
Tet1 Suppresses p21 to Ensure Proper Cell Cycle Progression in Embryonic Stem Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Embryonic Stem Cell Culture and Proliferation Assays
2.2. Cell Cycle Analysis and Apoptosis Assays
2.3. RT-qPCR and ChIP-qPCR
2.4. Western Blotting
2.5. Lentivirus Preparation for p21 Knockdown
2.6. Bioinformatic Analysis
3. Results
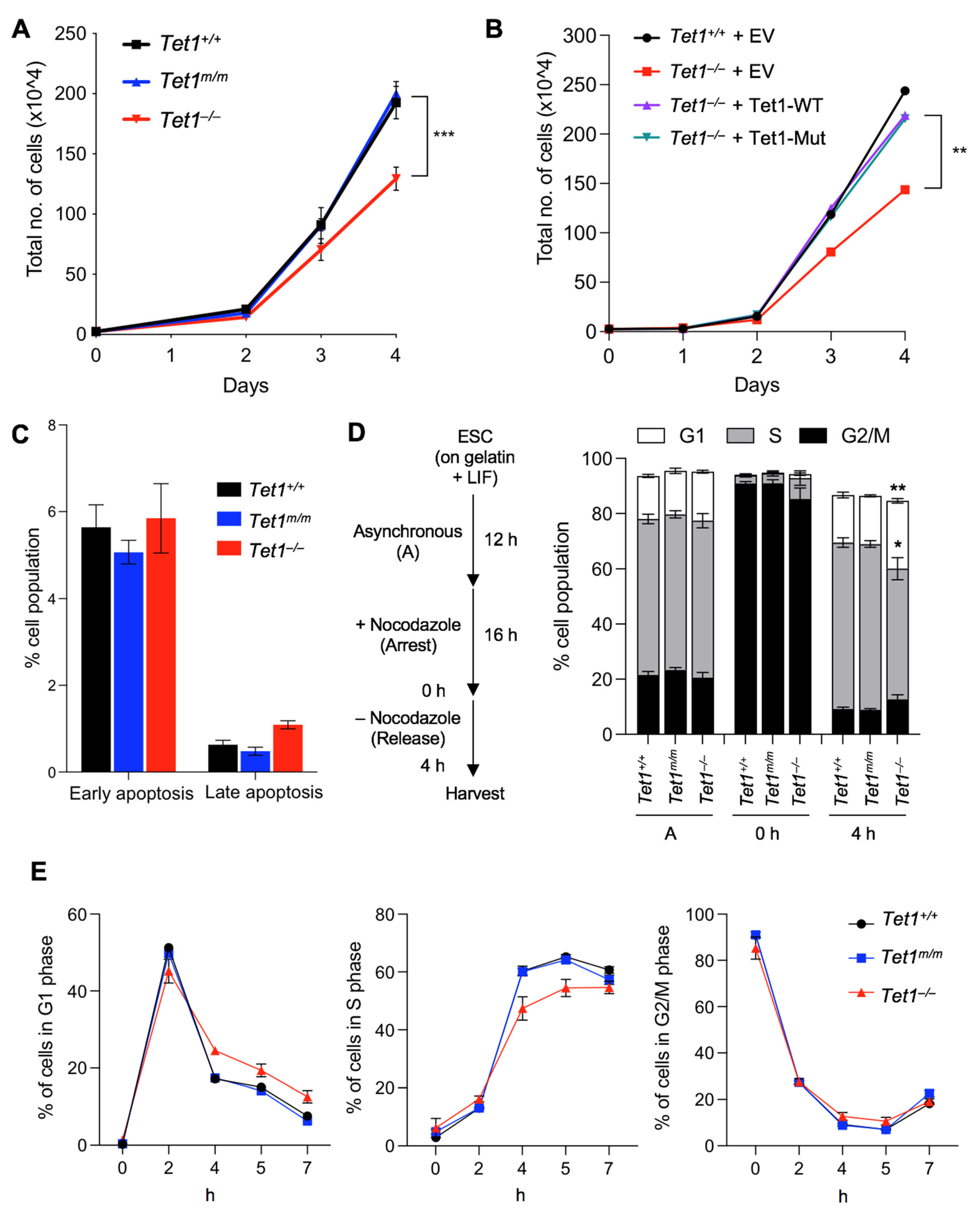
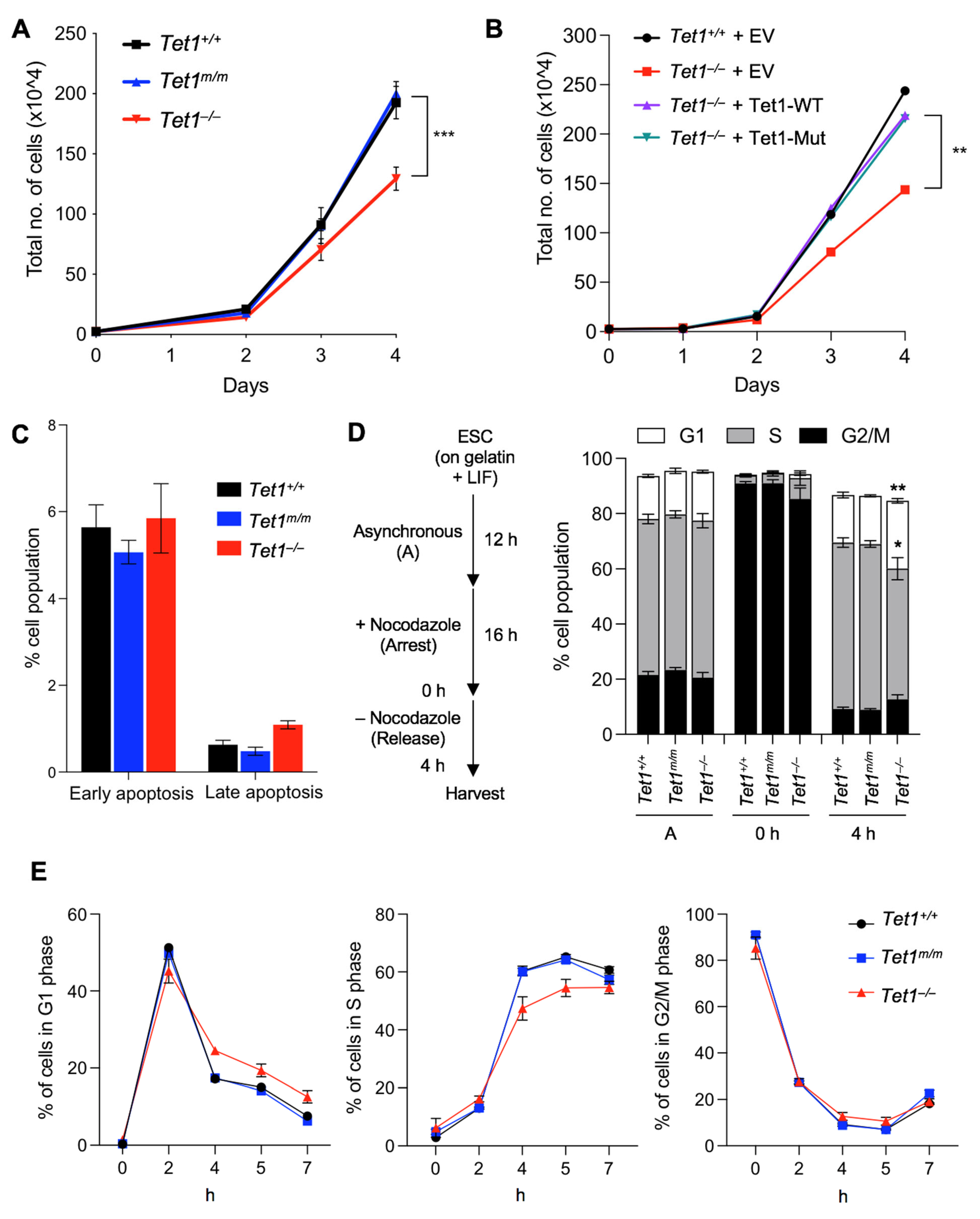
3.1. Deficiency of Tet1, but Not of Its Catalytic Activity, Leads to Reduced Proliferation and Extended G1 Phase in mESCs
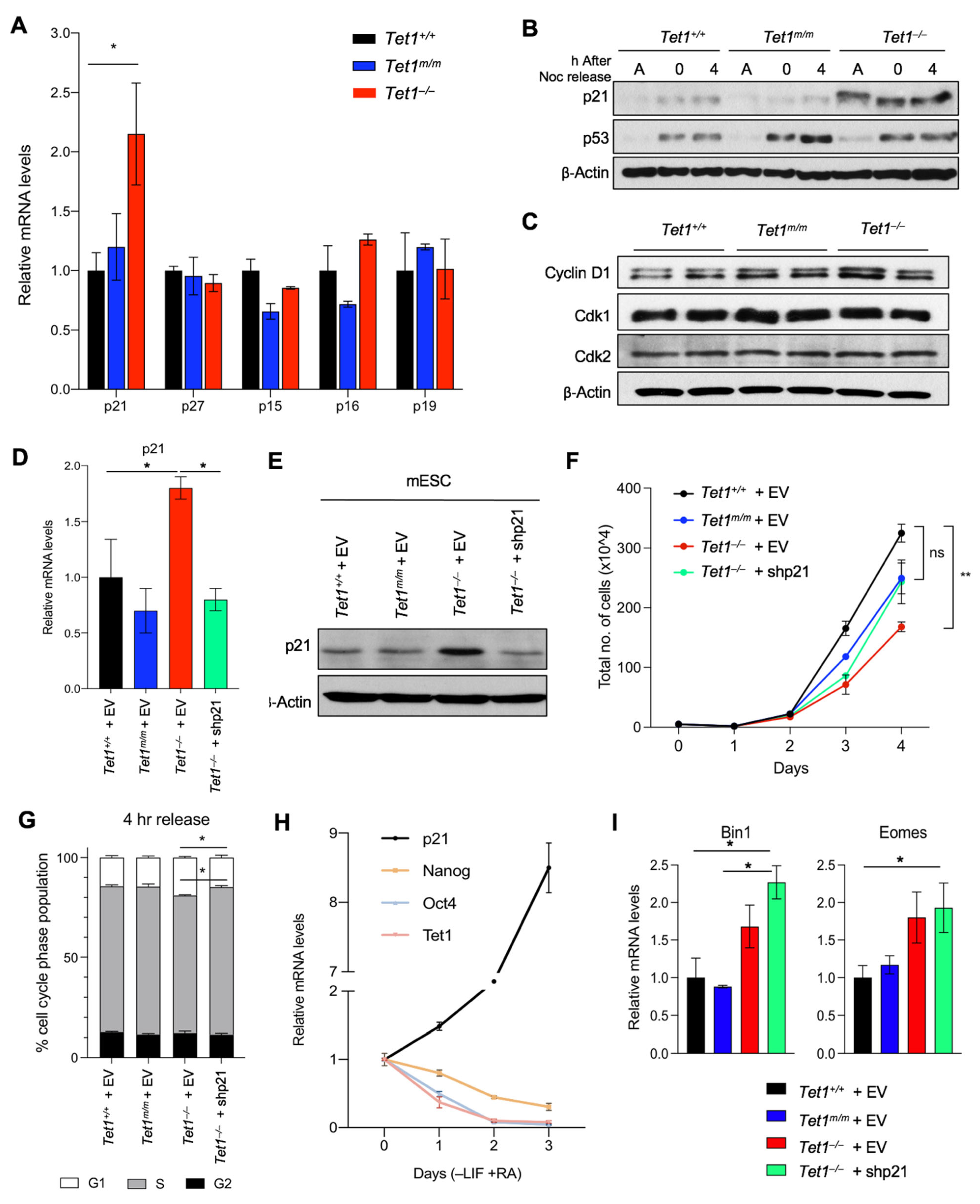
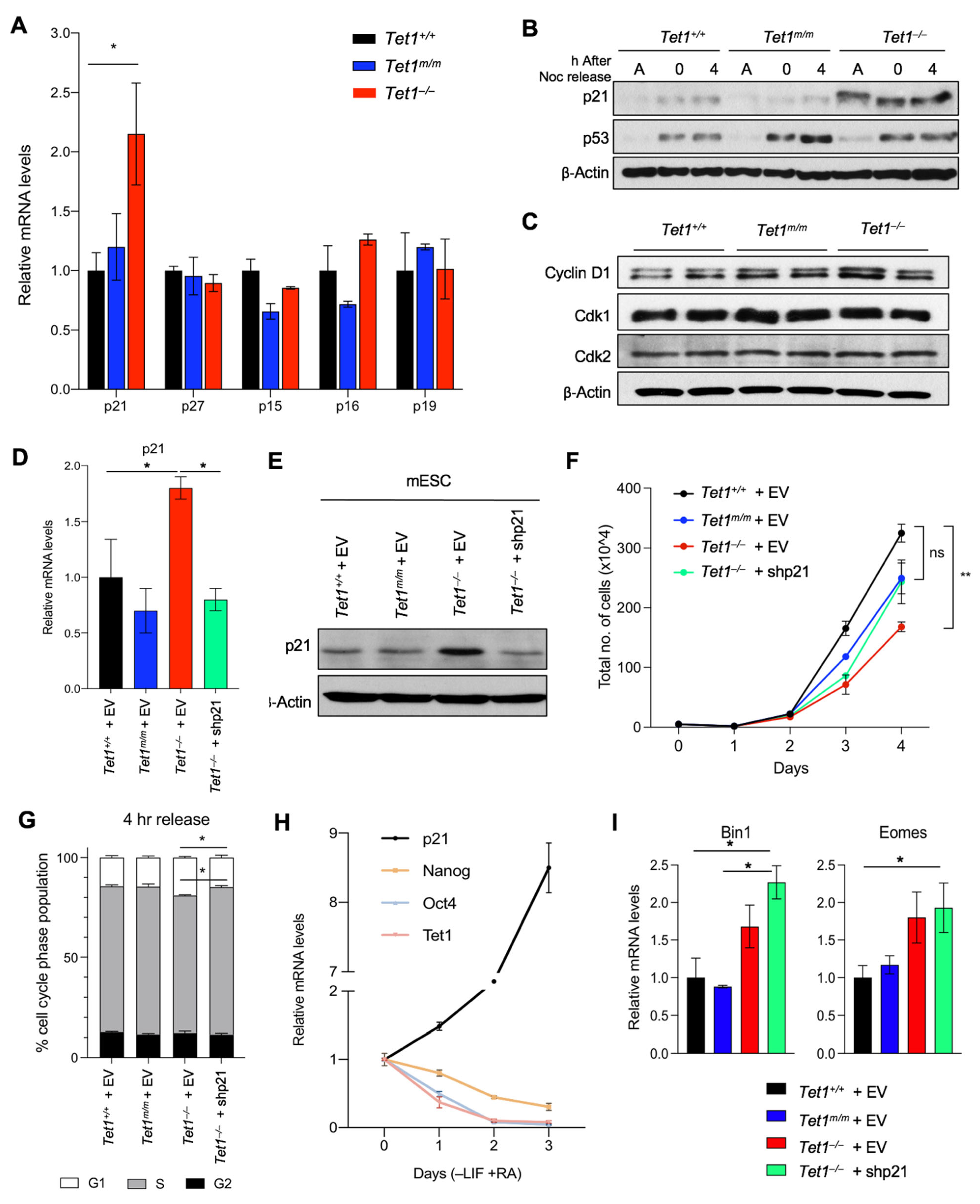
3.2. Upregulation of the Cyclin-Dependent Kinase Inhibitor p21 (Cdkn1a) Is Responsible for Delayed Cell Cycle Progression and Reduced Proliferation in Tet1−/− mESCs
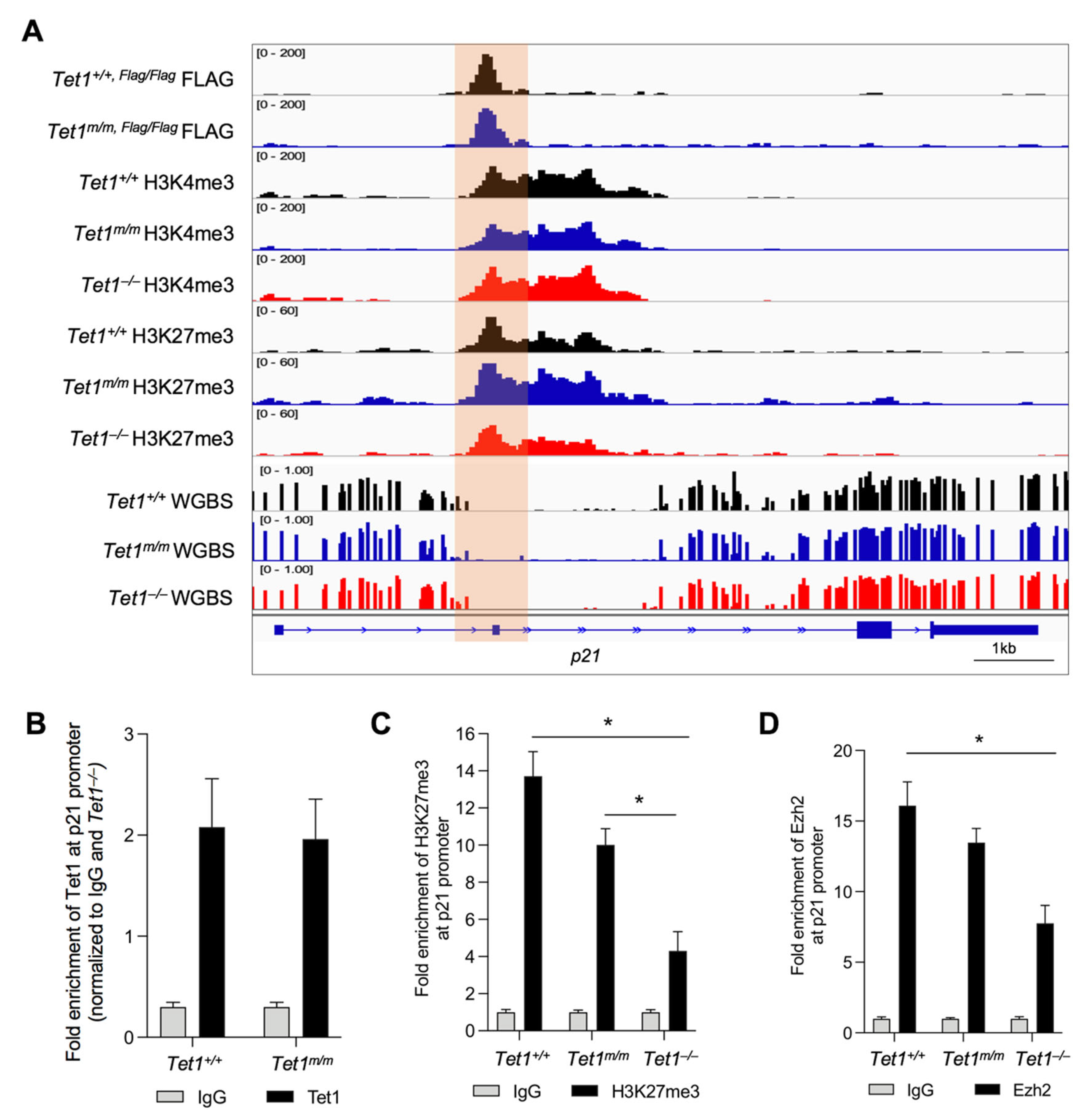
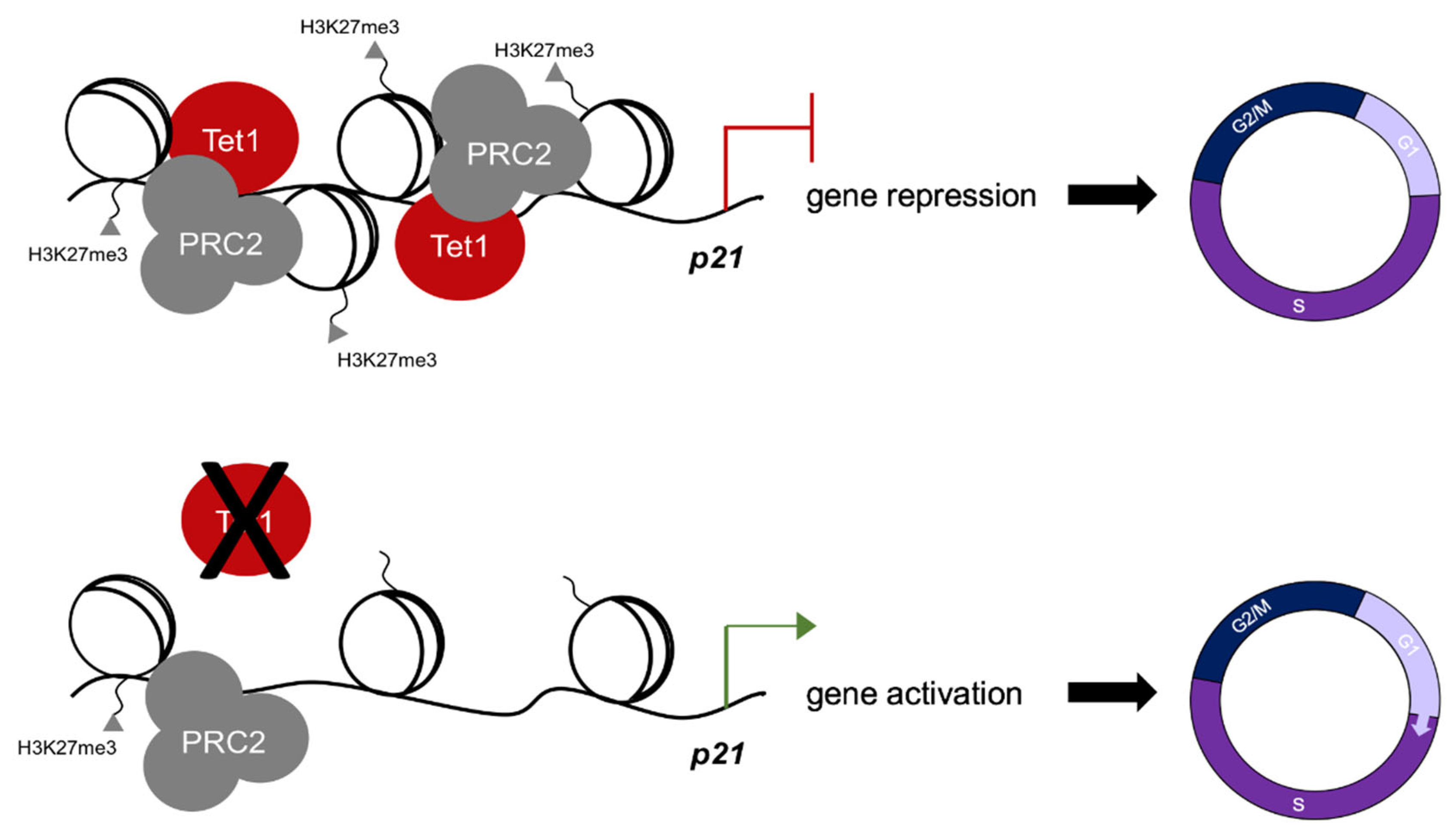
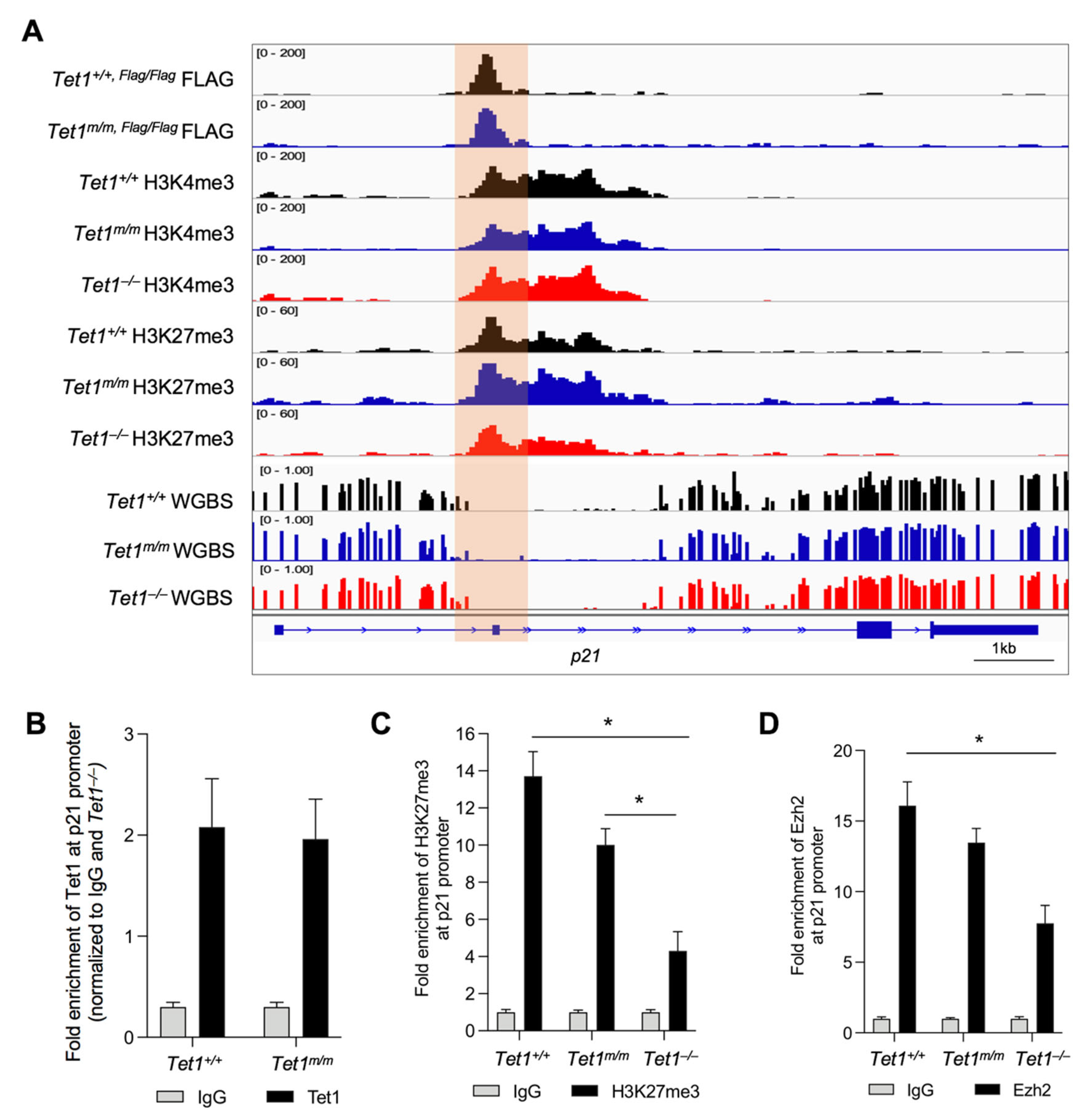
3.3. Tet1 Suppresses p21 Expression by Binding to Its Promoter and Facilitating PRC2 Recruitment and H3K27 Trimethylation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor, W.A.; Aravind, L.; Rao, A. TETonic shift: Biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; D’Alessio, A.C.; Ito, S.; Xia, K.; Wang, Z.; Cui, K.; Zhao, K.; Sun, Y.E.; Zhang, Y. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 2011, 473, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Christensen, J.; Pedersen, M.T.; Johansen, J.V.; Cloos, P.A.; Rappsilber, J.; Helin, K. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 2011, 473, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Chrysanthou, S.; Tang, Q.; Lee, J.; Taylor, S.J.; Zhao, Y.; Steidl, U.; Zheng, D.; Dawlaty, M.M. The DNA dioxygenase Tet1 regulates H3K27 modification and embryonic stem cell biology independent of its catalytic activity. Nucleic Acids Res. 2022, 50, 3169–3189. [Google Scholar] [CrossRef] [PubMed]
- Ficz, G.; Branco, M.R.; Seisenberger, S.; Santos, F.; Krueger, F.; Hore, T.A.; Marques, C.J.; Andrews, S.; Reik, W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 2011, 473, 398–402. [Google Scholar] [CrossRef]
- White, J.; Dalton, S. Cell cycle control of embryonic stem cells. Stem Cell Rev. 2005, 1, 131–138. [Google Scholar] [CrossRef]
- Ohtsuka, S.; Dalton, S. Molecular and biological properties of pluripotent embryonic stem cells. Gene Ther. 2008, 15, 74–81. [Google Scholar] [CrossRef]
- Li, V.C.; Ballabeni, A.; Kirschner, M.W. Gap 1 phase length and mouse embryonic stem cell self-renewal. Proc. Natl. Acad. Sci. USA 2012, 109, 12550–12555. [Google Scholar] [CrossRef] [Green Version]
