
Role of Respiratory Epithelial Cells in Allergic Diseases

 ,
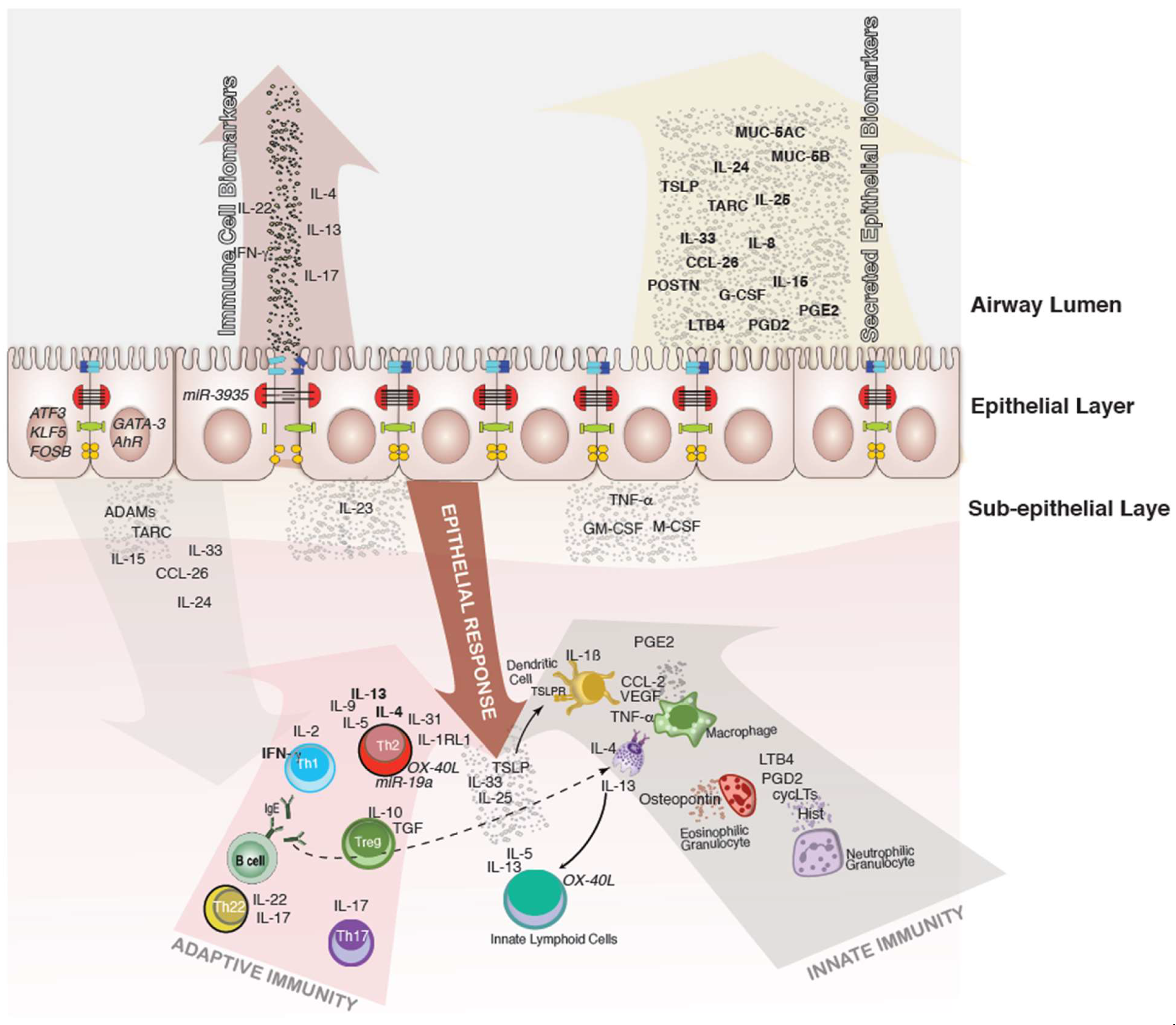
, 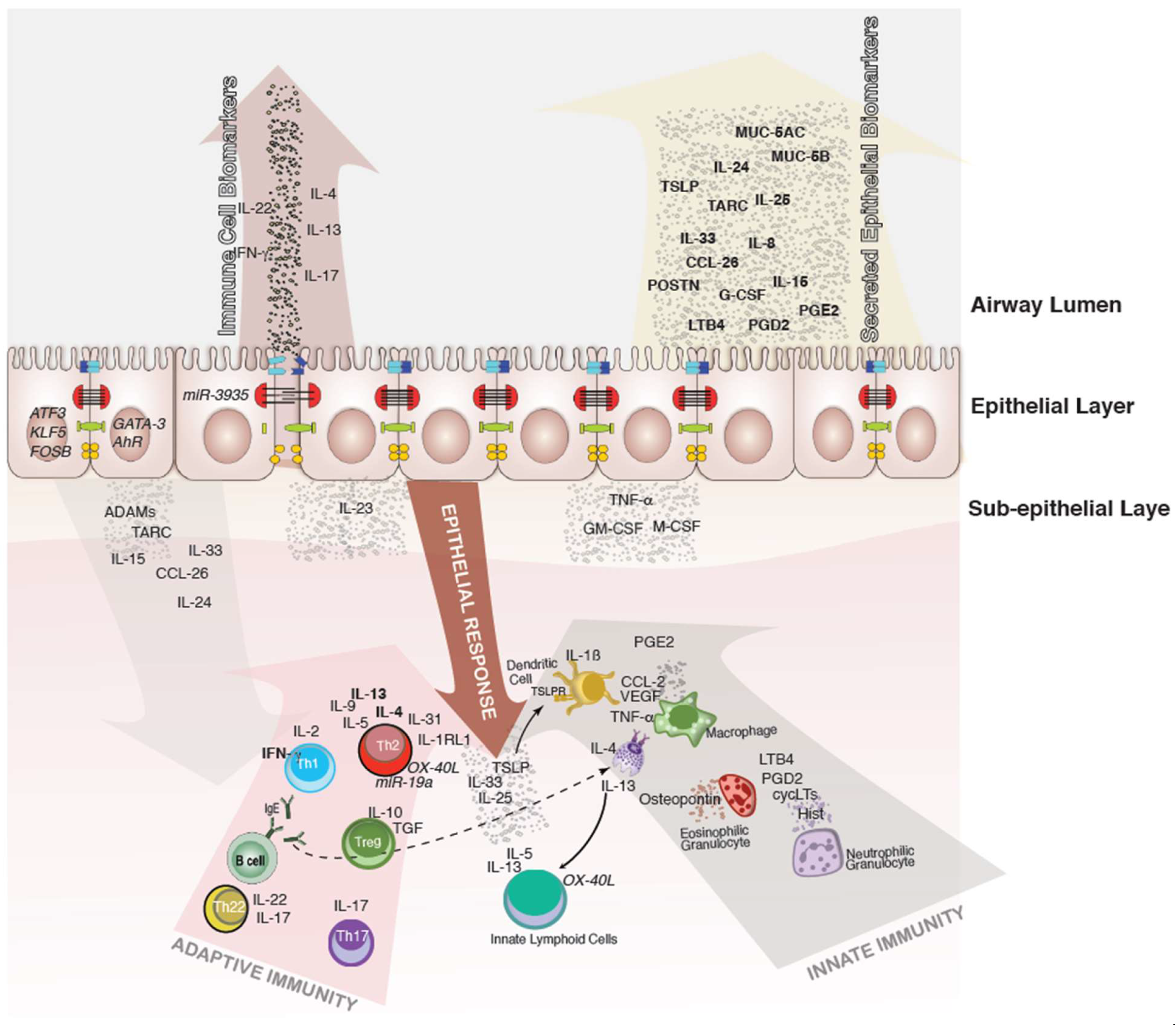{kind=link}
Abstract
:1. Introduction
2. Immune Cell Interactions with the Epithelial Surface
2.1. Overview
2.2. T Cells
2.3. Th17 Cells
2.4. Macrophages
2.5. Dendritic Cells
2.6. Mast Cells
3. Role of microRNAs in Epithelial Immunity
4. Epithelial Cytokines in Allergic Diseases
The Concept of Epithelial Polarization
5. Broader Distribution of Inflammatory Memory in Epithelial Barrier Tissues
6. Treatment Options for Inflammatory Status of Airway Epithelial Cells
7. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, D.Y.; Li, Y.; Yan, Y.; Li, C.; Shi, L. Upper airway stem cells: Understanding the nose and role for future cell therapy. Curr. Allergy Asthma Rep. 2015, 15, 490. [Google Scholar] [CrossRef]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl Acad Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [Green Version]
- Hiemstra, P.S.; McCray, P.B., Jr.; Bals, R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur. Respir. J. 2015, 45, 1150–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Tang, Z.; Huang, H.; Li, J.; Wang, Z.; Yu, Y.; Zhang, C.; Li, J.; Dai, H.; Wang, F.; et al. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc. Natl. Acad. Sci. USA 2018, 115, 2407–2412. [Google Scholar] [CrossRef] [Green Version]
- Madan, T.; Biswas, B.; Varghese, P.M.; Subedi, R.; Pandit, H.; Idicula-Thomas, S.; Kundu, I.; Rooge, S.; Agarwal, R.; Tripathi, D.M.; et al. A Recombinant Fragment of Human Surfactant Protein D Binds Spike Protein and Inhibits Infectivity and Replication of SARS-CoV-2 in Clinical Samples. Am. J. Respir. Cell Mol. Biol. 2021, 65, 41–53. [Google Scholar] [CrossRef]
- Luecken, M.D.; Zaragosi, L.E.; Madissoon, E.; Sikkema, L.; Firsova, A.B.; De Domenico, E.; Kummerle, L.; Saglam, A.; Berg, M.; Gay, A.C.A.; et al. The discovAIR project: A roadmap towards the Human Lung Cell Atlas. Eur. Respir. J. 2022, 59, 2102057. [Google Scholar] [CrossRef]
- Dean, C.H.; Snelgrove, R.J. New Rules for Club Development: New Insights into Human Small Airway Epithelial Club Cell Ontogeny and Function. Am. J. Respir. Crit. Care Med. 2018, 198, 1355–1356. [Google Scholar] [CrossRef]
