1. Introduction
Cardiac fibrosis may be considered an aberrant wound healing process. In non-cardiac tissues, the tissue injury response normally results in the synthesis of extracellular matrix (ECM) proteins balanced by the degradation of existing ECM proteins by matrix metalloproteinases (MMPs); this typically restores tissue function without causing fibrosis [
1]. In the myocardium, however, pathological stresses such as hypertension, valve disease, diabetes, or myocardial infarction result in a prolonged and/or dysregulated wound healing response that leads to scar formation or interstitial fibrosis, which interferes with cardiac function in a manner that frequently leads to heart failure or death [
1,
2]. Resident fibroblasts are primarily responsible for the synthesis of ECM components including collagens and proteoglycans; in response to pro-fibrotic stimuli, including growth factors, cytokines, and mechanical stretch, fibroblasts become activated to myofibroblasts that sharply upregulate ECM production [
1,
3,
4,
5,
6]. While cardiac fibrosis is now recognized as a major contributor to morbidity and mortality, there remain no therapeutic strategies for its management [
7].
The synthesis of ECM components, including large macromolecules such as fibrillar collagens, is an energy-intensive process. Metabolic reprogramming, including mitochondrial biogenesis, is required for the activation of human fetal lung fibroblasts to myofibroblasts [
8]. The underlying mechanisms governing energy metabolism in fibroblasts and myofibroblasts, however, remain poorly defined, and the metabolic processes themselves appear to vary across tissues. Glycolysis is increased in lung fibroblasts during activation to myofibroblasts [
9]. Conversely, the promotion of fatty acid oxidation attenuates tubulointerstitial fibrosis; thus, energy requirements in fibrosis likely require specific alterations in metabolism [
10]. Glutaminolysis utilizes glutamine metabolism for energy production via the conversion of glutamine to glutamate and ammonia by the rate-limiting enzyme glutaminase (GLS), and the subsequent conversion of glutamate to α-ketoglutarate (α-KG) by glutamate dehydrogenase (GDH), glutamate oxaloacetate transaminase (GOT), or the glutamate pyruvate transaminase (GPT). α-KG then enters the TCA cycle to meet energy demands [
11,
12]. Glutaminolysis has already been identified as a major target for the treatment of liver and pulmonary fibrosis [
13,
14]. A recent study in mouse embryonic fibroblasts identified increases in glycolysis and glutaminolysis as necessary to induce fibroblast activation and conversion to myofibroblasts, with α-KG-dependent histone demethylases in turn activating myofibroblast genes [
15]. The role of glutaminolysis specifically in cardiac fibroblasts remains unclear.
The transcription factor scleraxis plays a key role in the activation of fibroblasts to myofibroblasts via direct transcriptional activation of key fibrotic genes, leading to increased ECM synthesis [
5]. TGFβ is a potent inducer of fibrosis in many tissues, and it was recently shown that the activation of human lung fibroblasts to myofibroblasts required glutaminolysis, with GLS1 expression dependent on TGFβ and its downstream effector SMAD3 [
16]. Since we previously showed that TGFβ-mediated activation of cardiac fibroblasts and the induction of fibrotic gene expression require scleraxis, we examined whether scleraxis may play a role in cardiac fibroblast glutaminolysis [
5,
17,
18].
Here we report that, similar to lung fibroblasts, cardiac fibroblasts upregulated GLS1 expression in response to TGFβ, and that the GLS1 inhibitor CB-839 attenuated fibroblast activation. Scleraxis over-expression in cardiac fibroblasts induced the expression of multiple enzymes in the glutaminolysis pathway, including GLS1, GLS2, and GOT2. Conversely, GLS1 and GDH1 expression was decreased in scleraxis knockout cardiac fibroblasts. Scleraxis knockdown attenuated GLS1 expression and dramatically blunted GLS1 upregulation by TGFβ. Cellular concentrations of glutamate and glutamine increased with TGFβ treatment, indicative of increased throughput in the glutaminolysis pathway, but this effect was attenuated by scleraxis knockout. Scleraxis regulated GLS1 gene expression via a specific E-box binding site in the human GLS1 promoter. Scleraxis thus appears to be a key regulator of glutaminolysis in cardiac fibroblast activation via the transactivation of the GLS1 gene.
