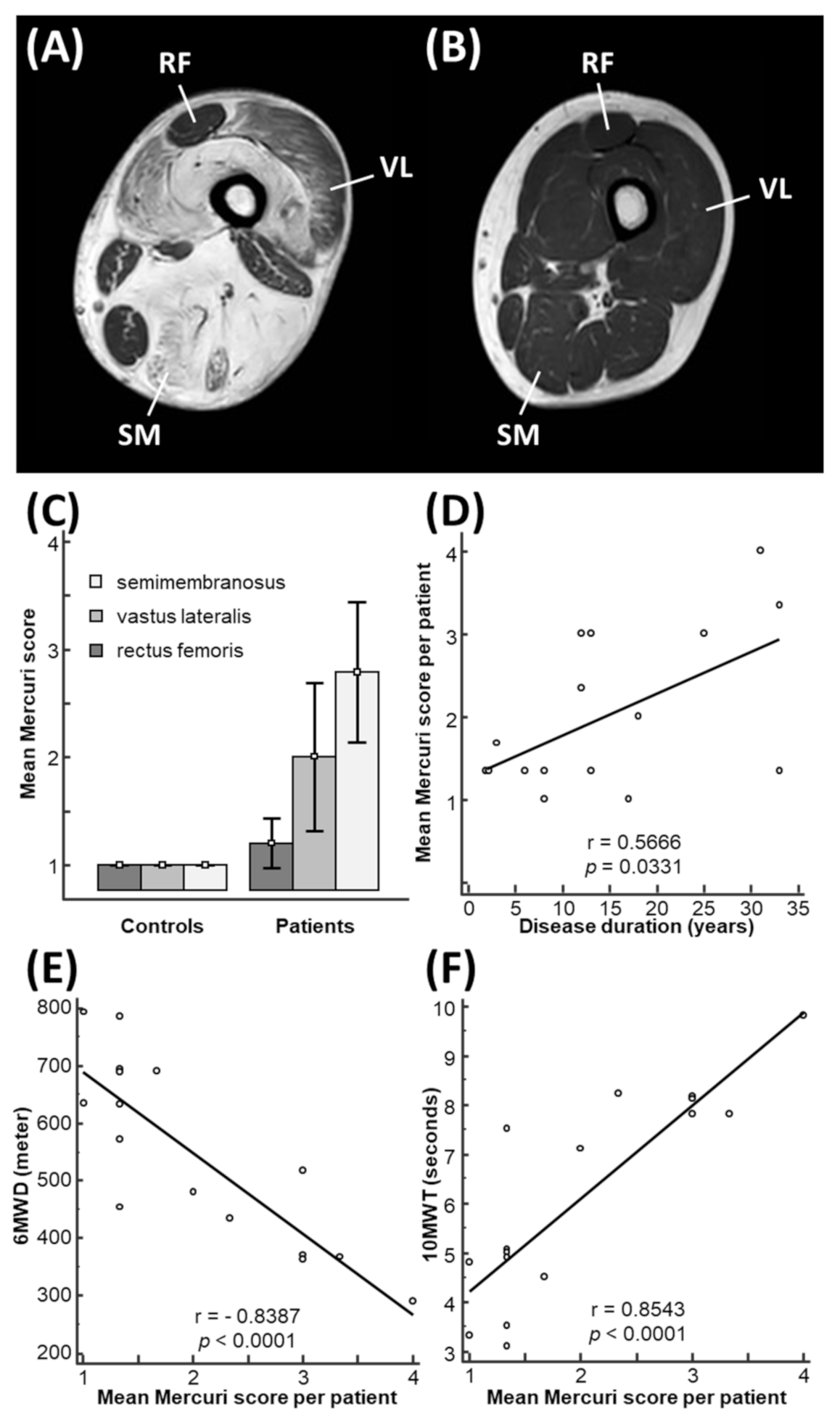
3.2. Mercuri Score Is Correlated with Functional Outcomes in LGMD-R12 Patients
3.3. Unbiased Analysis: Gene Expression Profiles Correlate with the Mercuri Score
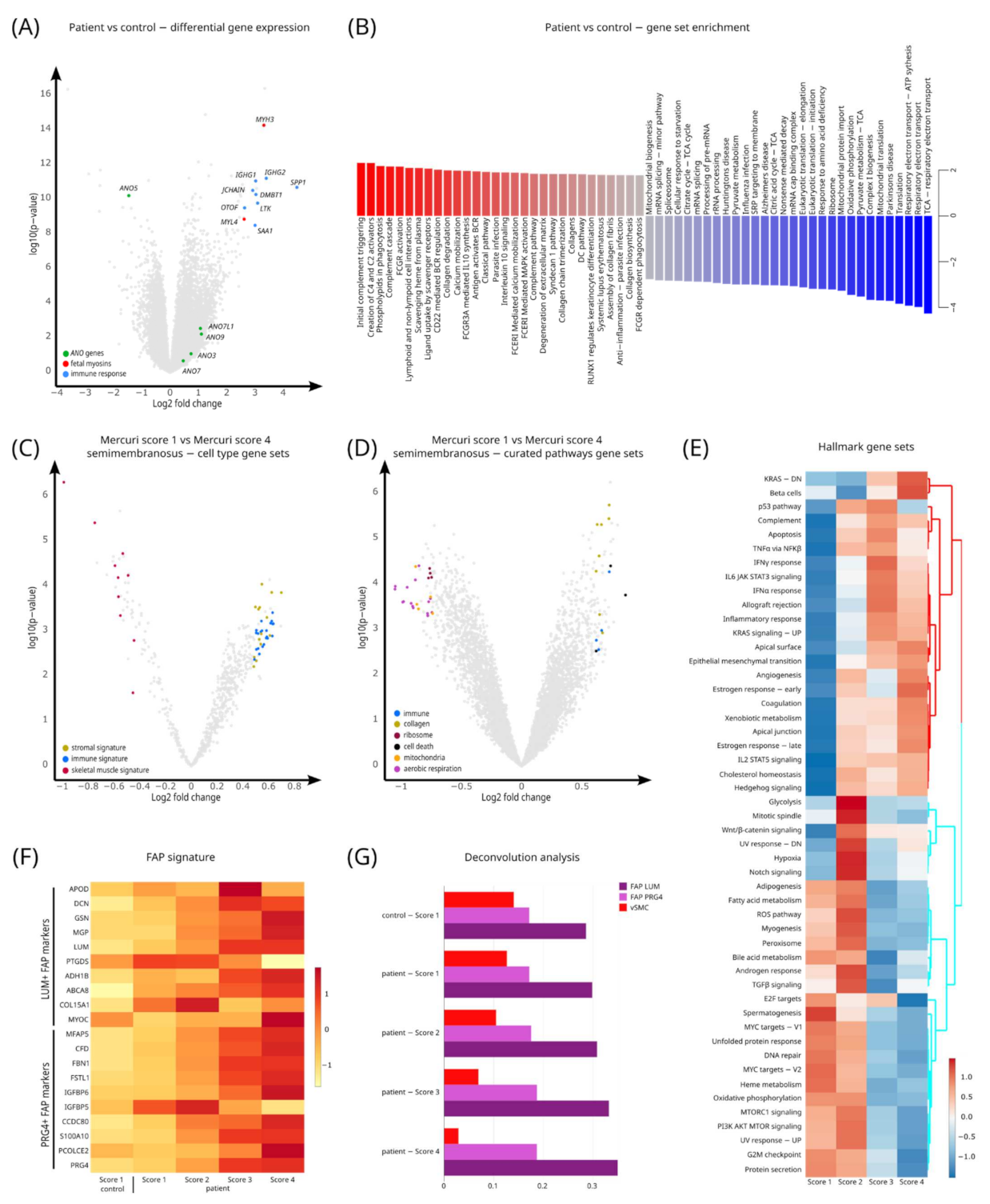
3.4. Quantification of Gene Signatures Associated with LGMD-R12
3.5. Pathway Analysis Characterizes Gene Sets for LGMD-R12 Progression
3.6. Deconvolution Analysis Shows an Increase in Fibroadipogenic Progenitor (FAP) Cells in LGMD-R12 Muscles
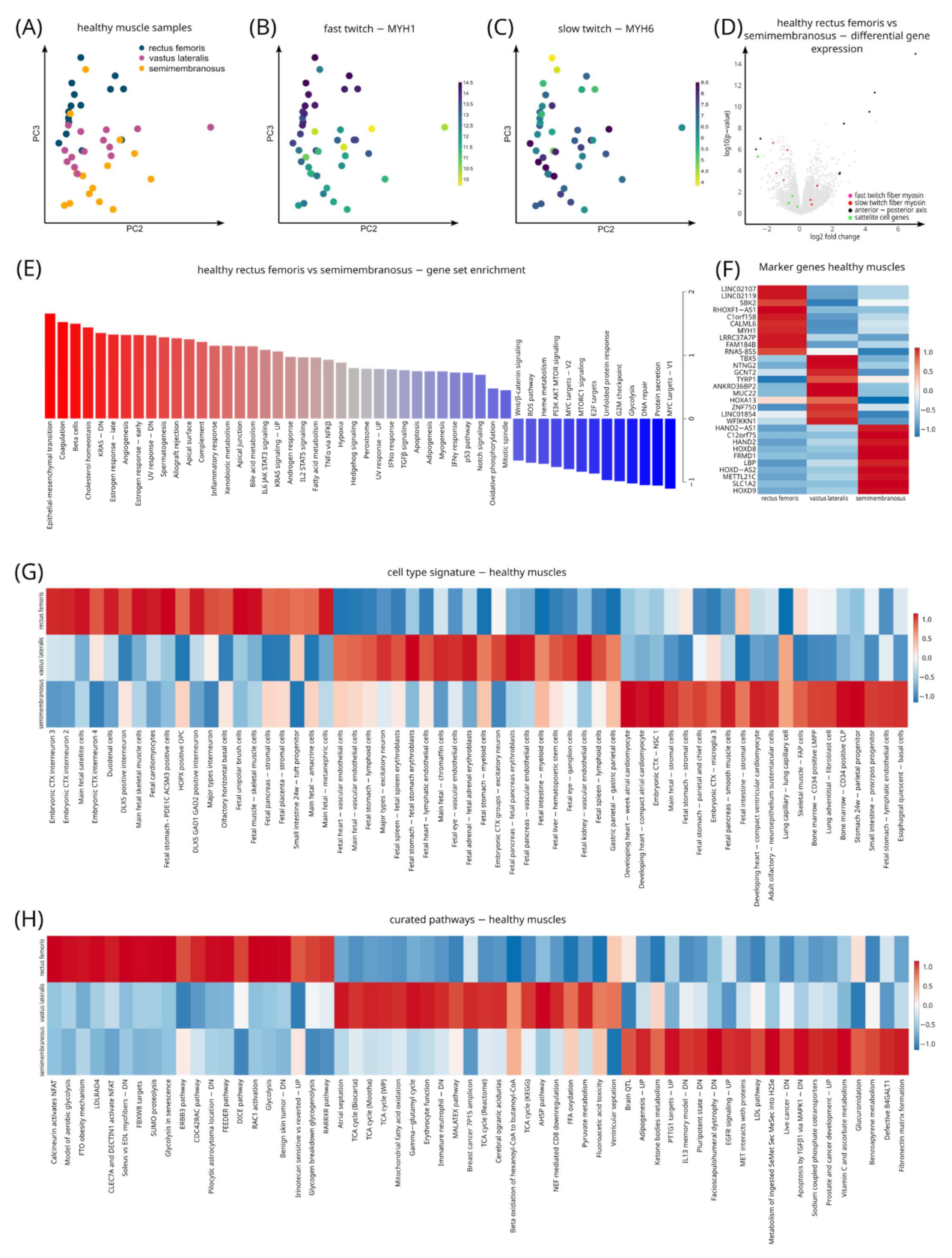
3.7. Distinct Gene Signatures in Different Muscles in Healthy Controls
3.8. Differential Gene Expression between Different Muscles: Identification of Genes Associated with Different Muscles
4. Discussion
4.1. Genes Involved in Inflammation Are Upregulated in Dystrophic LGMD-R12 Muscles
4.2. Fibroadipogenic Progenitor (FAP) Cells Are Upregulated in LGMD-R12 Muscles
4.3. Different Muscles Express Different Gene Profiles in Healthy Controls
4.4. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Straub, V.; Murphy, A.; Udd, B.; LGMD Workshop Study Group. 229th ENMC international workshop: Limb girdle muscular dystrophies—Nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolduc, V.; Marlow, G.; Boycott, K.M.; Saleki, K.; Inoue, H.; Kroon, J.; Itakura, M.; Robitaille, Y.; Parent, L.; Baas, F.; et al. Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am. J. Hum. Genet. 2010, 86, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, D.; Sarkozy, A.; Muelas, N.; Köehler, K.; Huebner, A.; Hudson, G.; Chinnery, P.F.; Barresi, R.; Eagle, M.; Polvikoski, T.; et al. A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy. Brain 2011, 134, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkozy, A.; Hicks, D.; Hudson, J.; Laval, S.H.; Barresi, R.; Hilton Jones, D.; Deschauer, M.; Harris, E.; Rufibach, L.; Hwang, E.; et al. ANO5 gene analysis in a large cohort of patients with anoctaminopathy: Confirmation of male prevalence and high occurrence of the common exon 5 gene mutation. Hum. Mutat. 2013, 34, 1111–1118. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, S.; Kamata, N.; Vokes, T.J.; Maruoka, Y.; Nakakuki, K.; Enomoto, S.; Omura, K.; Amagasa, T.; Nagayama, M.; Saito-Ohara, F.; et al. The novel gene encoding a putative transmembrane protein is mutated in gnathodiaphyseal dysplasia (GDD). Am. J. Hum. Genet. 2004, 74, 1255–1261. [Google Scholar] [CrossRef] [Green Version]
- Mizuta, K.; Tsutsumi, S.; Inoue, H.; Sakamoto, Y.; Miyatake, K.; Miyawaki, K.; Noji, S.; Kamata, N.; Itakura, M. Molecular characterization of GDD1/TMEM16E, the gene product responsible for autosomal dominant gnathodiaphyseal dysplasia. Biochem. Biophys. Res. Commun. 2007, 357, 126–132. [Google Scholar] [CrossRef]
- Whitlock, J.M.; Hartzell, H.C. Anoctamins/TMEM16 Proteins: Chloride Channels Flirting with Lipids and Extracellular Vesicles. Annu. Rev. Physiol. 2017, 79, 119–143. [Google Scholar] [CrossRef] [Green Version]
- Boccaccio, A.; Di Zanni, E.; Gradogna, A.; Scholz-Starke, J. Lifting the veils on TMEM16E function. Channels 2019, 13, 33–35. [Google Scholar] [CrossRef] [Green Version]
