
Secretory Immunoglobulin A Immunity in Chronic Obstructive Respiratory Diseases



{kind=link}
{kind=link}
Abstract
1. Introduction
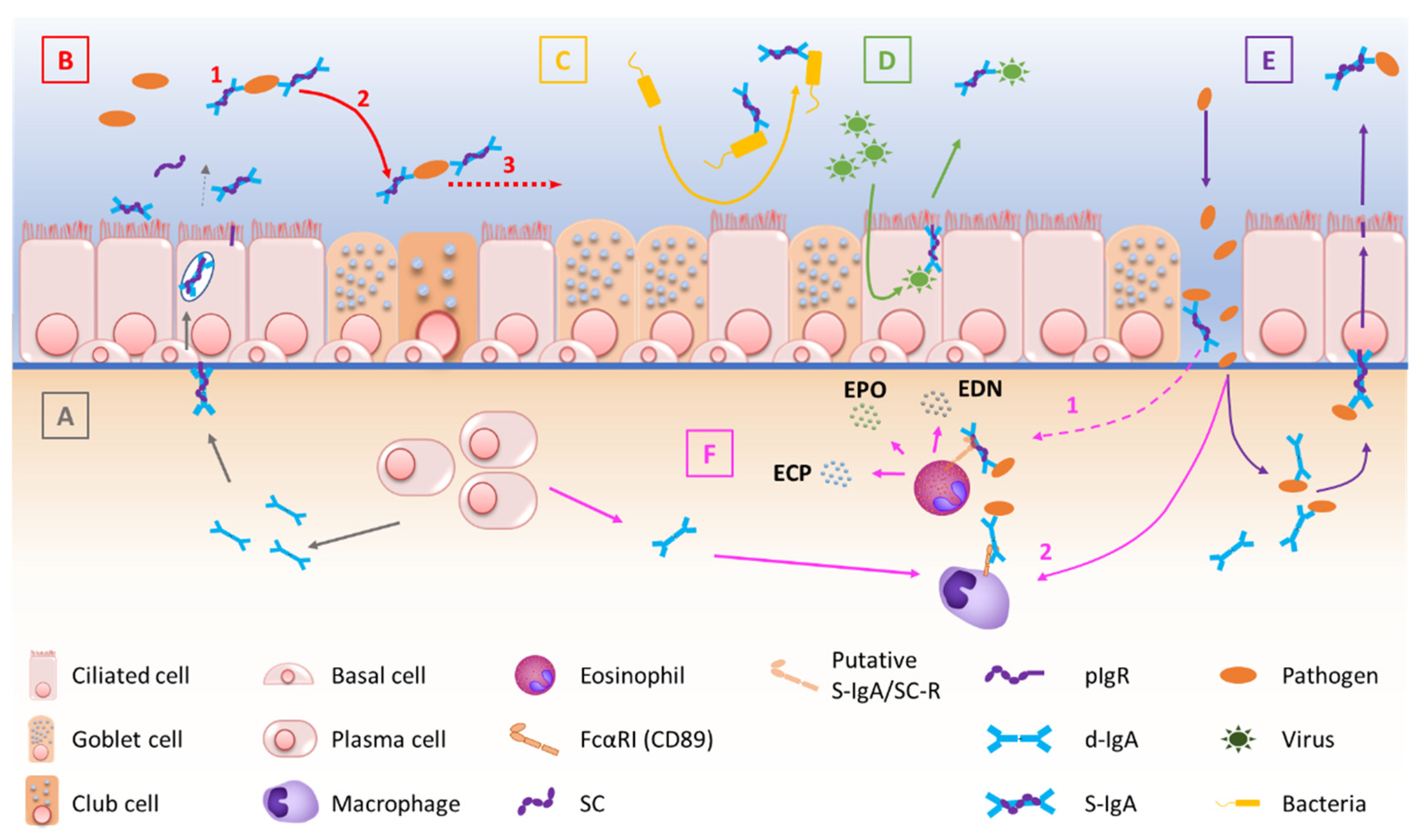
2. The Mucosal S-IgA System in Homeostasis
2.1. Production and Structure of S-IgA and pIgR
2.2. Transcytosis of d-IgA and Functions of S-IgA
2.3. Regulation of S-IgA Production
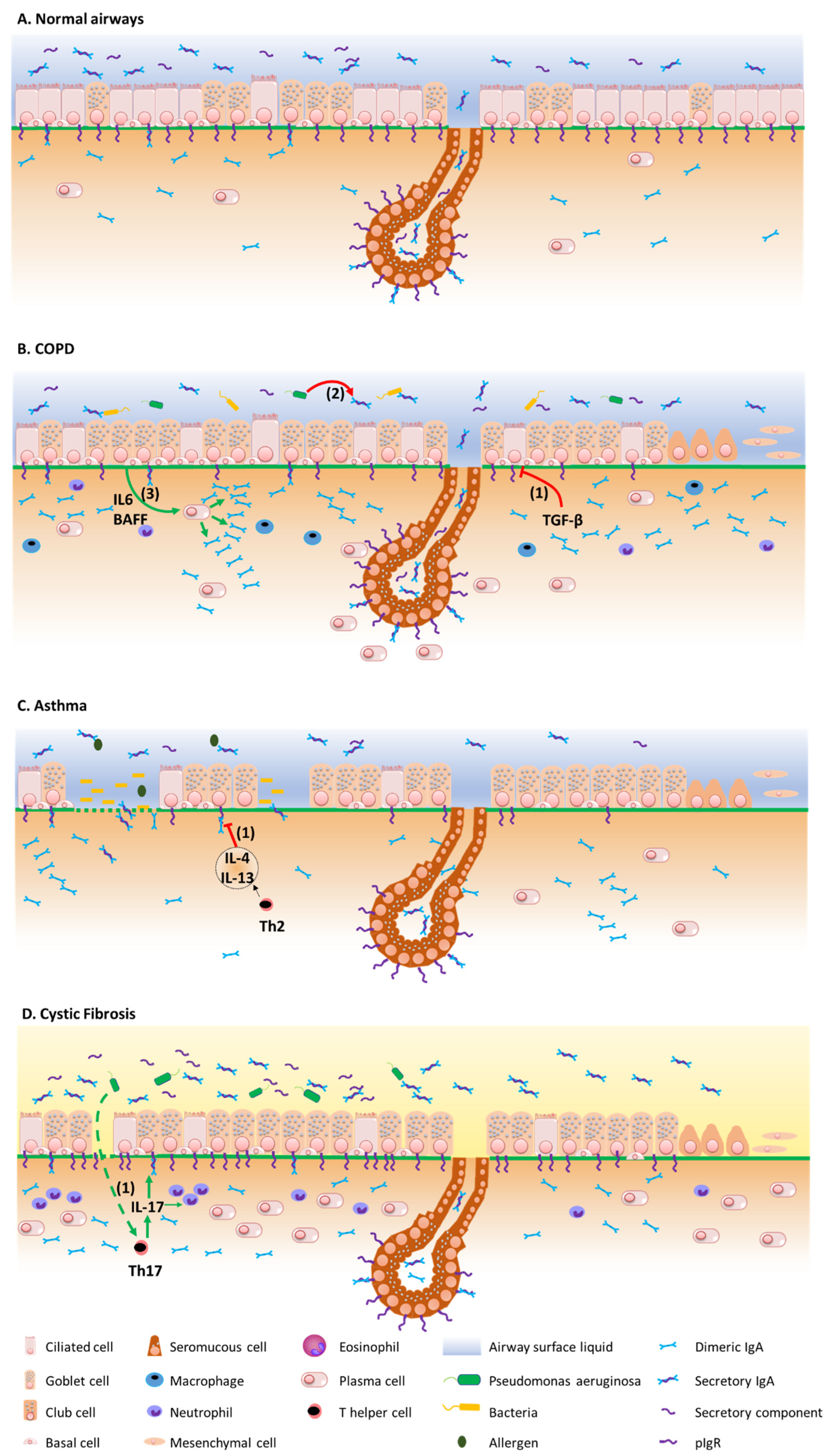
3. The Mucosal S-IgA System in Airway Disease
3.1. COPD
3.2. Asthma
3.3. Cystic Fibrosis
3.4. Bronchiolitis Obliterans Syndrome
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BAFF | B-cell activating factor |
| BALF | Bronchoalveolar lavage fluid |
| BOS | Bronchiolitis obliterans syndrome |
| CCL | C-C Motif Chemokine Ligand |
| CD | Cluster of differentiation |
| CF | Cystic Fibrosis |
| CLAD | Chronic lung allograft dysfunction |
| COPD | Chronic obstructive pulmonary disease |
| CS | Cigarette smoke |
| CXCL-8 | C-X-C motif chemokine ligand 8 |
| DC-SIGN | Dendritic cell specific ICAM-3 grabbing nonintegrin |
| d-IgA | Dimeric IgA |
| DLL4 | Delta-like canonical ligand 4 |
| ECP | Eosinophil cationic protein |
| EDN | Eosinophil-derived neurotoxin |
| EPO | Eosinophil peroxydase |
| Fab | Antigen-binding fragment |
| Fc | Antigen crystallizable fragment |
| FcαRI | Fc receptor for IgA |
| FEV | Forced expiratory volume |
| FVC | Forced vital capacity |
| IFN | Interferon |
| IFN-γ | Interferon γ |
| Ig | Immunoglobulin |
| IgA | Immunoglobulin A |
| IgG | Immunoglobulin G |
| IgM | Immunoglobulin M |
| IL-1 | Interleukin 1 |
| IL-4 | Interleukin 4 |
| IL-8 | Interleukin 8 |
| IRF-1 | IFN regulatory factor 1 |
| J chain | Joining chain |
| LT | Lung transplantation |
| MALT | Mucosa-associated lymphoid tissue |
| MAMP | Microbe-associated molecular pattern |
| MEF | Maximal expiratory flow |
| m-IgA | Monomeric IgA |
| NF-κB | Nuclear Factor κB |
| Pa | Pseudomonas aeruginosa |
| p-IgA | Polymeric IgA |
| pIgR | Polymeric immunoglobulin receptor |
| RAS | Restrictive allograft syndrome |
| RNA | Ribonucleic acid |
| SC | Secretory component |
| S-IgA | Secretory IgA |
| STAT6 | Signal transducer and activator of transcription 6 |
| TGF-β | Transforming growth factor β |
| TLR | Toll-like receptor |
| TNF-α | Tumour necrosis factor α |
| Treg | Regulatory T cell |
References
- Corthésy, B. Multi-Faceted Functions of Secretory IgA at Mucosal Surfaces. Front. Immunol. 2013, 4, 185. [Google Scholar] [CrossRef] [PubMed]
- Carlier, F.M.; Sibille, Y.; Pilette, C. The epithelial barrier and immunoglobulin A system in allergy. Clin. Exp. Allergy 2016, 46, 1372–1388. [Google Scholar] [CrossRef] [PubMed]
- Vos, T.; Lim, S.S.; Abbafati, C.; Abbas, K.M.; Abbasi, M.; Abbasifard, M.; Abbasi-Kangevari, M.; Abbastabar, H.; Abd-Allah, F.; Abdelalim, A. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Farrell, P.M. The prevalence of cystic fibrosis in the European Union. J. Cyst. Fibros. 2008, 7, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Mallia, P.; Contoli, M.; Caramori, G.; Pandit, A.; Johnston, S.; Papi, A. Exacerbations of Asthma and Chronic Obstructive Pulmonary Disease (COPD): Focus on Virus Induced Exacerbations. Curr. Pharm. Des. 2007, 13, 73–97. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.B.; Sullivan, S.D. The health economics of asthma and rhinitis. I. Assessing the economic impact. J. Allergy Clin. Immunol. 2001, 107, 3–8. [Google Scholar] [CrossRef]
- FitzGerald, J.M.; Barnes, P.J.; Chipps, B.E.; Jenkins, C.R.; O’Byrne, P.M.; Pavord, I.D.; Reddel, H.K. The burden of exacerbations in mild asthma: A systematic review. ERJ Open Res. 2020, 6, 00359–2019. [Google Scholar] [CrossRef]
- Viniol, C.; Vogelmeier, C.F. Exacerbations of COPD. Eur. Respir. Rev. 2018, 27, 170103. [Google Scholar] [CrossRef]
- Goss, C.H.; Burns, J.L. Exacerbations in cystic fibrosis. 1: Epidemiology and pathogenesis. Thorax 2007, 62, 360–367. [Google Scholar] [CrossRef]
- Sethi, S.; Murphy, T. Infection in the Pathogenesis and Course of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2008, 359, 2355–2365. [Google Scholar] [CrossRef]
- Putcha, N.; Paul, G.G.; Azar, A.; Wise, R.A.; O’Neal, W.K.; Dransfield, M.T.; Woodruff, P.G.; Curtis, J.L.; Comellas, A.P.; Drummond, M.B.; et al. Lower serum IgA is associated with COPD exacerbation risk in SPIROMICS. PLoS ONE 2018, 13, e0194924. [Google Scholar] [CrossRef] [PubMed]
- Burrows, P.D.; Cooper, M.D. IgA deficiency. Adv. Immunol. 1997, 65, 245–276. [Google Scholar] [PubMed]
- Yang, S.; Yu, M. Role of Goblet Cells in Intestinal Barrier and Mucosal Immunity. J. Inflamm. Res. 2021, 14, 3171–3183. [Google Scholar] [CrossRef] [PubMed]
- Boyaka, P.N.; Fujihashi, K. Host Defenses at Mucosal Surfaces. In Clinical Immunology, 5th ed.; Rich, R.R., Fleisher, T.A., Shearer, W.T., Schroeder, H.W., Frew, A.J., Weyand, C.M., Eds.; Elsevier: London, UK, 2019; pp. 285–298.e1. [Google Scholar]