- Koledova, Z.; Kramer, A.; Kafkova, L.R.; Divoky, V. Cell-cycle regulation in embryonic stem cells: Centrosomal decisions on self-renewal. Stem Cells Dev. 2010, 19, 1663–1678. [Google Scholar] [CrossRef] [PubMed]
- Itahana, Y.; Zhang, J.; Göke, J.; Vardy, L.A.; Han, R.; Iwamoto, K.; Cukuroglu, E.; Robson, P.; Pouladi, M.A.; Colman, A.; et al. Histone modifications and p53 binding poise the p21 promoter for activation in human embryonic stem cells. Sci. Rep. 2016, 6, 28112. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Tang, Q.; Gao, X.; Lee, J.; Lei, R.; Suzuki, M.; Zheng, D.; Ito, K.; Frenette, P.S.; Dawlaty, M.M. Tet-mediated DNA demethylation regulates specification of hematopoietic stem and progenitor cells during mammalian embryogenesis. Sci. Adv. 2022, 8, eabm3470. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Ganz, K.; Powell, B.E.; Hu, Y.C.; Markoulaki, S.; Cheng, A.W.; Gao, Q.; Kim, J.; Choi, S.W.; Page, D.C.; et al. Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 2011, 9, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawlaty, M.M.; Breiling, A.; Le, T.; Barrasa, M.I.; Raddatz, G.; Gao, Q.; Powell, B.E.; Cheng, A.W.; Faull, K.F.; Lyko, F.; et