- Tata, P.R.; Mou, H.; Pardo-Saganta, A.; Zhao, R.; Prabhu, M.; Law, B.M.; Vinarsky, V.; Cho, J.L.; Breton, S.; Sahay, A.; et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature 2013, 503, 218–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Q.; Yao, C.; Qi, X.; Stripp, B.R.; Tang, N. STK11 is required for the normal program of ciliated cell differentiation in airways. Cell Discov. 2019, 5, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.S.; Wells, J.M.; Zorn, A.M.; Wert, S.E.; Laubach, V.E.; Fernandez, L.G.; Whitsett, J.A. Transdifferentiation of ciliated cells during repair of the Respir.atory epithelium. Am. J. Respir. Cell Mol. Biol. 2006, 34, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Kondo, M.; Tamaoki, J.; Takeyama, K.; Nakata, J.; Nagai, A. Interleukin-13 induces goblet cell differentiation in primary cell culture from Guinea pig tracheal epithelium. Am. J. Respir. Cell Mol. Biol. 2002, 27, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Shamji, M.H.; Valenta, R.; Jardetzky, T.; Verhasselt, V.; Durham, S.R.; Wurtzen, P.A.; van Neerven, R.J.J. The role of allergen-specific IgE, IgG and IgA in allergic disease. Allergy 2021, 76, 3627–3641. [Google Scholar] [CrossRef] [PubMed]
- Heijink, I.H.; Kuchibhotla, V.N.S.; Roffel, M.P.; Maes, T.; Knight, D.A.; Sayers, I.; Nawijn, M.C. Epithelial cell dysfunction, a major driver of asthma development. Allergy 2020, 75, 1902–1917. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, O.; Bahmer, T.; Weckmann, M.; Dittrich, A.M.; Schaub, B.; Rosler, B.; Happle, C.; Brinkmann, F.; Ricklefs, I.; Konig, I.R.; et al. The all age asthma cohort (ALLIANCE)-from early beginnings to chronic disease: A longitudinal cohort study. BMC Pulm. Med. 2018, 18, 140. [Google Scholar] [CrossRef]
- Weckmann, M.; Bahmer, T.; Sand, J.M.; Rank Ronnow, S.; Pech, M.; Vermeulen, C.; Faiz, A.; Leeming, D.J.; Karsdal, M.A.; Lunding, L.; et al. COL4A3 is degraded in allergic asthma and degradation predicts response to anti-IgE therapy. Eur. Respir. J. 2021, 58, 2003969. [Google Scholar] [CrossRef]
- Zissler, U.M.; Ulrich, M.; Jakwerth, C.A.; Rothkirch, S.; Guerth, F.; Weckmann, M.; Schiemann, M.; Haller, B.; Schmidt-Weber, C.B.; Chaker, A.M. Biomatrix for upper and lower airway biomarkers in patients with allergic asthma. J. Allergy Clin. Immunol. 2018, 142, 1980–1983. [Google Scholar] [CrossRef] [Green Version]