- McNabb, P.C.; Tomasi, T.B. Host Defense Mechanisms at Mucosal Surfaces. Annu. Rev. Microbiol. 1981, 35, 477–496. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.D.; Wypych, T.P. Cellular and functional heterogeneity of the airway epithelium. Mucosal Immunol. 2021, 14, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Gohy, S.; Hupin, C.; Ladjemi, M.Z.; Hox, V.; Pilette, C. Key role of the epithelium in chronic upper airways diseases. Clin. Exp. Allergy 2019, 50, 135–146. [Google Scholar] [CrossRef]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020, 587, 619–625. [Google Scholar] [CrossRef]
- Knight, D.A.; Holgate, S.T. The airway epithelium: Structural and functional properties in health and disease. Respirology 2003, 8, 432–446. [Google Scholar] [CrossRef]
- Rock, J.R.; Randell, S.H.; Hogan, B.L.M. Airway basal stem cells: A perspective on their roles in epithelial homeostasis and remodeling. Dis. Model. Mech. 2010, 3, 545–556. [Google Scholar] [CrossRef]
- Hogan, B.L.; Barkauskas, C.E.; Chapman, H.A.; Epstein, J.A.; Jain, R.; Hsia, C.C.; Niklason, L.; Calle, E.; Le, A.; Randell, S.H. Repair and regeneration of the respiratory system: Complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 2014, 15, 123–138. [Google Scholar] [CrossRef]
- Boers, J.E.; Ambergen, A.W.; Thunnissen, F.B.J.M. Number and Proliferation of Basal and Parabasal Cells in Normal Human Airway Epithelium. Am. J. Respir. Crit. Care Med. 1998, 157, 2000–2006. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.-L.; Shenoy, S.A.; Li, S.; O’Beirne, S.L.; Strulovici-Barel, Y.; Leopold, P.L.; Wang, G.; Staudt, M.R.; Walters, M.S.; Mason, C.; et al. Ontogeny and Biology of Human Small Airway Epithelial Club Cells. Am. J. Respir. Crit. Care Med. 2018, 198, 1375–1388. [Google Scholar] [CrossRef]
- Hiemstra, P.S.; Bourdin, A. Club cells, CC10 and self-control at the epithelial surface. Eur. Respir. J. 2014, 44, 831–832. [Google Scholar] [CrossRef] [PubMed]
- Boers, J.E.; Brok, J.L.D.; Koudstaal, J.; Arends, J.W.; Thunnissen, F.B. Number and proliferation of neuroendocrine cells in normal human airway epithelium. Am. J. Respir. Crit. Care Med. 1996, 154, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Linnoila, R.I. Functional facets of the pulmonary neuroendocrine system. Lab. Investig. 2006, 86, 425–444. [Google Scholar] [CrossRef] [PubMed]
- Branchfield, K.; Nantie, L.; Verheyden, J.M.; Sui, P.; Wienhold, M.D.; Sun, X. Pulmonary neuroendocrine cells function as airway sensors to control lung immune response. Science 2016, 351, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef]
- He, W.-H.; Zhang, W.-D.; Cheng, C.-C.; Lu, J.; Liu, L.; Chen, Z.-H.; Wang, W.-H. Expression characteristics of polymeric immunoglobulin receptor in Bactrian camel (Camelus bactrianus) lungs. PLoS ONE 2022, 17, e0264815. [Google Scholar] [CrossRef]
- Blackburn, J.B.; Schaff, J.A.; Gutor, S.; Du, R.-H.; Nichols, D.; Sherrill, T.; Gutierrez, A.J.; Xin, M.K.; Wickersham, N.; Zhang, Y. Secretory cells are the primary source of pIgR in small airways. bioRxiv 2021. [Google Scholar] [CrossRef]
- Schiller, H.B.; Montoro, D.T.; Simon, L.M.; Rawlins, E.L.; Meyer, K.B.; Strunz, M.; Braga, F.A.V.; Timens, W.; Koppelman, G.H.; Budinger, G.R.S.; et al. The Human Lung Cell Atlas: A High-Resolution Reference Map of the Human Lung in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2019, 61, 31–41. [Google Scholar] [CrossRef]
- Woof, J.M.; Russell, M.W. Structure and function relationships in IgA. Mucosal Immunol. 2011, 4, 590–597. [Google Scholar] [CrossRef]
- Bakema, J.E.; van Egmond, M. Immunoglobulin A: A next generation of therapeutic antibodies? mAbs 2011, 3, 352–361. [Google Scholar] [CrossRef]
- Monteiro, R.C. The Role of IgA and IgA Fc Receptors as Anti-Inflammatory Agents. J. Clin. Immunol. 2010, 30, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Underdown, B.J.; Schiff, J.M. Immunoglobulin A: Strategic Defense Initiative at the Mucosal Surface. Annu. Rev. Immunol. 1986, 4, 389–417. [Google Scholar] [CrossRef]
- Li, Y.; Jin, L.; Chen, T. The Effects of Secretory IgA in the Mucosal Immune System. BioMed Res. Int. 2020, 2020, 2032057. [Google Scholar] [CrossRef] [PubMed]
- Pilette, C.; Ouadrhiri, Y.; Godding, V.; Vaerman, J.-P.; Sibille, Y. Lung mucosal immunity: Immunoglobulin-A revisited. Eur. Respir. J. 2001, 18, 571–588. [Google Scholar] [CrossRef] [PubMed]
- Conley, M.E.; Delacroix, D.L. Intravascular and Mucosal Immunoglobulin A: Two Separate but Related Systems of Immune Defense? Ann. Intern. Med. 1987, 106, 892. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.W.; Ding, J.L. The Unexplored Roles of Human Serum IgA. DNA Cell Biol. 2014, 33, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Burnett, D. Immunoglobulins in the lung. Thorax 1986, 41, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Delacroix, D.L.; Dive, C.; Rambaud, J.C.; Vaerman, J.P. IgA subclasses in various secretions and in serum. Immunology 1982, 47, 383–385. [Google Scholar]