- Griffin, D.A.; Johnson, R.W.; Whitlock, J.M.; Pozsgai, E.R.; Heller, K.N.; Grose, W.E.; Arnold, W.D.; Sahenk, Z.; Hartzell, H.C.; Rodino-Klapac, R. Defective membrane fusion and repair in Anoctamin5-deficient muscular dystrophy. Hum. Mol. Genet. 2016, 25, 1900–1911. [Google Scholar] [CrossRef] [Green Version]
- Whitlock, J.M.; Yu, K.; Cui, Y.Y.; Hartzell, H.C. Anoctamin 5/TMEM16E facilitates muscle precursor cell fusion. J. Gen. Physiol. 2018, 150, 1498–1509. [Google Scholar] [CrossRef] [Green Version]
- Chandra, G.; Sreetama, S.C.; Mazala, D.A.; Charton, K.; Van der Meulen, J.H.; Richard, I.; Jaiswal, J.K. Endoplasmic reticulum maintains ion homeostasis required for plasma membrane repair. J. Cell Biol. 2021, 220, e202006035. [Google Scholar] [CrossRef] [PubMed]
- Foltz, S.J.; Cui, Y.Y.; Choo, H.J.; Hartzell, H. ANO5 ensures trafficking of annexins in wounded myofibers. J. Cell Biol. 2021, 220, e202007059. [Google Scholar] [CrossRef] [PubMed]
- Thiruvengadam, G.; Sreetama, S.C.; Charton, K.; Hogarth, M.; Novak, J.S.; Suel-Petat, L.; Chandra, G.; Allard, B.; Richard, I.; Jaiswal, J.K. Anoctamin 5 Knockout Mouse Model Recapitulates LGMD2L Muscle Pathology and Offers Insight Into in vivo Functional Deficits. J. Neuromuscul. Dis. 2021, 8, S243–S255. [Google Scholar] [CrossRef] [PubMed]
- Dubowitz, V.; Sewry, C.A.; Oldfors, A. Muscle Biopsy E-Book: A Practical Approach, 4th ed.; Elsevier Health Sciences: New York, NY, USA, 2020. [Google Scholar]
- Silva, A.M.S.; Coimbra-Neto, A.R.; Souza, P.V.S.; Winckler, P.B.; Gonçalves, M.V.M.; Cavalcanti, E.B.U.; Carvalho, A.A.D.S.; Sobreira, C.F.D.R.; Camelo, C.G.; Mendonça, R.D.H.; et al. Clinical and molecular findings in a cohort of ANO5-related myopathy. Ann. Clin. Transl. Neurol. 2019, 6, 1225–1238. [Google Scholar] [CrossRef] [Green Version]
- Holm-Yildiz, S.; Witting, N.; de Stricker Borch, J.; Kass, K.; Khawajazada, T.; Krag, T.; Vissing, J. Muscle biopsy and MRI findings in ANO5-related myopathy. Muscle Nerve 2021, 64, 743–748. [Google Scholar] [CrossRef]
- Christiansen, J.; Güttsches, A.K.; Schara-Schmidt, U.; Vorgerd, M.; Heute, C.; Preusse, C.; Stenzel, W.; Roos, A. ANO5-related muscle diseases: From clinics and genetics to pathology and research strategies. Genes Diseases 2022. [Google Scholar] [CrossRef]
- Petrillo, S.; Pelosi, L.; Piemonte, F.; Travaglini, L.; Forcina, L.; Catteruccia, M.; Petrini, S.; Verardo, M.; D’Amico, A.; Musarò, A.; et al. Oxidative stress in Duchenne muscular dystrophy: Focus on the NRF2 redox pathway. Hum. Mol. Genet. 2017, 26, 2781–2790. [Google Scholar] [CrossRef]
- Vila, M.C.; Rayavarapu, S.; Hogarth, M.W.; Van der Meulen, J.H.; Horn, A.; Defour, A.; Takeda, S.; Brown, K.J.; Hathout, Y.; Nagaraju, K.; et al. Mitochondria mediate cell membrane repair and contribute to Duchenne muscular dystrophy. Cell Death Differ. 2017, 24, 330–342. [Google Scholar] [CrossRef] [Green Version]
- Tripodi, L.; Villa, C.; Molinaro, D.; Torrente, Y.; Farini, A. The Immune System in Duchenne Muscular Dystrophy Pathogenesis. Biomedicines 2021, 9, 1447. [Google Scholar] [CrossRef]
- Hogarth, M.W.; Uapinyoying, P.; Mázala, D.A.G.; Jaiswal, J.K. Pathogenic role and therapeutic potential of fibro-adipogenic progenitors in muscle disease. Trends Mol. Med. 2022, 28, 8–11. [Google Scholar] [CrossRef]