2. Materials and Methods
2.1. Cardiac Fibroblast Isolation and Culture
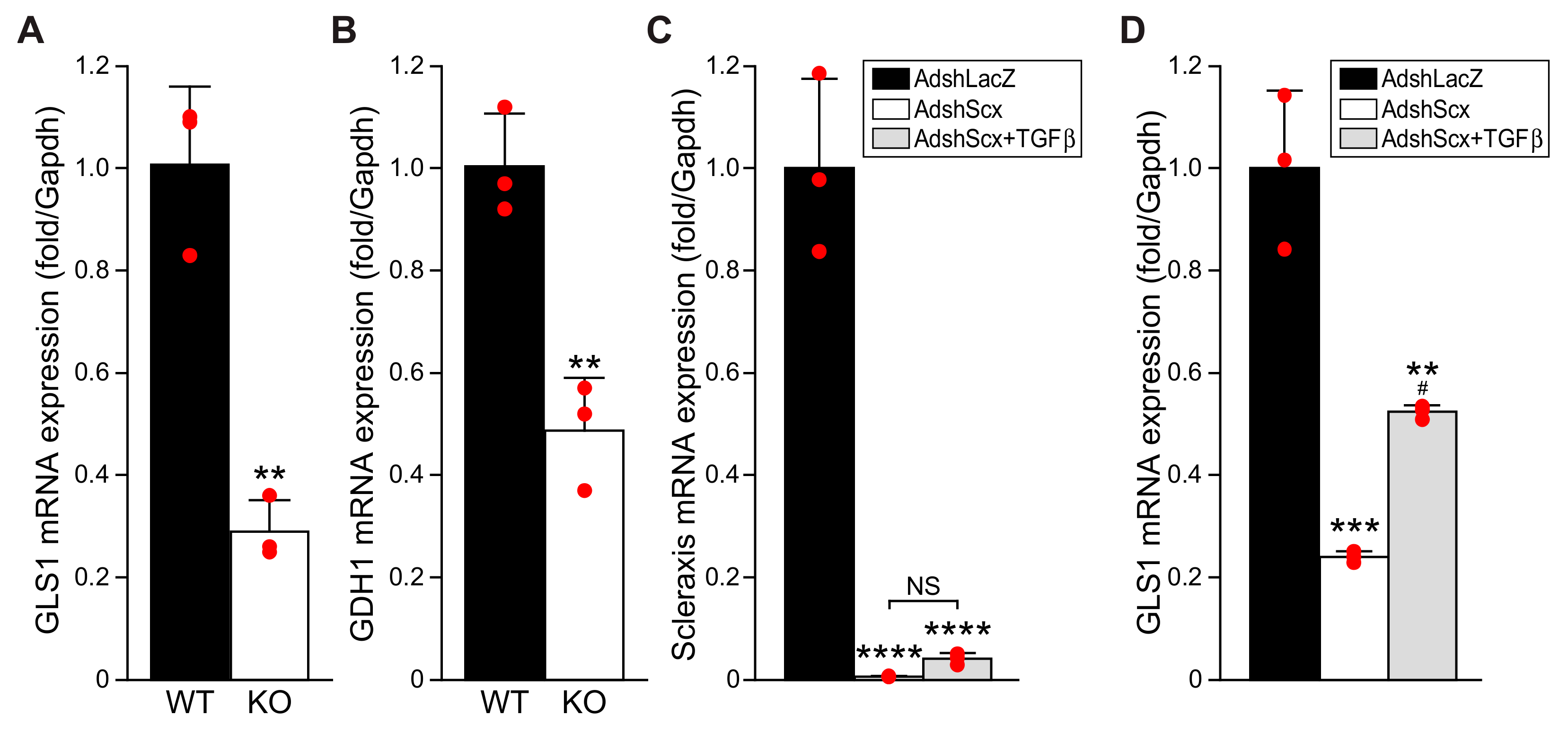
Primary rat cardiac fibroblasts (RCFs) from adult male Sprague Dawley rat hearts were isolated by enzymatic digestion as previously described [
19,
20]. Primary mouse cardiac fibroblasts (MCFs) were isolated using enzymatic digestion from scleraxis germline knockout (KO), or C57BL6 wild-type (WT) control mice as previously described [
5]. Primary adult RCFs and MCFs were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM)/F12 medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. NIH3T3 mouse fibroblasts were cultured in DMEM containing 10% fetal bovine serum and 1% penicillin-streptomycin. Adult human cardiac myofibroblasts (Cell Applications, San Diego, CA, USA) were cultured in DMEM/F12 medium supplemented with 20% fetal bovine serum and 1% penicillin-streptomycin. Primary rat/mouse cardiac fibroblasts harvested within 6 h of initial plating were used as zero passage (P0) cells or were passaged once at 48 h after initial plating and used as activated fibroblasts (P1) or were passaged a second time at 96 h after initial plating and used as myofibroblasts (P2) as described previously [
5]. All cells were maintained at 37 °C and 5% CO
2. For TGFβ
1 treatment, P1 cells were plated on either 35 mm or 6-well cell culture dishes and maintained in 10% serum-containing medium for 16 h to reach 70% confluence, then serum starved for 6 h prior to treatment with TGFβ
1 (10 ng/mL) or vehicle (PBS). After 24 h treatment, cells were harvested for processing. For inhibition of glutaminolysis, cells were grown to 70% confluence, serum-starved for 24 h, then treated with the GLS1 inhibitor CB-839 (0.3 μM) or vehicle (DMSO) followed one hour later by TGFβ
1 or vehicle treatment for 24 h, then cells were harvested and processed.