- Reboldi, A.; Arnon, T.I.; Rodda, L.B.; Atakilit, A.; Sheppard, D.; Cyster, J.G. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science 2016, 352, aaf4822. [Google Scholar] [CrossRef] [PubMed]
- Stumbles, P.A.; Upham, J.W.; Holt, P.G. Airway dendritic cells: Co-ordinators of immunological homeostasis and immunity in the respiratory tract. APMIS 2003, 111, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Gloudemans, A.K.; Plantinga, M.; Guilliams, M.; Willart, M.A.; Ozir-Fazalalikhan, A.; Van Der Ham, A.; Boon, L.; Harris, N.L.; Hammad, H.; Hoogsteden, H.C.; et al. The Mucosal Adjuvant Cholera Toxin B Instructs Non-Mucosal Dendritic Cells to Promote IgA Production via Retinoic Acid and TGF-β. PLoS ONE 2013, 8, e59822. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, E.V. Multiple Curricula for B Cell Developmental Programming. Immunity 2016, 45, 457–458. [Google Scholar] [CrossRef] [PubMed]
- Sanz, I.; Wei, C.; Jenks, S.A.; Cashman, K.S.; Tipton, C.; Woodruff, M.C.; Hom, J.; Lee, F.E.-H. Challenges and Opportunities for Consistent Classification of Human B Cell and Plasma Cell Populations. Front. Immunol. 2019, 10, 2458. [Google Scholar] [CrossRef]
- Takhar, P.; Corrigan, C.; Smurthwaite, L.; O’Connor, B.J.; Durham, S.R.; Lee, T.; Gould, H.J. Class switch recombination to IgE in the bronchial mucosa of atopic and nonatopic patients with asthma. J. Allergy Clin. Immunol. 2007, 119, 213–218. [Google Scholar] [CrossRef]
- Xu, Z.; Zan, H.; Pone, E.J.; Mai, T.; Casali, P. Immunoglobulin class-switch DNA recombination: Induction, targeting and beyond. Nat. Rev. Immunol. 2012, 12, 517–531. [Google Scholar] [CrossRef]
- Pilette, C.; Nouri-Aria, K.T.; Jacobson, M.R.; Wilcock, L.K.; Detry, B.; Walker, S.M.; Francis, J.N.; Durham, S.R. Grass pollen immunotherapy induces an allergen-specific IgA2 antibody response associated with mucosal TGF-beta expression. J. Immunol. 2007, 178, 4658–4666. [Google Scholar] [CrossRef]
- Asano, M.; Komiyama, K. Polymeric immunoglobulin receptor. J. Oral Sci. 2011, 53, 147–156. [Google Scholar] [CrossRef]
- Turula, H.; Wobus, C.E. The Role of the Polymeric Immunoglobulin Receptor and Secretory Immunoglobulins during Mucosal Infection and Immunity. Viruses 2018, 10, 237. [Google Scholar] [CrossRef]
- Johansen, F.-E.; Kaetzel, C. Regulation of the polymeric immunoglobulin receptor and IgA transport: New advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol. 2011, 4, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Mantis, N.J.; Rol, N.; Corthésy, B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 2011, 4, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Bustamante-Marin, X.M.; Ostrowski, L.E. Cilia and Mucociliary Clearance. Cold Spring Harb. Perspect. Biol. 2017, 9, a028241. [Google Scholar] [CrossRef] [PubMed]
- de Sousa-Pereira, P.; Woof, J.M. IgA: Structure, Function, and Developability. Antibodies 2019, 8, 57. [Google Scholar] [CrossRef]
- Yan, H.; Lamm, M.E.; Björling, E.; Huang, Y.T. Multiple Functions of Immunoglobulin A in Mucosal Defense against Viruses: An In Vitro Measles Virus Model. J. Virol. 2002, 76, 10972–10979. [Google Scholar] [CrossRef]
- Mazanec, M.B.; Kaetzel, C.S.; Lamm, M.E.; Fletcher, D.; Nedrud, J.G. Intracellular neutralization of virus by immuno-globulin A antibodies. Proc. Natl. Acad. Sci. USA 1992, 89, 6901–6905. [Google Scholar] [CrossRef]
- Wright, A.; Yan, H.; Lamm, M.E.; Huang, Y.T. Immunoglobulin A antibodies against internal HIV-1 proteins neutralize HIV-1 replication inside epithelial cells. Virology 2006, 356, 165–170. [Google Scholar] [CrossRef]
- Chu, P.G.; Arber, D.A. CD79: A review. Applied immunohistochemistry & molecular morphology. AIMM 2001, 9, 97–106. [Google Scholar]
- Diana, J.; Moura, I.; Vaugier, C.; Gestin, A.; Tissandie, E.; Beaudoin, L.; Corthésy, B.; Hocini, H.; Lehuen, A.; Monteiro, R. Secretory IgA Induces Tolerogenic Dendritic Cells through SIGNR1 Dampening Autoimmunity in Mice. J. Immunol. 2013, 191, 2335–2343. [Google Scholar] [CrossRef]
- Lamkhioued, B.; Gounni, A.S.; Gruart, V.; Pierce, A.; Capron, A.; Capron, M. Human eosinophils express a receptor for secretory component. Role in secretory IgA-dependent activation. Eur. J. Immunol. 1995, 25, 117–125. [Google Scholar] [CrossRef]
- Roos, A.; Bouwman, L.H.; Van Gijlswijk-Janssen, D.J.; Faber-Krol, M.C.; Stahl, G.; Daha, M.R. Human IgA Activates the Complement System Via the Mannan-Binding Lectin Pathway. J. Immunol. 2001, 167, 2861–2868. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, R.C.; van de Winkel, J.G.J. IgA Fc Receptors. Annu. Rev. Immunol. 2003, 21, 177–204. [Google Scholar] [CrossRef] [PubMed]