- Straub, V.; Carlier, P.G.; Mercuri, E. TREAT-NMD workshop: Pattern recognition in genetic muscle diseases using muscle MRI: 25-26 February 2011, Rome, Italy. Neuromuscul. Disord. 2012, 22, S42–S53. [Google Scholar] [CrossRef]
- Ten Dam, L.; van der Kooi, A.J.; Verhamme, C.; Wattjes, M.P.; de Visser, M. Muscle imaging in inherited and acquired muscle diseases. Eur. J. Neurol. 2016, 23, 688–703. [Google Scholar] [CrossRef] [PubMed]
- Mercuri, E.; Pichiecchio, A.; Allsop, J.; Messina, S.; Pane, M.; Muntoni, F. Muscle MRI in inherited neuromuscular disorders: Past, present, and future. J. Magn. Reson. Imaging 2007, 25, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Kinali, M.; Arechavala-Gomeza, V.; Cirak, S.; Glover, A.; Guglieri, M.; Feng, L.; Hollingsworth, K.G.; Hunt, D.; Jungbluth, H.; Roper, H.P.; et al. Muscle histology vs. MRI in Duchenne muscular dystrophy. Neurology 2011, 76, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Scotton, C.; Bovolenta, M.; Schwartz, E.; Falzarano, M.S.; Martoni, E.; Passarelli, C.; Armaroli, A.; Osman, H.; Rodolico, C.; Messina, S.; et al. Deep RNA profiling identified CLOCK and molecular clock genes as pathophysiological signatures in collagen VI myopathy. J. Cell Sci. 2016, 129, 1671–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carson, J.A.; Nettleton, D.; Reecy, J.M. Differential gene expression in the rat soleus muscle during early work overload-induced hypertrophy. FASEB J. 2002, 16, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, S.; Däpp, C.; Wittwer, M.; Vogt, M.; Hoppeler, H.; Flück, M. Endurance training modulates the muscular transcriptome response to acute exercise. Pflugers Arch. 2006, 451, 678–687. [Google Scholar] [CrossRef] [Green Version]
- Cummings, B.B.; Marshall, J.L.; Tukiainen, T.; Lek, M.; Donkervoort, S.; Foley, A.R.; Bolduc, V.; Waddell, L.B.; Sandaradura, S.A.; O’Grady, G.L.; et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med. 2017, 9, eaal5209. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.; Anders, S.; Lawrence, M.; Aboyoun, P.; Pagès, H.; Gentleman, R. ShortRead: A bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 2009, 25, 2607–2608. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risso, D.; Schwartz, K.; Sherlock, G.; Dudoit, S. GC-content normalization for RNA-Seq data. BMC Bioinformatics 2011, 12, 480. [Google Scholar] [CrossRef] [Green Version]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Park, J.; Susztak, K.; Zhang, N.R.; Li, M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 2019, 10, 380. [Google Scholar] [CrossRef] [Green Version]
- Rubenstein, A.B.; Smith, G.R.; Raue, U.; Begue, G.; Minchev, K.; Ruf-Zamojski, F.; Nair, V.D.; Wang, X.; Zhou, L.; Zaslavsky, E.; et al. Single-cell transcriptional profiles in human skeletal muscle. Sci. Rep. 2020, 10, 229. [Google Scholar] [CrossRef] [Green Version]
- Hong, F.; Breitling, R.; McEntee, C.W.; Wittner, B.S.; Nemhauser, J.L.; Chory, J. RankProd: A bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics 2006, 22, 2825–2827. [Google Scholar] [CrossRef] [Green Version]
- Hack, A.A.; Groh, M.E.; McNally, E.M. Sarcoglycans in muscular dystrophy. Microsc. Res. Tech. 2000, 48, 167–180. [Google Scholar] [CrossRef]