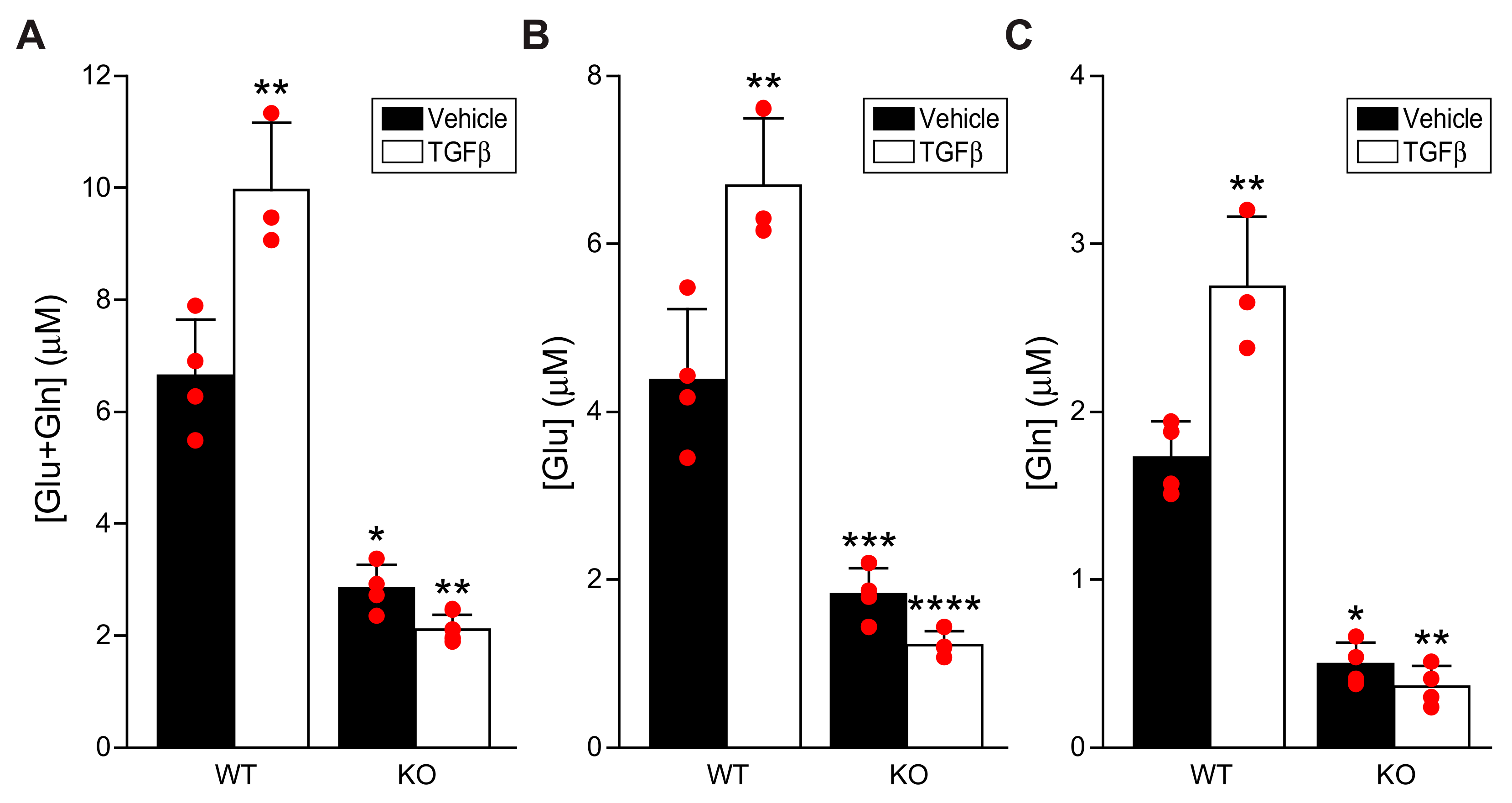
For scleraxis adenoviral overexpression experiments, RCFs at P1 were grown within 16 h to reach 70% confluence and transfected with adenoviruses encoding either scleraxis (AdScx) or Green Fluorescent Protein (AdGFP) at a multiplicity of infection of 100 in serum-free medium for 48 h. For scleraxis knockdown experiments, RCFs at P1 were grown for 16 h to reach 70% confluence, then serum starved for 6 h and transfected with scleraxis knockdown adenovirus (AdshScx) or control virus (AdshLacZ) at a multiplicity of infection of 100 in serum-free medium for 72 h, with or without TGFβ1 (10 ng/mL) added at 48 h after adenovirus addition. For luciferase assays, NIH3T3 fibroblasts were grown in 6-well plates to reach 60–70% confluence and maintained in low serum (0.5%) for 2 h prior to transfection. For chromatin immunoprecipitation assays, adult human cardiac myofibroblasts were grown in 150 mm dishes for 16 h to reach 70–80% confluence, then serum starved for 30 h prior to harvest for further analysis. For intracellular glutamine/glutamate assay, freshly isolated wild type or scleraxis knockout MCFs were plated on 12-well dishes to reach 60–70% confluence, followed by serum starvation for 24 h prior to treatment with TGFβ1 (10 ng/mL) or vehicle.