- Pilette, C.; Ouadrhiri, Y.; Dimanche, F.; Vaerman, J.P.; Sibille, Y. Secretory component is cleaved by neutrophil serine proteinases but its epithelial production is increased by neutrophils through NF-kappa B- and p38 mitogen-activated protein kinase-dependent mechanisms. Am. J. Respir Cell Mol. Biol. 2003, 28, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, S.; Talay, S.R.; Brandtzaeg, P.; Chhatwal, G.S. SpsA, a novel pneumococcal surface protein with specific binding to secretory Immunoglobulin A and secretory component. Mol. Microbiol. 1997, 25, 1113–1124. [Google Scholar] [CrossRef]
- Mathias, A.; Corthésy, B. Recognition of Gram-positive Intestinal Bacteria by Hybridoma- and Colostrum-derived Secretory Immunoglobulin A Is Mediated by Carbohydrates. J. Biol. Chem. 2011, 286, 17239–17247. [Google Scholar] [CrossRef]
- Perrier, C.; Sprenger, N.; Corthésy, B. Glycans on Secretory Component Participate in Innate Protection against Mucosal Pathogens. J. Biol. Chem. 2006, 281, 14280–14287. [Google Scholar] [CrossRef]
- Gohy, S.T. Polymeric immunoglobulin receptor down-regulation in chronic obstructive pulmonary disease. Persistence in the cultured epithelium and role of transforming growth factor-beta. Am. J. Respir Crit. Care Med. 2014, 190, 509–521. [Google Scholar] [CrossRef]
- Loman, S.; Jansen, H.M.; Out, T.A.; Lutter, R. Interleukin-4 and interferon-gamma synergistically increase secretory component gene expression, but are additive in stimulating secretory immunoglobulin A release by Calu-3 airway epithelial cells. Immunology 1999, 96, 537. [Google Scholar] [CrossRef]
- Ladjemi, M.Z.; Gras, D.; Dupasquier, S.; Detry, B.; Lecocq, M.; Garulli, C.; Fregimilicka, C.; Bouzin, C.; Gohy, S.; Chanez, P. Bronchial Epithelial IgA Secretion Is Impaired in Asthma. Role of IL-4/IL-13. Am. J. Res-Piratory Crit. Care Med. 2018, 197, 1396–1409. [Google Scholar] [CrossRef]
- Ratajczak, C.; Guisset, A.; Detry, B.; Sibille, Y.; Pilette, C. Dual effect of neutrophils on pIgR/secretory component in human bronchial epithelial cells: Role of TGF-beta. J. Biomed. Biotechnol. 2010, 2010, 428618. [Google Scholar] [CrossRef]
- Collin, A.M.; Lecocq, M.; Noel, S.; Detry, B.; Carlier, F.M.; Nana, F.A.; Bouzin, C.; Leal, T.; Vermeersch, M.; De Rose, V.; et al. Lung immunoglobulin A immunity dysregulation in cystic fibrosis. eBioMedicine 2020, 60, 102947. [Google Scholar] [CrossRef] [PubMed]
- Gohy, S.; Moeremans, A.; Pilette, C.; Collin, A. Immunoglobulin A Mucosal Immunity and Altered Respiratory Epithelium in Cystic Fibrosis. Cells 2021, 10, 3603. [Google Scholar] [CrossRef]
- Carlier, F.M.; Detry, B.; Lecocq, M.; Collin, A.M.; Verleden, S.E.; Stanciu-Pop, C.M.; Janssens, W.; Ambroise, J.; Vanaudenaerde, B.M.; Gohy, S.T. The memory of airway epithelium damage in smokers and COPD patients. bioRxiv 2021. [Google Scholar] [CrossRef]
- Guo, M.-Y.; Chen, H.-K.; Ying, H.-Z.; Qiu, F.-S.; Wu, J.-Q. The Role of Respiratory Flora in the Pathogenesis of Chronic Respiratory Diseases. BioMed Res. Int. 2021, 2021, 6431862. [Google Scholar] [CrossRef]
- Sato, S.; Kiyono, H. The mucosal immune system of the respiratory tract. Curr. Opin. Virol. 2012, 2, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Carlier, F.M.; de Fays, C.; Pilette, C. Epithelial Barrier Dysfunction in Chronic Respiratory Diseases. Front. Physiol. 2021, 12, 691227. [Google Scholar] [CrossRef]
- Salvi, S.S.; Barnes, P.J. Chronic obstructive pulmonary disease in non-smokers. Lancet 2009, 374, 733–743. [Google Scholar] [CrossRef]
- Vogelmeier, C.F.; Criner, G.J.; Martinez, F.J.; Anzueto, A.; Barnes, P.J.; Bourbeau, J.; Celli, B.R.; Chen, R.; Decramer, M.; Fabbri, L.M.; et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report. GOLD Executive Summary. Am. J. Respir. Crit. Care Med. 2017, 195, 557–582. [Google Scholar] [CrossRef]
- Ohlmeier, S.; Mazur, W.; Linja-Aho, A.; Louhelainen, N.; Rönty, M.; Toljamo, T.; Bergmann, U.; Kinnula, V.L. Sputum Proteomics Identifies Elevated PIGR levels in Smokers and Mild-to-Moderate COPD. J. Proteome Res. 2011, 11, 599–608. [Google Scholar] [CrossRef]
- Pilette, C.; Godding, V.; Kiss, R.; Delos, M.; Verbeken, E.; Decaestecker, C.; De Paepe, K.; Vaerman, J.-P.; Decramer, M.; Sibille, Y. Reduced Epithelial Expression of Secretory Component in Small Airways Correlates with Airflow Obstruction in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2001, 163, 185–194. [Google Scholar] [CrossRef]
- Zuo, W.-L.; Rostami, M.R.; Shenoy, S.A.; LeBlanc, M.G.; Salit, J.; Strulovici-Barel, Y.; O’Beirne, S.L.; Kaner, R.J.; Leopold, P.L.; Mezey, J.G.; et al. Cell-specific expression of lung disease risk-related genes in the human small airway epithelium. Respir. Res. 2020, 21, 200. [Google Scholar] [CrossRef] [PubMed]