- Csapo, R.; Gumpenberger, M.; Wessner, B. Skeletal Muscle Extracellular Matrix—What Do We Know About Its Composition, Regulation, and Physiological Roles? A Narrative Review. Front. Physiol. 2020, 11, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, M.P.; Milne, K.J.; Hawke, T.J. Adiponectin-Consideration for its Role in Skeletal Muscle Health. Int. J. Mol. Sci. 2019, 20, 1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capote, J.; Kramerova, I.; Martinez, L.; Vetrone, S.; Barton, E.R.; Sweeney, H.L.; Miceli, M.C.; Spencer, M.J. Osteopontin ablation ameliorates muscular dystrophy by shifting macrophages to a pro-regenerative phenotype. J. Cell Biol. 2016, 213, 275–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasgewatte Wijesinghe, D.K.; Mackie, E.J.; Pagel, C.N. Normal inflammation and regeneration of muscle following injury require osteopontin from both muscle and non-muscle cells. Skelet. Muscle 2019, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- Schiaffino, S.; Rossi, A.C.; Smerdu, V.; Leinwand, L.A.; Reggiani, C. Developmental myosins: Expression patterns and functional significance. Skelet. Muscle 2015, 5, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, T.; Xu, L.; Lau, Y.S.; Liu, D.; Liu, T.; Gao, Y.; Lai, L.; Han, R.; Li, Z. Development of muscular dystrophy in a CRISPR-engineered mutant rabbit model with frame-disrupting ANO5 mutations. Cell Death Dis. 2018, 9, 609. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Liu, Z. A benchmark for RNA-seq deconvolution analysis under dynamic testing environments. Genome Biol. 2021, 22, 102. [Google Scholar] [CrossRef]
- Stuart, C.A.; Stone, W.L.; Howell, M.E.; Brannon, M.F.; Hall, H.K.; Gibson, A.L.; Stone, M.H. Myosin content of individual human muscle fibers isolated by laser capture microdissection. Am. J. Physiol. Cell Physiol. 2016, 310, C381–C389. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.A.; Karabina, A.; Broadwell, L.J.; Leinwand, L.A. The ancient sarcomeric myosins found in specialized muscles. Skelet Muscle 2019, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Breitling, R.; Armengaud, P.; Amtmann, A.; Herzyk, P. Rank products: A simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 2004, 573, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Zoabi, M.; Zhang, L.; Li, T.M.; Elias, J.E.; Carlson, S.M.; Gozani, O. Methyltransferase-like 21C (METTL21C) methylates alanine tRNA synthetase at Lys-943 in muscle tissue. J. Biol. Chem. 2020, 295, 11822–11832. [Google Scholar] [CrossRef] [PubMed]
- Juban, G.; Saclier, M.; Yacoub-Youssef, H.; Kernou, A.; Arnold, L.; Boisson, C.; Ben Larbi, S.; Magnan, M.; Cuvellier, S.; Théret, M.; et al. AMPK Activation Regulates LTBP4-Dependent TGF-β1 Secretion by Pro-inflammatory Macrophages and Controls Fibrosis in Duchenne Muscular Dystrophy. Cell Rep. 2018, 25, 2163–2176.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Chazaud, B. Benefits and pathologies associated with the inflammatory response. Exp. Cell Res. 2021, 409, 112905. [Google Scholar] [CrossRef]
- Spencer, M.J.; Tidball, J.G. Do immune cells promote the pathology of dystrophin-deficient myopathies? Neuromuscul. Disord. 2001, 11, 556–564. [Google Scholar] [CrossRef]