2.2. Quantitative Real-Time PCR
Total RNA from primary adult rat or mouse cardiac fibroblasts were harvested using a Monarch Total RNA Miniprep Kit (New England Biolabs, Whitby, ON, Canada) according to the manufacturer’s instructions. cDNA was generated using an iScript cDNA Synthesis kit (Bio-Rad, Mississauga, ON, Canada), and SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) was used for qPCR amplification according to the manufacturer’s instructions. PCR amplification was performed in a CFX384 Touch Real-Time PCR (Bio-Rad, Canada). The cycling conditions were 95 °C (3 min), followed by 40 cycles of denaturation at 95 °C (15 s) and extension at 62 °C (30 s). After amplification, a continuous melt curve was generated from 60 to 95 °C to confirm the amplification of single amplicons. Relative gene expression was calculated using the 2
−ΔΔCt method with normalization to Gapdh. Primer sequences are listed in
Table S1.
2.3. Western Blotting
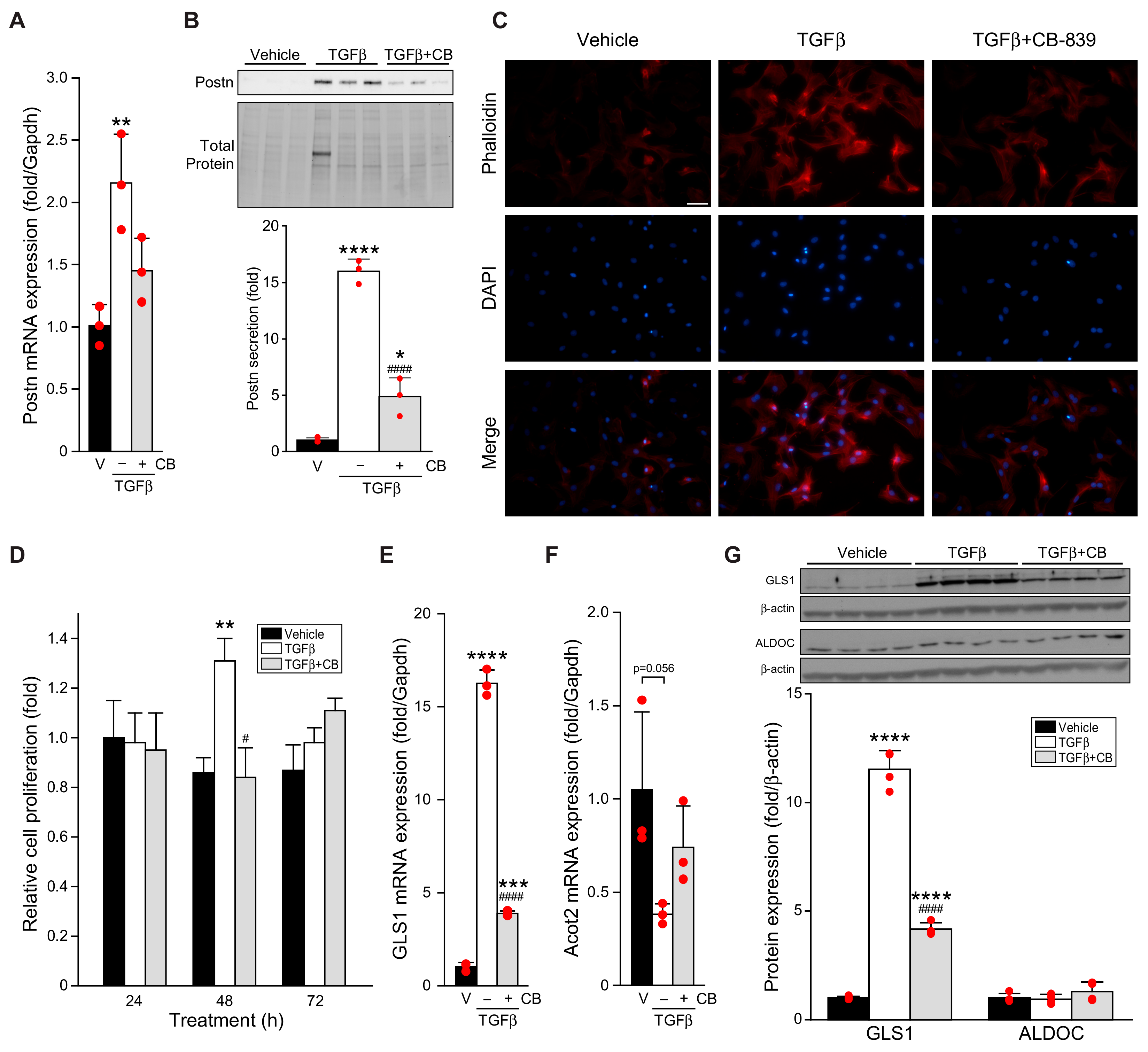
For whole-cell lysates, 30 μg protein samples were resolved on 10% SDS-PAGE gels and transferred to PVDF membranes, which were then blocked with 5% nonfat milk in Tris-Buffered Saline with 0.1% Tween-20 (TBS-T) for 1 h at room temperature. For detection of proteins of interest, the membranes were incubated with primary antibodies for GLS1 (1:1000; PA5-35365; ThermoFisher