- Zissler, U.M.; Jakwerth, C.A.; Guerth, F.; Lewitan, L.; Rothkirch, S.; Davidovic, M.; Ulrich, M.; Oelsner, M.; Garn, H.; Schmidt-Weber, C.B.; et al. Allergen-specific immunotherapy induces the suppressive secretoglobin 1A1 in cells of the lower airways. Allergy 2021, 76, 2461–2474. [Google Scholar] [CrossRef]
- Jakwerth, C.A.; Chaker, A.M.; Guerth, F.; Oelsner, M.; Pechtold, L.; Zur Bonsen, L.S.; Ullmann, J.T.; Krauss-Etschmann, S.; Erb, A.; Kau, J.; et al. Sputum microRNA-screening reveals Prostaglandin EP3 receptor as selective target in allergen-specific immunotherapy. Clin. Exp. Allergy 2021, 51, 1577–1591. [Google Scholar] [CrossRef]
- Schupp, J.C.; Khanal, S.; Gomez, J.L.; Sauler, M.; Adams, T.S.; Chupp, G.L.; Yan, X.; Poli, S.; Zhao, Y.; Montgomery, R.R.; et al. Single-Cell Transcriptional Archetypes of Airway Inflammation in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 202, 1419–1429. [Google Scholar] [CrossRef]
- Morse, C.; Tabib, T.; Sembrat, J.; Buschur, K.L.; Bittar, H.T.; Valenzi, E.; Jiang, Y.; Kass, D.J.; Gibson, K.; Chen, W.; et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur. Respir. J. 2019, 54, 1802441. [Google Scholar] [CrossRef]
- Vieira Braga, F.A.; Kar, G.; Berg, M.; Carpaij, O.A.; Polanski, K.; Simon, L.M.; Brouwer, S.; Gomes, T.; Hesse, L.; Jiang, J.; et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 2019, 25, 1153–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raredon, M.S.B.; Adams, T.S.; Suhail, Y.; Schupp, J.C.; Poli, S.; Neumark, N.; Leiby, K.L.; Greaney, A.M.; Yuan, Y.; Horien, C.; et al. Single-cell connectomic analysis of adult mammalian lungs. Sci Adv. 2019, 5, eaaw3851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, R.J.; Lloyd, C.M. Regulation of immune responses by the airway epithelial cell landscape. Nat. Rev. Immunol. 2021, 21, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, S. Immunologic influences on allergy and the TH1/TH2 balance. J. Allergy Clin. Immunol. 2004, 113, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Zissler, U.M.; Esser-von Bieren, J.; Jakwerth, C.A.; Chaker, A.M.; Schmidt-Weber, C.B. Current and future biomarkers in allergic asthma. Allergy 2016, 71, 475–494. [Google Scholar] [CrossRef] [Green Version]