- Polosukhin, V.V.; Cates, J.M.; Lawson, W.E.; Zaynagetdinov, R.; Milstone, A.P.; Massion, P.P.; Ocak, S.; Ware, L.B.; Lee, J.W.; Bowler, R.P. Bronchial secretory immunoglobulin a deficiency correlates with airway inflammation and progression of chronic obstructive pulmonary disease. Am. J. Respir Crit. Care Med. 2011, 184, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Polosukhin, V.V.; Richmond, B.W.; Du, R.-H.; Cates, J.; Wu, P.; Nian, H.; Massion, P.P.; Ware, L.B.; Lee, J.W.; Kononov, A.; et al. Secretory IgA Deficiency in Individual Small Airways Is Associated with Persistent Inflammation and Remodeling. Am. J. Respir. Crit. Care Med. 2017, 195, 1010–1021. [Google Scholar] [CrossRef] [PubMed]
- Atiş, S.; Tutluoğlu, B.; Salepçi, B.; Ocal, Z. Serum IgA and secretory IgA levels in bronchial lavages from patients with a variety of respiratory diseases. J. Investig. Allergy Clin. Immunol. 2001, 11, 112–117. [Google Scholar]
- Du, R.-H.; Richmond, B.W.; Blackwell, T.S.; Cates, J.; Massion, P.P.; Ware, L.B.; Lee, J.W.; Kononov, A.; Lawson, W.E.; Polosukhin, V.V. Secretory IgA from submucosal glands does not compensate for its airway surface deficiency in chronic obstructive pulmonary disease. Virchows Arch. 2015, 467, 657–665. [Google Scholar] [CrossRef]
- Madissoon, E.; Oliver, A.J.; Kleshchevnikov, V.; Wilbrey-Clark, A.; Polanski, K.; Orsi, A.R.; Mamanova, L.; Bolt, L.; Richoz, N.; Elmentaite, R. A spatial multi-omics atlas of the human lung reveals a novel immune cell survival niche. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ladjemi, M.Z.; Lecocq, M.; Weynand, B.; Bowen, H.; Gould, H.J.; Van Snick, J.; Detry, B.; Pilette, C. Increased IgA production by B-cells in COPD via lung epithelial interleukin-6 and TACI pathways. Eur. Respir. J. 2015, 45, 980–993. [Google Scholar] [CrossRef]
- Murphy, T.F.; Kirkham, C.; Jones, M.M.; Sethi, S.; Kong, Y.; Pettigrew, M.M. Expression of IgA Proteases by Haemophilus influenzae in the Respiratory Tract of Adults With Chronic Obstructive Pulmonary Disease. J. Infect. Dis. 2015, 212, 1798–1805. [Google Scholar] [CrossRef]
- Murphy, T.F.; Kirkham, C.; Gallo, M.C.; Yang, Y.; Wilding, G.E.; Pettigrew, M.M. Immunoglobulin A Protease Variants Facilitate Intracellular Survival in Epithelial Cells By Nontypeable Haemophilus influenzae That Persist in the Human Respiratory Tract in Chronic Obstructive Pulmonary Disease. J. Infect. Dis. 2017, 216, 1295–1302. [Google Scholar] [CrossRef]
- Gallo, M.C.; Kirkham, C.; Eng, S.; Bebawee, R.S.; Kong, Y.; Pettigrew, M.M.; Tettelin, H.; Murphy, T.F. Changes in IgA Protease Expression Are Conferred by Changes in Genomes during Persistent Infection by Nontypeable Haemophilus influenzae in Chronic Obstructive Pulmonary Disease. Infect. Immun. 2018, 86, e00313–e00318. [Google Scholar] [CrossRef]
- Brandsma, C.-A.; Kerstjens, H.A.; Van Geffen, W.H.; Geerlings, M.; Postma, D.S.; Hylkema, M.N.; Timens, W. Differential switching to IgG and IgA in active smoking COPD patients and healthy controls. Eur. Respir. J. 2012, 40, 313–321. [Google Scholar] [CrossRef]
- Ladjemi, M.Z.; Martin, C.; Lecocq, M.; Detry, B.; Nana, F.A.; Moulin, C.; Weynand, B.; Fregimilicka, C.; Bouzin, C.; Thurion, P.; et al. Increased IgA Expression in Lung Lymphoid Follicles in Severe Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2019, 199, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Shimada, S.; Kawaguchi-Miyashita, M.; Kushiro, A.; Sato, T.; Nanno, M.; Sako, T.; Matsuoka, Y.; Sudo, K.; Tagawa, Y.-I.; Iwakura, Y.; et al. Generation of polymeric immunoglobulin receptor-deficient mouse with marked reduction of secretory IgA. J. Immunol. 1999, 163, 5367–5373. [Google Scholar] [PubMed]
- Tjärnlund, A.; Rodríguez, A.; Cardona, P.-J.; Guirado, E.; Ivanyi, J.; Singh, M.; Troye-Blomberg, M.; Fernández, C.; Duan, X.; Hisaeda, H.; et al. Polymeric IgR knockout mice are more susceptible to mycobacterial infections in the respiratory tract than wild-type mice. Int. Immunol. 2006, 18, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Richmond, B.W.; Brucker, R.M.; Han, W.; Du, R.-H.; Zhang, Y.; Cheng, D.-S.; Gleaves, L.; Abdolrasulnia, R.; Polosukhina, D.; Clark, P.E.; et al. Airway bacteria drive a progressive COPD-like phenotype in mice with polymeric immunoglobulin receptor deficiency. Nat. Commun. 2016, 7, 11240. [Google Scholar] [CrossRef]
- Kuruvilla, M.E.; Lee, F.E.-H.; Lee, G.B. Understanding Asthma Phenotypes, Endotypes, and Mechanisms of Disease. Clin. Rev. Allergy Immunol. 2019, 56, 219–233. [Google Scholar] [CrossRef]
- Wenzel, S.E. Asthma phenotypes: The evolution from clinical to molecular approaches. Nat. Med. 2012, 18, 716–725. [Google Scholar] [CrossRef]