Scientific, Ottawa, ON, Canada), ALDOC (1:1000; PA5-27659; ThermoFisher Scientific, Canada), or Acot2 (1:1000; 15633-1-AP; ThermoFisher Scientific, Canada) in 2% nonfat milk in TBS-T at 4 °C overnight, followed by washing and incubation with secondary horseradish peroxidase-conjugated goat anti-rabbit antibody (1:10,000; 111-035-003; Jackson Laboratories, Bar Harbor, ME, USA) in TBS-T with 1% nonfat milk for 1 h at room temperature. Protein bands were visualized using Pierce ECL Western blotting chemiluminescent substrate (ThermoFisher Scientific, Canada) and exposure to CL-XPosure film (Mandel Scientific, Guelph, ON, Canada). Protein quantification was conducted using Image J (NIH, Bethesda, MD, USA), with normalization to horseradish peroxidase-conjugated β-actin antibody (1:65,000; SC-47778; Santa Cruz Biotechnology Inc., Dallas, TX, USA). Secreted periostin was similarly assessed using culture medium collected from plated cells, with proteins concentrated using Amicon Ultra-15 centrifugal filters (Millipore Sigma, Oakville, ON, Canada) and resolved on 4–15% TGX Stain-Free Precast Gels (Bio-Rad, Canada). Proteins were transferred to PVDF membranes and probed with primary antibodies for periostin (1:1000; ab14041; Abcam, Toronto, ON, Canada), with normalization to total protein given the lack of a specific loading control for secreted proteins.