- Zissler, U.M.; Chaker, A.M.; Effner, R.; Ulrich, M.; Guerth, F.; Piontek, G.; Dietz, K.; Regn, M.; Knapp, B.; Theis, F.J.; et al. Interleukin-4 and interferon-gamma orchestrate an epithelial polarization in the airways. Mucosal Immunol. 2016, 9, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Geha, R.S.; Jabara, H.H.; Brodeur, S.R. The regulation of immunoglobulin E class-switch recombination. Nat. Rev. Immunol. 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Zissler, U.M.; Jakwerth, C.A.; Guerth, F.M.; Pechtold, L.; Aguilar-Pimentel, J.A.; Dietz, K.; Suttner, K.; Piontek, G.; Haller, B.; Hajdu, Z.; et al. Early IL-10 producing B-cells and coinciding Th/Tr17 shifts during three year grass-pollen AIT. EBioMedicine 2018, 36, 475–488. [Google Scholar] [CrossRef] [Green Version]
- Tieu, D.D.; Peters, A.T.; Carter, R.G.; Suh, L.; Conley, D.B.; Chandra, R.; Norton, J.; Grammer, L.C.; Harris, K.E.; Kato, A.; et al. Evidence for diminished levels of epithelial psoriasin and calprotectin in chronic rhinosinusitis. J. Allergy Clin. Immunol. 2010, 125, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Alagha, K.; Bourdin, A.; Vernisse, C.; Garulli, C.; Tummino, C.; Charriot, J.; Vachier, I.; Suehs, C.; Chanez, P.; Gras, D. Goblet cell hyperplasia as a feature of neutrophilic asthma. Clin. Exp. Allergy 2019, 49, 781–788. [Google Scholar] [CrossRef]
- Corry, D.B.; Kheradmand, F. Biology and therapeutic potential of the interleukin-4/interleukin-13 signaling pathway in asthma. Am. J. Respir. Med. 2002, 1, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Zissler, U.M.; Buettner, M.; Heine, S.; Heldner, A.; Kotz, S.; Pechtold, L.; Kau, J.; Plaschke, M.; Ullmann, J.T.; et al. An exhausted phenotype of TH 2 cells is primed by allergen exposure, but not reinforced by allergen-specific immunotherapy. Allergy 2021, 76, 2827–2839. [Google Scholar] [CrossRef] [PubMed]
- Bacher, P.; Heinrich, F.; Stervbo, U.; Nienen, M.; Vahldieck, M.; Iwert, C.; Vogt, K.; Kollet, J.; Babel, N.; Sawitzki, B.; et al. Regulatory T Cell Specificity Directs Tolerance versus Allergy against Aeroantigens in Humans. Cell 2016, 167, 1067–1078.e1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakae, S.; Komiyama, Y.; Nambu, A.; Sudo, K.; Iwase, M.; Homma, I.; Sekikawa, K.; Asano, M.; Iwakura, Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 2002, 17, 375–387. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Carson, W.F.; Cavassani, K.A.; Connett, J.M.; Kunkel, S.L. CCR6 as a mediator of immunity in the lung and gut. Exp. Cell Res. 2011, 317, 613–619. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Kang, S.G.; Lee, J.; Sun, Z.; Kim, C.H. The roles of CCR6 in migration of Th17 cells and regulation of effector T-cell balance in the gut. Mucosal Immunol. 2009, 2, 173–183. [Google Scholar] [CrossRef]