- Alhamwe, B.A.; Miethe, S.; Von Strandmann, E.P.; Potaczek, D.P.; Garn, H. Epigenetic Regulation of Airway Epithelium Immune Functions in Asthma. Front. Immunol. 2020, 11, 1747. [Google Scholar] [CrossRef]
- Peters, U.; Dixon, A.E.; Forno, E. Obesity and asthma. J. Allergy Clin. Immunol. 2018, 141, 1169–1179. [Google Scholar] [CrossRef]
- Potaczek, D.P.; Miethe, S.; Schindler, V.; Alhamdan, F.; Garn, H. Role of airway epithelial cells in the development of different asthma phenotypes. Cell. Signal. 2020, 69, 109523. [Google Scholar] [CrossRef]
- Frey, A.; Lunding, L.P.; Ehlers, J.C.; Weckmann, M.; Zissler, U.M.; Wegmann, M. More than Just a Barrier: The Immune Functions of the Airway Epithelium in Asthma Pathogenesis. Front. Immunol. 2020, 11, 761. [Google Scholar] [CrossRef] [PubMed]
- Nahm, D.-H.; Kim, H.-Y.; Park, H.-S. Elevation of specific immunoglobulin A antibodies to both allergen and bacterial antigen in induced sputum from asthmatics. Eur. Respir. J. 1998, 12, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Peebles, R.S.; Hamilton, R.G.; Lichtenstein, L.M.; Schlosberg, M.; Liu, M.C.; Proud, D.; Togias, A. Antigen-specific IgE and IgA antibodies in bronchoalveolar lavage fluid are associated with stronger antigen-induced late phase reactions. Clin. Exp. Allergy 2001, 31, 239–248. [Google Scholar] [CrossRef]
- Xu, W.; Santini, P.A.; Matthews, A.J.; Chiu, A.; Plebani, A.; He, B.; Chen, K.; Cerutti, A. Viral Double-Stranded RNA Triggers Ig Class Switching by Activating Upper Respiratory Mucosa B Cells through an Innate TLR3 Pathway Involving BAFF. J. Immunol. 2008, 181, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Stavnezer, J.; Guikema, J.E.; Schrader, C.E. Mechanism and Regulation of Class Switch Recombination. Annu. Rev. Immunol. 2008, 26, 261–292. [Google Scholar] [CrossRef] [PubMed]
- Ruane, D.; Chorny, A.; Lee, H.; Faith, J.J.; Pandey, G.; Shan, M.; Simchoni, N.; Rahman, A.; Garg, A.; Weinstein, E.G.; et al. Microbiota regulate the ability of lung dendritic cells to induce IgA class-switch recombination and generate protective gastrointestinal immune responses. J. Exp. Med. 2015, 213, 53–73. [Google Scholar] [CrossRef] [PubMed]
- Orivuori, L.; Loss, G.; Roduit, C.; Dalphin, J.C.; Depner, M.; Genuneit, J.; Lauener, R.; Pekkanen, J.; Pfefferle, P.; Riedler, J. Soluble immunoglobulin A in breast milk is inversely associated with atopic dermatitis at early age: The PASTURE cohort study. Clin. Exp. Allergy 2014, 44, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, F.M.; Monteiro, R.C.; Volanakis, J.E.; Cooper, M.D. IgA deficiency. Immunodefic. Rev. 1991, 3, 15–44. [Google Scholar]
- Kim, W.-J.; Choi, I.S.; Kim, C.S.; Lee, J.-H.; Kang, H.-W. Relationship between serum IgA level and allergy/asthma. Korean J. Intern. Med. 2017, 32, 137–145. [Google Scholar] [CrossRef]
- Orivuori, L.; Mustonen, K.; Roduit, C.; Braun-Fahrländer, C.; Dalphin, J.-C.; Genuneit, J.; Lauener, R.; Pfefferle, P.; Riedler, J.; Weber, J.; et al. Immunoglobulin A and immunoglobulin G antibodies against β-lactoglobulin and gliadin at age 1 associate with immunoglobulin E sensitization at age 6. Pediatric Allergy Immunol. 2014, 25, 329–337. [Google Scholar] [CrossRef]
- Suzuki, K.; Ha, S.-A.; Tsuji, M.; Fagarasan, S. Intestinal IgA synthesis: A primitive form of adaptive immunity that regulates microbial communities in the gut. Semin. Immunol. 2007, 19, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.L.; Gold, M.J.; Hartmann, M.; Willing, B.P.; Thorson, L.; Wlodarska, M.; Gill, N.; Blanchet, M.-R.; Mohn, W.W.; McNagny, K.M.; et al. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 2012, 13, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Noverr, M.C.; Falkowski, N.R.; McDonald, R.A.; McKenzie, A.N.; Huffnagle, G.B. Development of allergic airway disease in mice following antibiotic therapy and fungal microbiota increase: Role of host genetics, antigen, and interleukin-13. Infect. Immun. 2005, 73, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Mkaddem, S.B.; Christou, I.; Rossato, E.; Berthelot, L.; Lehuen, A.; Monteiro, R.C. IgA, IgA Receptors, and Their Anti-inflammatory Properties. In Fc Receptors; Daeron, M., Nimmerjahn, F., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 221–235. [Google Scholar]
- Shen, C.; Detry, B.; Lecocq, M.; Pilette, C. A novel IgA/Delta-like 4/Notch axis induces immunosuppressive activity in human dendritic cells. Clin. Immunol. 2016, 168, 37–46. [Google Scholar] [CrossRef]
- Wang, W.; Wei, C.; Cheng, Z.; Yang, J. Aberrant Th2 Immune Responses Are Associated With a Reduced Frequency of IL-35-Induced Regulatory T Cells After Allergen Exposure in Patients With Allergic Asthma. Allergy Asthma Immunol. Res. 2020, 12, 1029–1045. [Google Scholar] [CrossRef]