2.4. Plasmid Constructs and Mutagenesis
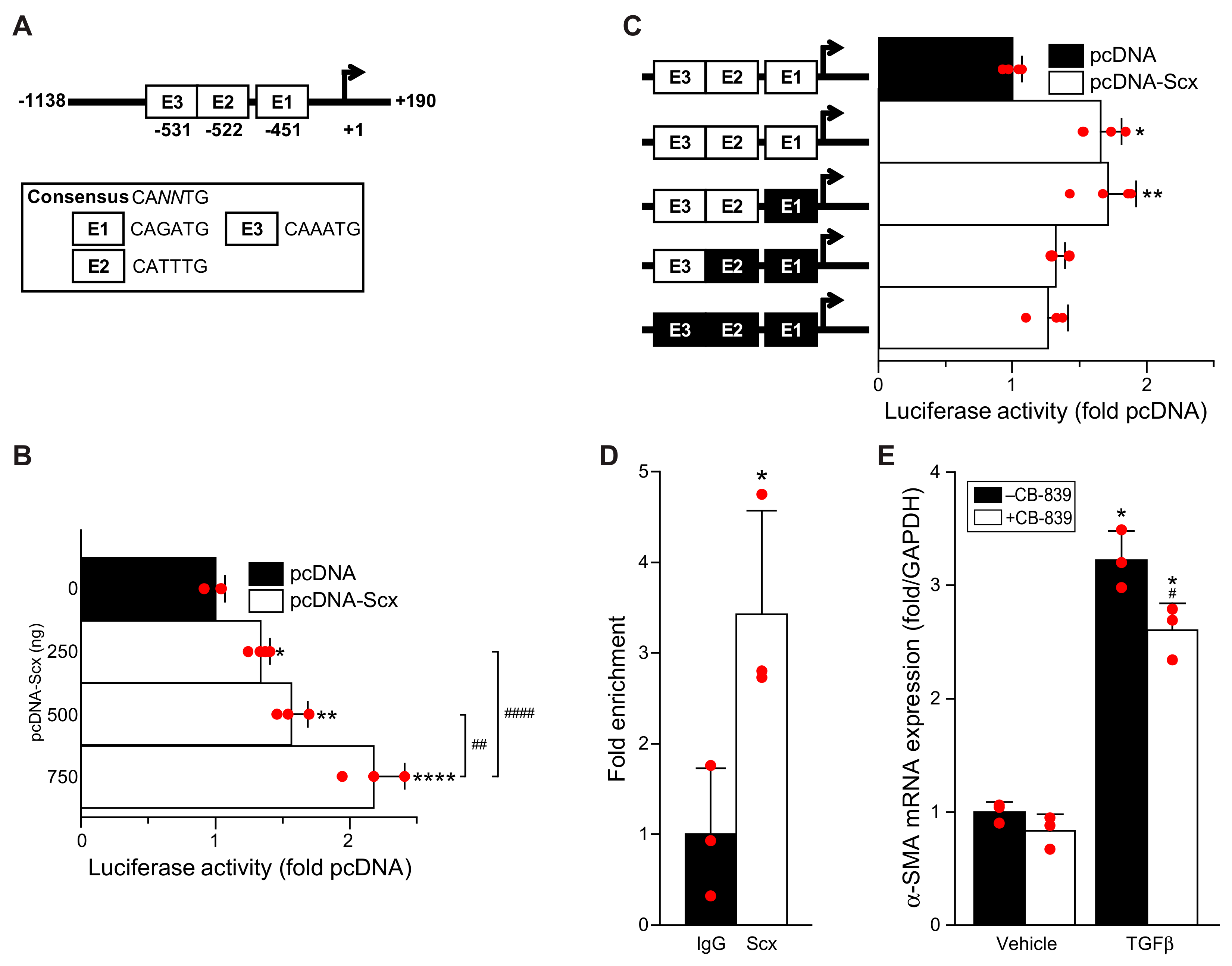
The proximal human GLS1 promoter (−1138 to +190 relative to the transcription start site, 1329 base pairs total length) was cloned by PCR amplification from the HPRM39644 pEZX-PG02.1 vector (GeneCopoeia, Rockville, MD, USA) using the primers listed in
Table S2, and incorporating restriction sites for SacI (forward primer) and EcoRV (reverse primer). The promoter was then subcloned into the luciferase reporter vector pGL4.10-luc2 (Promega, Madison, WI, USA), generating the construct pGL4.10-hGLS1.
Site-directed mutagenesis of three putative E-box sites was performed sequentially using a QuikChange II XL kit (Agilent Technologies, Santa Clara, CA, USA) and primers listed in
Table S3. All constructs were confirmed by sequencing.
2.5. Luciferase Assay
NIH3T3 fibroblasts were co-transfected with scleraxis expression vector pcDNA-Scx or empty control vector pcDNA (pcDNA6, ThermoFisher Scientific, Canada), and pGL4.10-hGLS1 with or without E-box mutants using Lipofectamine 3000 (ThermoFisher Scientific, Canada) for 24 h. Renilla luciferase vector pRL was co-transfected as an internal transfection control. Luciferase activity was measured using the Dual Luciferase Reporter Assay System kit (Promega, USA) and a GloMax-Multi+ Multimode Plate Reader (Promega, USA) according to manufacturer’s directions.