- Power, C.A.; Church, D.J.; Meyer, A.; Alouani, S.; Proudfoot, A.E.; Clark-Lewis, I.; Sozzani, S.; Mantovani, A.; Wells, T.N. Cloning and characterization of a specific receptor for the novel CC chemokine MIP-3alpha from lung dendritic cells. J. Exp. Med. 1997, 186, 825–835. [Google Scholar] [CrossRef] [Green Version]
- Williams, I.R. Chemokine receptors and leukocyte trafficking in the mucosal immune system. Immunol. Res. 2004, 29, 283–292. [Google Scholar] [CrossRef]
- Mizutani, N.; Nabe, T.; Yoshino, S. IL-17A promotes the exacerbation of IL-33-induced airway hyperresponsiveness by enhancing neutrophilic inflammation via CXCR2 signaling in mice. J. Immunol. 2014, 192, 1372–1384. [Google Scholar] [CrossRef] [Green Version]
- Jaffar, Z.; Ferrini, M.E.; Herritt, L.A.; Roberts, K. Cutting edge: Lung mucosal Th17-mediated responses induce polymeric Ig receptor expression by the airway epithelium and elevate secretory IgA levels. J. Immunol. 2009, 182, 4507–4511. [Google Scholar] [CrossRef] [Green Version]
- Halwani, R.; Al-Kufaidy, R.; Vazquez-Tello, A.; Pureza, M.A.; BaHammam, A.S.; Al-Jahdali, H.; Alnassar, S.A.; Hamid, Q.; Al-Muhsen, S. IL-17 Enhances Chemotaxis of Primary Human B Cells during Asthma. PLoS ONE 2014, 9, e114604. [Google Scholar] [CrossRef] [Green Version]
- Lerch, E.; Muller, U.R. Long-term protection after stopping venom immunotherapy: Results of re-stings in 200 patients. J. Allergy Clin. Immunol. 1998, 101, 606–612. [Google Scholar] [CrossRef]
- Jacobsen, L.; Niggemann, B.; Dreborg, S.; Ferdousi, H.A.; Halken, S.; Host, A.; Koivikko, A.; Norberg, L.A.; Valovirta, E.; Wahn, U.; et al. Specific immunotherapy has long-term preventive effect of seasonal and perennial asthma: 10-year follow-up on the PAT study. Allergy 2007, 62, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef]
- Gagliani, N.; Amezcua Vesely, M.C.; Iseppon, A.; Brockmann, L.; Xu, H.; Palm, N.W.; de Zoete, M.R.; Licona-Limon, P.; Paiva, R.S.; Ching, T.; et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015, 523, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Schiller, H.B.; Montoro, D.T.; Simon, L.M.; Rawlins, E.L.; Meyer, K.B.; Strunz, M.; Vieira Braga, F.A.; Timens, W.; Koppelman, G.H.; Budinger, G.R.S.; et al. The Human Lung Cell Atlas: A High-Resolution Reference Map of the Human Lung in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2019, 61, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e1821. [Google Scholar] [CrossRef]