al. Loss of Tet enzymes compromises proper differentiation of embryonic stem cells. Dev. Cell 2014, 29, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Vella, P.; Scelfo, A.; Jammula, S.; Chiacchiera, F.; Williams, K.; Cuomo, A.; Roberto, A.; Christensen, J.; Bonaldi, T.; Helin, K.; et al. Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol. Cell 2013, 49, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Koh, K.P.; Yabuuchi, A.; Rao, S.; Huang, Y.; Cunniff, K.; Nardone, J.; Laiho, A.; Tahiliani, M.; Sommer, C.A.; Mostoslavsky, G.; et al. Tet1 and Tet2 Regulate 5-Hydroxymethylcytosine Production and Cell Lineage Specification in Mouse Embryonic Stem Cells. Cell Stem Cell 2011, 8, 200–213. [Google Scholar] [CrossRef] [Green Version]
- Rosello-Diez, A.; Madisen, L.; Bastide, S.; Zeng, H.; Joyner, A.L. Cell-nonautonomous local and systemic responses to cell arrest enable long-bone catch-up growth in developing mice. PLoS Biol. 2018, 16, e2005086. [Google Scholar] [CrossRef]
- Tie, G.; Yan, J.; Khair, L.; Tutto, A.; Messina, L.M. Hypercholesterolemia Accelerates the Aging Phenotypes of Hematopoietic Stem Cells by a Tet1-Dependent Pathway. Sci. Rep. Investig. 2020, 10, 3567. [Google Scholar] [CrossRef]