- Arahata, K.; Ishihara, T.; Fukunaga, H.; Orimo, S.; Lee, J.H.; Goto, K.; Nonaka, I. Inflammatory response in facioscapulohumeral muscular dystrophy (FSHD): Immunocytochemical and genetic analyses. Muscle Nerve 1995, 18, S56–S66. [Google Scholar] [CrossRef]
- Gallardo, E.; Rojas-García, R.; De Luna, N.; Pou, A.; Brown, R.H. Inflammation in dysferlin myopathy: Immunohistochemical characterization of 13 patients. Neurology 2001, 57, 2136–2138. [Google Scholar] [CrossRef]
- Panicucci, C.; Baratto, S.; Raffaghello, L.; Tonin, P.; D’Amico, A.; Tasca, G.; Traverso, M.; Fiorillo, C.; Minetti, C.; Previtali, S.C.; et al. Muscle inflammatory pattern in alpha- and gamma-sarcoglycanopathies. Clin. Neuropathol. 2021, 40, 310–318. [Google Scholar] [CrossRef]
- Dahlqvist, J.R.; Andersen, G.; Khawajazada, T.; Vissing, C.; Thomsen, C.; Vissing, J. Relationship between muscle inflammation and fat replacement assessed by MRI in facioscapulohumeral muscular dystrophy. J. Neurol. 2019, 266, 1127–1135. [Google Scholar] [CrossRef]
- van den Heuvel, A.; Lassche, S.; Mul, K.; Greco, A.; San León Granado, D.; Heerschap, A.; Küsters, B.; Tapscott, S.J.; Voermans, N.C.; van Engelen, B.G.M.; et al. Facioscapulohumeral dystrophy transcriptome signatures correlate with different stages of disease and are marked by different MRI biomarkers. Sci. Rep. 2022, 12, 1426. [Google Scholar] [CrossRef] [PubMed]
- Tidball, J.G.; Welc, S.S.; Wehling-Henricks, M. Immunobiology of Inherited Muscular Dystrophies. Compr. Physiol. 2018, 8, 1313–1356. [Google Scholar] [CrossRef] [PubMed]
- Saclier, M.; Ben Larbi, S.; My Ly, H.; Moulin, E.; Mounier, R.; Chazaud, B.; Juban, G. Interplay between myofibers and pro-inflammatory macrophages controls muscle damage in mdx mice. J. Cell Sci. 2021, 134, jcs258429. [Google Scholar] [CrossRef]
- Milone, M.; Liewluck, T.; Winder, T.L.; Pianosi, P.T. Amyloidosis and exercise intolerance in ANO5 muscular dystrophy. Neuromuscul. Disord. 2012, 22, 13–15. [Google Scholar] [CrossRef]
- Liewluck, T.; Winder, T.L.; Dimberg, E.L.; Crum, B.A.; Heppelmann, C.J.; Wang, Y.; Bergen, H.R.3rd; Milone, M. ANO5-muscular dystrophy: Clinical, pathological and molecular findings. Eur. J. Neurol. 2013, 20, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Liu, Y.; Dai, J.; Li, L.; Ding, X.; Xu, Z.; Mori, M.; Miyahara, H.; Sawashita, J.; Higuchi, K. Apolipoprotein A-II induces acute-phase response associated AA amyloidosis in mice through conformational changes of plasma lipoprotein structure. Sci. Rep. 2018, 8, 5620. [Google Scholar] [CrossRef]
- Joe, A.W.; Yi, L.; Natarajan, A.; Le Grand, F.; So, L.; Wang, J.; Rudnicki, M.A.; Rossi, F.M. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010, 12, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Uezumi, A.; Ito, T.; Morikawa, D.; Shimizu, N.; Yoneda, T.; Segawa, M.; Yamaguchi, M.; Ogawa, R.; Matev, M.M.; Miyagoe-Suzuki, Y.; et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J. Cell Sci. 2011, 124, 3654–3664. [Google Scholar] [CrossRef] [Green Version]