2.6. Glutamine/Glutamate Assay
Mouse cardiac fibroblasts were harvested after treatment with TGFβ1, and the cell lysates were processed as per the manufacturer’s instructions (Glutamine/Glutamate-Glo™ Assay; J8021; Promega, USA) for determining the intracellular glutamine and glutamate concentrations. Two reactions were performed in duplicate per sample in an opaque, white 96-well plate (Corning Costar; 3912; Millipore-Sigma, Canada). For one reaction, the cell lysates were incubated with glutaminase enzyme to provide total glutamine + glutamate concentration; for the second reaction, the cell lysates were incubated without glutaminase enzyme, thereby providing the glutamate concentration alone. Subtracting these two values yielded the glutamine concentration. Luminescence was recorded using a GloMax-Multi+ Multimode Plate Reader (Promega, USA).
2.7. Chromatin Immunoprecipitation Assay
Adult human cardiac myofibroblasts were serum starved for 30 h, then cross-linked with 4% paraformaldehyde, and chromatin was sheared into 300–500 base pair fragments by sonication. After sonication, cell lysates were processed using an EZ-Magna ChIP HiSens Chromatin Immunoprecipitation Kit (Millipore-Sigma, Canada), and the experiment was carried out according to the manufacturer’s instructions. For the chromatin pull-down, either anti-scleraxis antibody or non-specific IgG was used [
20]. Specific primers were used to amplify the region of the scleraxis-bound E-boxes (E1/E2/E3) in the proximal human GLS1 gene promoter, with assessment by qPCR (
Table S4).
2.8. Phalloidin Staining
Freshly isolated adult rat cardiac fibroblasts were plated on acid-washed 18 mm glass coverslips in 12-well plates, then treated with CB-839 and/or TGFβ1 as above. Cells were fixed with 4% paraformaldehyde for 30 min at room temperature followed by 3 washes in PBS, then permeabilized in 0.1% Triton X-100 in PBS for 5 min followed by 3 washes in PBS. Coverslips were incubated for 1 h at room temperature with CytoPainter Phalloidin-iFluor 594 Reagent (ab176757; Abcam, Canada) as per the manufacturer’s instructions. Staining solution was removed, and coverslips were washed 3 times with PBS. Coverslips were mounted onto slides using ProLong Gold Antifade Mountant with DAPI (ThermoFisher Scientific, Canada). Cells were imaged on a Nikon Eclipse TE2000S fluorescent microscope using a 20× objective. NIS software (Nikon, Melville, NY, USA) was used for image capture, and Image J (NIH, USA) was used to merge captured images.
2.9. CCK8 Cell Proliferation Assay
Cell proliferation was analyzed using a Cell Counting Kit-8 (CCK-8) (CK04-011; Dojindo Laboratories, Rockville, MD, USA), according to the manufacturer’s directions. Briefly, rat cardiac fibroblasts were plated in 96-well plates and treated with or without TGFβ1 and/or CB-839 as above for 24, 48, or 72 h as above. CCK-8 solution was added to each well, except for 3 baseline control wells from each of the treated groups. Plates were incubated at 37 °C for 1 h, then optical density at 450 nm was measured for each well using a microplate reader, baseline mean was subtracted from each sample value, and results were then normalized to vehicle-treated cells.
2.10. Statistical Analysis
Data are reported as mean ± standard deviation for a minimum of three independent biological replicates. To reduce variability, all cells, including control and treatment groups, were isolated and cultured on the same day. Results were analyzed by a two-tailed Student’s t-test or a one-way or two-way analysis of variance (ANOVA), with Tukey post hoc analysis as appropriate, using GraphPad Prism v.9.3.1 (GraphPad Software, San Diego, CA, USA) with p < 0.05 considered to be statistically significant. Normality of distribution was confirmed using the Shapiro–Wilk test; non-normally distributed data were analyzed by the Kruskal–Wallis test, followed by Dunn’s multiple comparisons test.
4. Discussion
Interstitial fibroblasts normally synthesize the extracellular matrix components of the healthy heart [
25,
26]. During wound healing or in response to stress, however, it is the myofibroblasts that play a major role through the accelerated synthesis of extracellular matrix components such as fibrillar collagens. Persistent fibrosis contributes to structural and functional remodeling of the heart, with increased arrhythmogenesis due to altered conduction characteristics, leading to further functional and structural derangements that may cause heart failure and death [
1,
8]. At present, cardiac fibrosis remains without existing interventions for clinical management.
Despite the significant impact of fibrosis on cardiac function and its key role in patient morbidity and mortality, surprisingly little is known of the energy metabolism of cardiac fibroblasts and myofibroblasts, although some advances have been made in other tissues. Increased glycolysis enhances myofibroblast activation and contributes to lung fibrosis [
8,
9]. Recent studies in mouse embryonic fibroblasts showed that fibrotic signaling increased α-KG availability, with increases in glycolysis and glutaminolysis to provide TCA cycle intermediates [
15]. Glutaminolysis has also been identified as a key energy source for liver and lung fibrosis [
13,
14]. Our results suggest that a similar requirement for glutaminolysis occurs when cardiac fibroblasts are activated to become myofibroblasts (
Figure 6).