- Lee, H.-C.; Headley, M.; Loo, Y.-M.; Berlin, A.; Gale, M.; Debley, J.S.; Lukacs, N.W.; Ziegler, S.F. Thymic stromal lymphopoietin is induced by respiratory syncytial virus–infected airway epithelial cells and promotes a type 2 response to infection. J. Allergy Clin. Immunol. 2012, 130, 1187–1196.e5. [Google Scholar] [CrossRef]
- Lei, L.; Zhang, Y.; Yao, W.; Kaplan, M.H.; Zhou, B. Thymic Stromal Lymphopoietin Interferes with Airway Tolerance by Suppressing the Generation of Antigen-Specific Regulatory T Cells. J. Immunol. 2011, 186, 2254–2261. [Google Scholar] [CrossRef]
- Nguyen, K.D.; Vanichsarn, C.; Nadeau, K.C. TSLP directly impairs pulmonary Treg function: Association with aberrant tolerogenic immunity in asthmatic airway. Allergy, Asthma Clin. Immunol. 2010, 6, 4. [Google Scholar] [CrossRef]
- van Heerden, D.; van Binnendijk, R.S.; Tromp, S.A.M.; Savelkoul, H.F.J.; van Neerven, R.J.J.; den Hartog, G. Asth-ma-Associated Long TSLP Inhibits the Production of IgA. Int. J. Mol. Sci. 2021, 22, 3592. [Google Scholar] [CrossRef]
- McBrien, C.N.; Menzies-Gow, A. The Biology of Eosinophils and Their Role in Asthma. Front. Med. 2017, 4, 93. [Google Scholar] [CrossRef]
- Motegi, Y.; Kita, H.; Kato, M.; Morikawa, A. Role of secretory IgA, secretory component, and eosinophils in mucosal inflammation. Int. Arch. Allergy Immunol. 2000, 122, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Nakagome, K.; Nagata, M. Involvement and Possible Role of Eosinophils in Asthma Exacerbation. Front. Immunol. 2018, 9, 2220. [Google Scholar] [CrossRef] [PubMed]
- Bartemes, K.R.; Cooper, K.M.; Drain, K.L.; Kita, H. Secretory IgA induces antigen-independent eosinophil survival and cytokine production without inducing effector functions. J. Allergy Clin. Immunol. 2005, 116, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Van Epps, D.E.; Williams, R.C. Suppression of leukocyte chemotaxis by human IgA myeloma components. J. Exp. Med. 1976, 144, 1227–1242. [Google Scholar] [CrossRef] [PubMed]
- van Egmond, M.; Damen, C.A.; van Spriel, A.B.; Vidarsson, G.; van Garderen, E.; van de Winkel, J.G.J. IgA and the IgA Fc receptor. Trends Immunol. 2001, 22, 205–211. [Google Scholar] [CrossRef]
- Hupin, C.; Rombaux, P.; Bowen, H.; Gould, H.; Lecocq, M.; Pilette, C. Downregulation of polymeric immunoglobulin receptor and secretory IgA antibodies in eosinophilic upper airway diseases. Allergy 2013, 68, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Bell, S.C.; Rowe, S.M.; Goss, C.H.; Quittner, A.L.; Bush, A. Cystic fibrosis. Nat. Rev. Dis. Prim. 2015, 1, 15010. [Google Scholar] [CrossRef]
- Collin, A.M.; Lecocq, M.; Detry, B.; Carlier, F.M.; Bouzin, C.; de Sany, P.; Hoton, D.; Verleden, S.; Froidure, A.; Pilette, C.; et al. Loss of ciliated cells and altered airway epithelial integrity in cystic fibrosis. J. Cyst. Fibros. 2021, 20, e129–e139. [Google Scholar] [CrossRef]
- Marshall, L.J.; Perks, B.; Bodey, K.; Suri, R.; Bush, A.; Shute, J.K. Free Secretory Component from Cystic Fibrosis Sputa Displays the Cystic Fibrosis Glycosylation Phenotype. Am. J. Respir. Crit. Care Med. 2004, 169, 399–406. [Google Scholar] [CrossRef]
- Hodson, M.E.; Morris, L.; Batten, J.C. Serum immunoglobulins and immunoglobulin G subclasses in cystic fibrosis related to the clinical state of the patient. Eur. Respir. J. 1988, 1, 701–705. [Google Scholar]
- Hassan, J.; Feighery, C.; Bresnihan, B.; Keogan, M.; Fitzgerald, M.X.; Whelan, A. Serum IgA and IGg Subclasses during Treatment for Acute Respiratory Exacerbation in Cystic Fibrosis: Analysis of Patients Colonised with Mucoid or Non-Mucoid Strains of Pseudomonas Aeruginosa. Immunol. Investig. 1994, 23, 1–13. [Google Scholar] [CrossRef]
- Van Bever, H.P.; Gigase, P.L.; De Clerck, L.S.; Bridts, C.H.; Franckx, H.; Stevens, W.J. Immune complexes and Pseudomonas aeruginosa antibodies in cystic fibrosis. Arch. Dis. Child. 1988, 63, 1222–1228. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Konstan, M.W.; Hilliard, K.A.; Norvell, T.M.; Berger, M. Bronchoalveolar lavage findings in cystic fibrosis patients with stable, clinically mild lung disease suggest ongoing infection and inflammation. Am. J. Respir Crit. Care Med. 1994, 150, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; McGarry, D.P.; Joseph, N.; Peppers, B.; Hostoffer, R. Salivary IgA deficiency in a patient with cystic fibrosis (genotype M470V/V520F). Ann. Allergy, Asthma Immunol. 2018, 121, 619–620. [Google Scholar] [CrossRef]
- Verleden, G.M.; Raghu, G.; Meyer, K.C.; Glanville, A.R.; Corris, P. A new classification system for chronic lung allograft dysfunction. J. Hear. Lung Transplant 2014, 33, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Verleden, G.M.; Glanville, A.R.; Lease, E.D.; Fisher, A.J.; Calabrese, F.; Corris, P.A.; Ensor, C.R.; Gottlieb, J.; Hachem, R.R.; Lama, V. Chronic lung allograft dysfunction: Definition, diagnostic criteria, and approaches to treatment-A consensus report from the Pulmonary Council of the ISHLT. J. Heart Lung Transplant 2019, 38, 493–503. [Google Scholar] [CrossRef]