- Uezumi, A.; Ikemoto-Uezumi, M.; Tsuchida, K. Roles of nonmyogenic mesenchymal progenitors in pathogenesis and regeneration of skeletal muscle. Front. Physiol. 2014, 5, 68. [Google Scholar] [CrossRef] [Green Version]
- Uezumi, A.; Fukada, S.; Yamamoto, N.; Takeda, S.; Tsuchida, K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010, 12, 143–152. [Google Scholar] [CrossRef]
- Giuliani, G.; Rosina, M.; Reggio, A. Signaling pathways regulating the fate of fibro/adipogenic progenitors (FAPs) in skeletal muscle regeneration and disease. FEBS J. 2021. [Google Scholar] [CrossRef]
- Biferali, B.; Proietti, D.; Mozzetta, C.; Madaro, L. Fibro-Adipogenic Progenitors Cross-Talk in Skeletal Muscle: The Social Network. Front. Physiol. 2019, 10, 1074. [Google Scholar] [CrossRef]
- Reggio, A.; Rosina, M.; Palma, A.; Cerquone Perpetuini, A.; Petrilli, L.L.; Gargioli, C.; Fuoco, C.; Micarelli, E.; Giuliani, G.; Cerretani, M.; et al. Adipogenesis of skeletal muscle fibro/adipogenic progenitors is affected by the WNT5a/GSK3/β-catenin axis. Cell Death Differ. 2020, 27, 2921–2941. [Google Scholar] [CrossRef] [PubMed]
- Mázala, D.A.; Novak, J.S.; Hogarth, M.W.; Nearing, M.; Adusumalli, P.; Tully, C.B.; Habib, N.F.; Gordish-Dressman, H.; Chen, Y.W.; Jaiswal, J.K.; et al. TGF-β-driven muscle degeneration and failed regeneration underlie disease onset in a DMD mouse model. JCI Insight 2020, 5, e135703. [Google Scholar] [CrossRef] [PubMed]
- Hogarth, M.W.; Defour, A.; Lazarski, C.; Gallardo, E.; Diaz Manera, J.; Partridge, T.A.; Nagaraju, K.; Jaiswal, J.K. Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nat. Commun. 2019, 10, 2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosnakovski, D.; Shams, A.S.; Yuan, C.; da Silva, M.T.; Ener, E.T.; Baumann, C.W.; Lindsay, A.J.; Verma, M.; Asakura, A.; Lowe, D.A.; et al. Transcriptional and cytopathological hallmarks of FSHD in chronic DUX4-expressing mice. J. Clin. Investig. 2020, 130, 2465–2477. [Google Scholar] [CrossRef] [PubMed]
- Polgar, J.; Johnson, M.A.; Weightman, D.; Appleton, D. Data on fibre size in thirty-six human muscles. An autopsy study. J. Neurol. Sci. 1973, 19, 307–318. [Google Scholar] [CrossRef]
- Garrett, W.E., Jr.; Califf, J.C.; Bassett, F.H. 3rd. Histochemical correlates of hamstring injuries. Am. J. Sports Med. 1984, 12, 98–103. [Google Scholar] [CrossRef]
- Terry, E.E.; Zhang, X.; Hoffmann, C.; Hughes, L.D.; Lewis, S.A.; Li, J.; Wallace, M.J.; Riley, L.A.; Douglas, C.M.; Gutierrez-Monreal, M.A.; et al. Transcriptional profiling reveals extraordinary diversity among skeletal muscle tissues. eLife 2018, 7, e34613. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Number | Gender | Age at Symptom Onset (y) | Age at Study Inclusion (y) | Disease Duration at Inclusion (y) | 6MWD (m) | 10MWT (s) | Mercuri Score SM | Mercuri Score VL | Mercuri Score RF | ANO5 Mutations |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 30 | 63 | 33 | 366 | 7.8 | 4 | 4 | 2 | c.191dupA (p.Asn64Lysfs*15); c.2317A>G (p.Met773Val) |
| 2 | M | 31 | 33 | 2 | 785 | 3.5 | 2 | 1 | 1 | c.191dupA (p.Asn64Lysfs*15); c.191dupA (p.Asn64Lysfs*15) |
| 3 | M | 34 | 37 | 3 | 689 | 4.5 | 2 | 2 | 1 | c.191dupA (p.Asn64Lysfs*15); c.191dupA (p.Asn64Lysfs*15) |
| 4 | M | 30 | 38 | 8 | 632 | 4.8 | 1 | 1 | 1 | c.191dupA (p.Asn64Lysfs*15); c.1961G>A (p.Arg654Gln) and c.155A>G (p.Asn52Ser) |