- Vuong, L.; Brobst, D.E.; Saadi, A.; Ivanovic, I.; Al-Ubaidi, M.R. Pattern of expression of p53, its family members, and regulators during early ocular development and in the post-mitotic retina. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4821–4831. [Google Scholar] [CrossRef]
- Medeiros, L.A.; Dennis, L.M.; Gill, M.E.; Houbaviy, H.; Markoulaki, S.; Fu, D.; White, A.C.; Kirak, O.; Sharp, P.A.; Page, D.C.; et al. Mir-290-295 deficiency in mice results in partially penetrant embryonic lethality and germ cell defects. Proc. Natl. Acad. Sci. USA 2011, 108, 14163–14168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome-wide mapping of in vivo protein-DNA interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasano, C.A.; Dimos, J.T.; Ivanova, N.B.; Lowry, N.; Lemischka, I.R.; Temple, S. shRNA knockdown of Bmi-1 reveals a critical role for p21-Rb pathway in NSC self-renewal during development. Cell Stem Cell 2007, 1, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Dolezalova, D.; Mraz, M.; Barta, T.; Plevova, K.; Vinarsky, V.; Holubcova, Z.; Jaros, J.; Dvorak, P.; Pospisilova, S.; Hampl, A. MicroRNAs regulate p21(Waf1/Cip1) protein expression and the DNA damage response in human embryonic stem cells. Stem Cells 2012, 30, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Baskerville, S.; Shenoy, A.; Babiarz, J.E.; Baehner, L.; Blelloch, R. Embryonic stem cell-specific microRNAs regulate the G1-S transition and promote rapid proliferation. Nat. Genet. 2008, 40, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Lichner, Z.; Pall, E.; Kerekes, A.; Pallinger, E.; Maraghechi, P.; Bosze, Z.; Gocza, E. The miR-290-295 cluster promotes pluripotency maintenance by regulating cell cycle phase distribution in mouse embryonic stem cells. Differentiation 2011, 81, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L.; Tyner, A.L. Transcriptional regulation of the p21((WAF1/CIP1)) gene. Exp. Cell Res. 1999, 246, 280–289. [Google Scholar] [CrossRef]
- Roccio, M.; Schmitter, D.; Knobloch, M.; Okawa, Y.; Sage, D.; Lutolf, M.P. Predicting stem cell fate changes by differential cell cycle progression patterns. Development 2013, 140, 459–470. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Takahashi, K.; Ichisaka, T.; Aoi, T.; Kanagawa, O.; Nakagawa, M.; Okita, K.; Yamanaka, S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 2009, 460, 1132–1135. [Google Scholar] [CrossRef]
- Hanna, J.; Saha, K.; Pando, B.; van Zon, J.; Lengner, C.J.; Creyghton, M.P.; van Oudenaarden, A.; Jaenisch, R. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 2009, 462, 595–601. [Google Scholar] [CrossRef]