- Pedersen, S.S.; Espersen, F.; Hoiby, N.; Jensen, T. Immunoglobulin A and immunoglobulin G antibody responses to alginates from Pseudomonas aeruginosa in patients with cystic fibrosis. J. Clin. Microbiol. 1990, 28, 747–755. [Google Scholar] [CrossRef]
- Kronborg, G.; Fomsgaard, A.; Galanos, C.; Freudenberg, M.A.; Hoiby, N. Antibody responses to lipid A, core, and O sugars of the Pseudomonas aeruginosa lipopolysaccharide in chronically infected cystic fibrosis patients. J. Clin. Microbiol. 1992, 30, 1848–1855. [Google Scholar] [CrossRef]
- Aanaes, K.; Johansen, H.K.; Poulsen, S.S.; Pressler, T.; Buchwald, C.; Høiby, N. Secretory IgA as a diagnostic tool for Pseudomonas aeruginosa respiratory colonization. J. Cyst. Fibros. 2012, 12, 81–87. [Google Scholar] [CrossRef]
- Hallberg, K.; Mattsson-Rydberg, A.; Fändriks, L.; Strandvik, B. Gastric IgA in Cystic Fibrosis in Relation to the Migrating Motor Complex. Scand. J. Gastroenterol. 2001, 36, 843–848. [Google Scholar] [CrossRef]
- Wallwork, J.C.; McFARLANE, H. The SIgA system and hypersensitivity in patients with cystic fibrosis. Clin. Exp. Allergy 1976, 6, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Martinu, T.; Howell, D.N.; Davis, R.D.; Steele, M.P.; Palmer, S.M. Pathologic correlates of bronchiolitis obliterans syn-drome in pulmonary retransplant recipients. Chest 2006, 129, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Le Pavec, J.; Pradère, P.; Gigandon, A.; Dauriat, G.; Dureault, A.; Aguilar, C.; Henry, B.; Lanternier, F.; Savale, L.; Dolidon, S.; et al. Risk of Lung Allograft Dysfunction Associated With Aspergillus Infection. Transplant. Direct 2021, 7, e675. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.E.; Preiksaitis, C.M.; Lease, E.D.; Edelman, J.; Kirby, K.A.; Leisenring, W.M.; Raghu, G.; Boeckh, M.; Limaye, A.P. Symptomatic Respiratory Virus Infection and Chronic Lung Allograft Dysfunction. Clin. Infect. Dis. 2015, 62, 313–319. [Google Scholar] [CrossRef]
- Gregson, A.L. Infectious Triggers of Chronic Lung Allograft Dysfunction. Curr. Infect. Dis. Rep. 2016, 18, 21. [Google Scholar] [CrossRef]
- Bastian, A.; Tunkel, C.; Lins, M.; Bottcher, H.; Hirt, S.W.; Cremer, J.; Bewig, B. Immunoglobulin A and secretory immunoglobulin A in the bronchoalveolar lavage from patients after lung transplantation. Clin. Transplant. 2000, 14, 580–585. [Google Scholar] [CrossRef]
- Vandermeulen, E.; Verleden, S.E.; Bellon, H.; Ruttens, D.; Lammertyn, E.; Claes, S.; Vandooren, J.; Ugarte-Berzal, E.; Schols, D.; Emonds, M.-P.; et al. Humoral immunity in phenotypes of chronic lung allograft dysfunction: A broncho-alveolar lavage fluid analysis. Transpl. Immunol. 2016, 38, 27–32. [Google Scholar] [CrossRef]
- Murthy, S.C.; Avery, R.K.; Budev, M.; Gupta, S.; Pettersson, G.B.; Nowicki, E.R.; Mehta, A.; Chapman, J.T.; Rajeswaran, J.; Blackstone, E.H. Low pretransplant IgA level is associated with early post–lung transplant seromucous infection. J. Thorac. Cardiovasc. Surg. 2018, 156, 882–891.e8. [Google Scholar] [CrossRef]
- Chambers, D.C.; Davies, B.; Mathews, A.; Yerkovich, S.T.; Hopkins, P.M. Bronchiolitis obliterans syndrome, hypogammaglobulinemia, and infectious complications of lung transplantation. J. Hear. Lung Transplant. 2013, 32, 36–43. [Google Scholar] [CrossRef]
- Sun, L.K.; Fung, M.S.; Sun, W.N.; Sun, C.R.; Chang, W.-I.; Chang, T.W. Human IgA Monoclonal Antibodies Specific for a Major Ragweed Pollen Antigen. Bio/Technology 1995, 13, 779–786. [Google Scholar] [CrossRef]
- Kamenov, B.; Kamenov, S.; Kamenov, A.; Stamenkovic, H.; Stankovic, T. Clinical manifestations of selective IgA immu-nodeficiency in childhood. Eur. Respir. J. 2013, 42 (Suppl. 57), P1220. [Google Scholar]
- Singh, K.; Chang, C.; Gershwin, M.E. IgA deficiency and autoimmunity. Autoimmun. Rev. 2013, 13, 163–177. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Fays, C.; Carlier, F.M.; Gohy, S.; Pilette, C. Secretory Immunoglobulin A Immunity in Chronic Obstructive Respiratory Diseases. Cells 2022, 11, 1324. https://doi.org/10.3390/cells11081324
de Fays C, Carlier FM, Gohy S, Pilette C. Secretory Immunoglobulin A Immunity in Chronic Obstructive Respiratory Diseases. Cells. 2022; 11(8):1324. https://doi.org/10.3390/cells11081324
Chicago/Turabian Stylede Fays, Charlotte, François M. Carlier, Sophie Gohy, and Charles Pilette. 2022. "Secretory Immunoglobulin A Immunity in Chronic Obstructive Respiratory Diseases" Cells 11, no. 8: 1324. https://doi.org/10.3390/cells11081324
APA Stylede Fays, C., Carlier, F. M., Gohy, S., & Pilette, C. (2022). Secretory Immunoglobulin A Immunity in Chronic Obstructive Respiratory Diseases. Cells, 11(8), 1324. https://doi.org/10.3390/cells11081324