TGFβ is a potent driver of fibroblast activation to myofibroblasts, contributing to fibrosis in many tissue types, and we observed that TGFβ increased the expression and secretion of the myofibroblast-specific marker periostin, increased stress fiber formation, and induced a transient increase in cell proliferation concomitant with a dramatic increase in GLS1 expression. Intriguingly, all of these effects could be attenuated by the GLS1 inhibitor CB-839, suggesting that the blockade of glutaminolysis prevented fibroblast activation and metabolic reprogramming. Although we did not assess fatty acid β-oxidation, it is notable that CB-839 also restored the loss of Acot2 expression induced by TGFβ, suggesting that an increase in glutaminolysis may be accompanied by a decrease in this metabolic pathway. While increased glycolysis has been implicated in lung fibroblast activation, we observed no change in ALDOC expression by either TGFβ or CB-839, suggesting the cell type specificity of this pathway. Passaging fibroblasts to induce activation to myofibroblasts, or overexpressing scleraxis to induce activation, caused similar increases in GLS1 expression, and scleraxis also increased the expression of other glutaminolysis pathway components, including GLS2, GOT2, and GDH1. The activation of cardiac fibroblasts to myofibroblasts, regardless of the inducer, thus increases gene expression indicative of an increase in glutaminolysis. We also observed increases in intracellular glutamate and glutamine, indicative of greater flux through the glutaminolysis pathway, in response to TGFβ. Together, these findings support a central role for the induction of glutaminolysis during cardiac fibroblast activation, similar to observations in the liver and lung.
TGFβ potently induces downstream pro-fibrotic gene programs via both the canonical Smad pathway and a variety of non-canonical signal transduction mechanisms. We have previously shown that TGFβ induces scleraxis expression via both mechanisms, i.e., Smad3 and Erk1/2 [
27]. We have also reported that scleraxis not only induces the expression of myriad pro-fibrotic genes via direct promoter transactivation, but also is critically required for TGFβ to regulate the expression of these genes, ostensibly via the physical interaction of scleraxis with Smad3 [
5,
17,
18,
19,
20]. Our data suggest that a similar mechanism is involved in the induction of glutaminolysis, since the TGFβ-mediated upregulation of GLS1 or increases in intracellular glutamine and glutamate required scleraxis. As a basic helix-loop-helix transcription factor, scleraxis regulates gene expression by binding to E-box sequences within target gene promoters, including fibrillar collagens, fibronectin, MMP2, αSMA, Twist1, Snai1, and vimentin [
5,
17,
18,
19,
20,
28]. Luciferase assays confirm that, while the proximal human GLS1 promoter contains three sites that conform to the E-box consensus sequence CA
NNTG, only one of these sites (E2) appears to be critical for scleraxis-mediated promoter transactivation. The control of GLS1 expression by scleraxis thus exhibits the same hallmarks of many other scleraxis target genes involved in fibroblast activation and ECM expression, suggesting that GLS1 upregulation (and increased glutaminolysis) may be part of this pro-fibrotic gene program. While we did not examine other glutaminolysis genes in the same detail as GLS1, our data suggest that scleraxis may similarly regulate the expression of GOT2 and GDH1, providing multiple sites of control over glutaminolysis.
Given its central role in patient morbidity and mortality, interfering with fibrosis is a critical, unmet clinical need in many tissue types, including the heart. The present work suggests two approaches that may help to address this gap—interfering with scleraxis to attenuate GLS1 expression or blocking GLS1 function by pharmacologic inhibition. Removing glutaminolysis as a key source of cellular energy has already been demonstrated to be effective in cancer therapy [
29,
30]. GLS1 is abnormally upregulated in a variety of cancers, and depleting or pharmacologically inhibiting GLS1 with CB-839 reduced tumour proliferation and metastasis [
31,
32,
33]. CB-839 has also been effective in reducing pulmonary or hepatic fibrosis [
13,
14]. Our data suggest that this approach could be similarly effective in the heart. Knocking down scleraxis dramatically reduced GLS1 expression, and scleraxis knockout cells failed to increase glutamine and glutamate levels in response to TGFβ. CB-839 blocked the TGFβ-mediated upregulation of both GLS1 and periostin, suggesting a blockade of fibroblast activation. An important next step will be to determine whether CB-839 administration in vivo attenuates cardiac fibrosis in preclinical models.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}