- Costa, Y.; Ding, J.; Theunissen, T.W.; Faiola, F.; Hore, T.A.; Shliaha, P.V.; Fidalgo, M.; Saunders, A.; Lawrence, M.; Dietmann, S.; et al. NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 2013, 495, 370–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Guo, L.; Zhang, L.; Wu, H.; Yang, J.; Liu, H.; Wang, X.; Hu, X.; Gu, T.; Zhou, Z.; et al. Vitamin C modulates TET1 function during somatic cell reprogramming. Nat. Genet. 2013, 45, 1504–1509. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, J.; Li, K.; Wu, T.; Huang, B.; Liu, W.; Kou, X.; Zhang, Y.; Huang, H.; Jiang, Y.; et al. Replacement of Oct4 by Tet1 during iPSC induction reveals an important role of DNA methylation and hydroxymethylation in reprogramming. Cell Stem Cell 2013, 12, 453–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrysanthou, S.; Senner, C.E.; Woods, L.; Fineberg, E.; Okkenhaug, H.; Burge, S.; Perez-Garcia, V.; Hemberger, M. A Critical Role of TET1/2 Proteins in Cell-Cycle Progression of Trophoblast Stem Cells. Stem Cell Rep. 2018, 10, 1355–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Zhu, Z.; Wang, Y.; Wang, Y.; Xu, L.; Chen, X.; Xu, Q.; Zhang, Q.; Zhao, X.; Yu, Y.; et al. Tet1 is required for Rb phosphorylation during G1/S phase transition. Biochem. Biophys. Res. Commun. 