| 5 | M | 47 | 59 | 12 | 369 | 8.1 | 4 | 4 | 1 | c.172C>T (p.Arg58Trp); c.172C>T (p.Arg58Trp) |
| 6 | M | 30 | 48 | 18 | 479 | 7.1 | 4 | 3 | 1 | c.191dupA (p.Asn64Lysfs*15); c.692G>T (p.Gly231Val) |
| 7 | M | 38 | 55 | 17 | 792 | 3.3 | 2 | 1 | 1 | c.1213C>T (p.Gln405X); c.1733T>C (p.Phe578Ser) |
| 8 | M | 39 | 64 | 25 | 362 | 8.1 | 4 | 4 | 2 | c.191dupA (p.Asn64Lysfs*15); c.191dupA (p.Asn64Lysfs*15) |
| 9 | M | 35 | 43 | 8 | 452 | 7.5 | 2 | 1 | 1 | c.1210C>T (p.Arg404X); c.2387C>T (p.Ser796Leu) |
| 10 | M | 33 | 46 | 13 | 516 | 7.8 | 4 | 3 | 2 | c.191dupA (p.Asn64Lysfs*15); c.294+1G>A (p.?) |
| 11 | M | 15 | 48 | 33 | 630 | 5.0 | 2 | 1 | 1 | c.649-2A>G (p.?); c.679-2A>G (p.?) |
| 12 | M | 33 | 64 | 31 | 288 | 9.8 | 4 | 4 | 4 | c.41-1G>A (p.?); c.752C>T (p.Pro251Leu) |
| 13 | M | 13 | 26 | 13 | 570 | 5.0 | 2 | 1 | 1 | c.191dupA (p.Asn64Lysfs*15); c.191dupA (p.Asn64Lysfs*15) |
| 14 | M | 34 | 36 | 2 | 692 | 4.9 | 2 | 1 | 1 | c.191dupA (p.Asn64Lysfs*15); c.242A>G (p.Asp81Gly) |
| 15 | M | 28 | 40 | 12 | 433 | 8.2 | 4 | 2 | 1 | c.191dupA (p.Asn64Lysfs*15); c.1213C>T (p.Gln405X) |
| 16(#) | M | 25 | 31 | 6 | 687 | 3.1 | 2 | 1 | 1 | c.2411G>C (p.Cys804Ser); c.1627dupA (p.Met543Asnfs*11) |
| Marker Gene Analysis across Healthy Muscles | |||
|---|---|---|---|
| Rank | Semimembranosus | Rectus Femoris | Vastus Lateralis |
| 1 | HAND2-AS1 | TBX5 | LINC02107 |
| 2 | C12orf75 | NTNG2 | LINC02119 |
| 3 | HAND2 | GCNT2 | SBK2 |
| 4 | HOXD8 | TYRP1 | RHOXF1-AS1 |
| 5 | FRMD1 | ANKRD36BP2 | C1orf158 |
| 6 | LBP | MUC22 | CALML6 |
| 7 | HOXD-AS2 | HOXA13 | MYH1 |
| 8 | METTL21C | ZNF750 | LRRC37A7P |
| 9 | SLC1A2 | LINC01854 | FAM184B |
| 10 | HOXD9 | WFIKKN1 | RNA5-8S5 |
| 11 | IL31RA | TBX5-AS1 | LINC01886 |
| 12 | KIF1A | FNDC10 | FEZF1-AS1 |
| 13 | ANGPTL8 | IPCEF1 | CRNDE |
| 14 | CFAP57 | JCHAIN | CPXM1 |
| 15 | DNAH3 | CD300LB | TCF24 |
| 16 | IL22RA1 | SATB2-AS1 | CDH22 |
| 17 | BDNF | TUBB1 | PAX3 |
| 18 | LRRC52 | DIRAS1 | SNORD115-30 |
| 19 | C10orf67 | SPTA1 | GGT7 |
| 20 | CAPN8 | LINC01968 | MYHAS |
| 21 | CROCC2 | HMGCS2 | RGS10 |
| 22 | LAD1 | TREM1 | SLITRK3 |
| 23 | ANKRD18B | CCDC189 | PLCH1 |
| 24 | GLYAT | COL9A1 | IGFN1 |
| 25 | SCD | P2RX3 | ATRNL1 |
| 26 | IRX6 | RRM2 | ACTN3 |
| 27 | PAQR9-AS1 | IGHM | SHISA2 |
| 28 | LINC01484 | IGHV3-7 | GADD45G |
| 29 | C6orf132 | SPAG17 | MIR503HG |
| 30 | HOXD3 | RPS27AP9 | FGF10 |
| 31 | TMC1 | PAX1 | GREM2 |
| 32 | FOS | IRX4 | GDA |
| 33 | SCRT1 | CXCR2P1 | OPRD1 |
| 34 | RNY4P10 | DNAH11 | RN7SL813P |
| 35 | SLC26A9 | SIM2 | UBASH3B |
| 36 | CCDC78 | TMEM163 | SNORD115-23 |
| 37 | COMP | CDH20 | GDNF |
| 38 | ADRB1 | TMEM105 | NANOS1 |
| 39 | PLPPR1 | SKA3 | LINC01773 |
| 40 | GPR39 | SLC7A11-AS1 | HSD52 |
| 41 | LINC00877 | EPB42 | NPR3 |
| 42 | SLC29A4 | SLC30A8 | NME9 |
| 43 | RSPO1 | HS6ST2 | CHAD |
| 44 | BARX2 | FGD5P1 | MKRN3 |
| 45 | LOXL1-AS1 | SEL1L2 | SCT |
| 46 | GPA33 | NLRP12 | RN7SL267P |
| 47 | SDR42E2 | SLC4A10 | KHDRBS2 |
| 48 | CERS3 | CD160 | MYH4 |
| 49 | C2CD4B | ETF1P2 | RN7SL541P |
| 50 | TRPM1 | RYR3 | SH2D1B |
| Marker Gene Analysis across Patient Muscles | |||
|---|---|---|---|
| Rank | Semimembranosus | Rectus Femoris | Vastus Lateralis |
| 1 | COMP | LINC02107 | SIM2 |
| 2 | HAND2 | MYH1 | P2RX3 |
| 3 | HAND2-AS1 | LRRC37A7P | HMGCS2 |
| 4 | COL20A1 | AQP4 | CHAC1 |
| 5 | AQP6 | LINC01773 | LINC01854 |
| 6 | ADIPOQ | LINC02119 | KCTD8 |
| 7 | PLEKHG4B | C1orf158 | TBX5-AS1 |
| 8 | FRMD1 | PVALB | ZNF750 |
| 9 | CIDEC | ACTN3 | IGLV3-21 |
| 10 | TNMD | CALML6 | IGLC3 |
| 11 | HYDIN | MYLK4 | HOXA13 |
| 12 | SCD | FBP2 | IGHD |
| 13 | SALL1 | HCN1 | ADAMTS19-AS1 |
| 14 | LRRC74A | UNC13C | LAMC3 |
| 15 | LEP | B3GALT1 | MAPT-AS1 |
| 16 | SLC1A6 | ERBB4 | FBP2 |
| 17 | PLA2G2A | GREM2 | HAND2-AS1 |
| 18 | KCNQ2 | ATP2A1 | NEU4 |
| 19 | LGALS12 | RHOXF1-AS1 | SNCB |
| 20 | CHI3L1 | LINC01886 | IL20RA |
| 21 | GRM5 | NANOS1 | AQP4 |
| 22 | SLC5A10 | SHISA2 | HOXC12 |
| 23 | CCL18 | UGT3A1 | DIRAS1 |
| 24 | CUX2 | MLF1 | SLC16A3 |
| 25 | KLB | NRG4 | TSHR |
| 26 | MUC16 | SH2D1B | GDNF |
| 27 | GRIN2B | PLCH1 | KLHDC7B |
| 28 | PIEZO2 | LRRC3B | LINC01018 |
| 29 | SAA1 | MYHAS | SLC51A |
| 30 | MKX | MYLK2 | GHRL |
| 31 | GRM4 | FEZF1-AS1 | TBX1 |
| 32 | TSPEAR | FAM166B | IGHV1-3 |
| 33 | TNC | SNORD23 | TYRP1 |
| 34 | MYEOV | ENO3 | CRYM |
| 35 | PCK1 | ENSAP2 | PIANP |
| 36 | PLIN1 | IRX3 | IGKV1-5 |
| 37 | CACNA1I | CDH22 | TMEM26 |
| 38 | USH2A | ATRNL1 | MTND4P24 |
| 39 | MUC6 | LANCL1-AS1 | RN7SKP276 |
| 40 | COL22A1 | NEK10 | FAM166B |
| 41 | SCUBE1 | PDE4DIPP1 | HPN |
| 42 | SCRG1 | TMEM266 | OSCAR |
| 43 | S100A3 | FABP7 | HES7 |
| 44 | KRT7 | KLHL38 | PAX1 |
| 45 | OPRM1 | AGMAT | ASB12 |
| 46 | DUX4L19 | SMCO1 | DDX11L2 |
| 47 | MYBL2 | ASB14 | ANKRD20A21P |
| 48 | AMZ1 | NPSR1-AS1 | SPAG17 |
| 49 | RASAL1 | PITX1 | TNNI3 |
| 50 | MROH4P | DDIT4L | C1orf105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Depuydt, C.E.; Goosens, V.; Janky, R.; D’Hondt, A.; De Bleecker, J.L.; Noppe, N.; Derveaux, S.; Thal, D.R.; Claeys, K.G. Unraveling the Molecular Basis of the Dystrophic Process in Limb-Girdle Muscular Dystrophy LGMD-R12 by Differential Gene Expression Profiles in Diseased and Healthy Muscles. Cells 2022, 11, 1508. https://doi.org/10.3390/cells11091508
Depuydt CE, Goosens V, Janky R, D’Hondt A, De Bleecker JL, Noppe N, Derveaux S, Thal DR, Claeys KG. Unraveling the Molecular Basis of the Dystrophic Process in Limb-Girdle Muscular Dystrophy LGMD-R12 by Differential Gene Expression Profiles in Diseased and Healthy Muscles. Cells. 2022; 11(9):1508. https://doi.org/10.3390/cells11091508
Chicago/Turabian StyleDepuydt, Christophe E., Veerle Goosens, Rekin’s Janky, Ann D’Hondt, Jan L. De Bleecker, Nathalie Noppe, Stefaan Derveaux, Dietmar R. Thal, and Kristl G. Claeys. 2022. "Unraveling the Molecular Basis of the Dystrophic Process in Limb-Girdle Muscular Dystrophy LGMD-R12 by Differential Gene Expression Profiles in Diseased and Healthy Muscles" Cells 11, no. 9: 1508. https://doi.org/10.3390/cells11091508
APA StyleDepuydt, C. E., Goosens, V., Janky, R., D’Hondt, A., De Bleecker, J. L., Noppe, N., Derveaux, S., Thal, D. R., & Claeys, K. G. (2022). Unraveling the Molecular Basis of the Dystrophic Process in Limb-Girdle Muscular Dystrophy LGMD-R12 by Differential Gene Expression Profiles in Diseased and Healthy Muscles. Cells, 11(9), 1508. https://doi.org/10.3390/cells11091508