2013, 434, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Filipczak, P.T.; Leng, S.; Tellez, C.S.; Do, K.C.; Grimes, M.J.; Thomas, C.L.; Walton-Filipczak, S.R.; Picchi, M.A.; Belinsky, S.A. p53-Suppressed Oncogene TET1 Prevents Cellular Aging in Lung Cancer. Cancer Res. 2019, 79, 1758–1768. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrysanthou, S.; Flores, J.C.; Dawlaty, M.M. Tet1 Suppresses p21 to Ensure Proper Cell Cycle Progression in Embryonic Stem Cells. Cells 2022, 11, 1366. https://doi.org/10.3390/cells11081366
Chrysanthou S, Flores JC, Dawlaty MM. Tet1 Suppresses p21 to Ensure Proper Cell Cycle Progression in Embryonic Stem Cells. Cells. 2022; 11(8):1366. https://doi.org/10.3390/cells11081366
Chicago/Turabian StyleChrysanthou, Stephanie, Julio C. Flores, and Meelad M. Dawlaty. 2022. "Tet1 Suppresses p21 to Ensure Proper Cell Cycle Progression in Embryonic Stem Cells" Cells 11, no. 8: 1366. https://doi.org/10.3390/cells11081366
APA StyleChrysanthou, S., Flores, J. C., & Dawlaty, M. M. (2022). Tet1 Suppresses p21 to Ensure Proper Cell Cycle Progression in Embryonic Stem Cells. Cells, 11(8), 1366. https://doi.org/10.3390/cells11081366