- Adam Gayoso, R.L.; Xing, G.; Boyeau, P.; Wu, K.; Michael Jayasuriya, E.M.; Langevin, M.; Liu, Y.; Jules Samaran, G.M.; Nazaret, A.; Clivio, O.; et al. Jeffrey Regier, and Nir Yosef scvi-tools: A library for deep probabilistic analysis of single-cell omics data. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lotfollahi, M.; Naghipourfar, M.; Luecken, M.D.; Khajavi, M.; Buttner, M.; Wagenstetter, M.; Avsec, Z.; Gayoso, A.; Yosef, N.; Interlandi, M.; et al. Mapping single-cell data to reference atlases by transfer learning. Nat. Biotechnol. 2022, 40, 121–130. [Google Scholar] [CrossRef]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef]
- Haimerl, P.; Bernhardt, U.; Schindela, S.; Henkel, F.D.R.; Lechner, A.; Zissler, U.M.; Pastor, X.; Thomas, D.; Cecil, A.; Ge, Y.; et al. Inflammatory macrophage memory in nonsteroidal anti-inflammatory drug-exacerbated Respir.atory disease. J. Allergy Clin. Immunol. 2021, 147, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Puttur, F.; Gregory, L.G.; Lloyd, C.M. Airway macrophages as the guardians of tissue repair in the lung. Immunol. Cell Biol. 2019, 97, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Ural, B.B.; Yeung, S.T.; Damani-Yokota, P.; Devlin, J.C.; de Vries, M.; Vera-Licona, P.; Samji, T.; Sawai, C.M.; Jang, G.; Perez, O.A.; et al. Identification of a nerve-associated, lung-resident interstitial macrophage subset with distinct localization and immunoregulatory properties. Sci. Immunol. 2020, 5, eaax8756. [Google Scholar] [CrossRef]
- Westphalen, K.; Gusarova, G.A.; Islam, M.N.; Subramanian, M.; Cohen, T.S.; Prince, A.S.; Bhattacharya, J. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature 2014, 506, 503–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.G.; Kim, S.J.; Jeong, J.J.; Han, S.S.; Jarjour, N.N.; Lee, H.; Abboud-Werner, S.L.; Chung, S.; Choi, H.S.; Natarajan, V.; et al. Airway Epithelial Cell-Derived Colony Stimulating Factor-1 Promotes Allergen Sensitization. Immunity 2018, 49, 275–287.e275. [Google Scholar] [CrossRef] [Green Version]
- Gschwend, J.; Sherman, S.P.M.; Ridder, F.; Feng, X.; Liang, H.E.; Locksley, R.M.; Becher, B.; Schneider, C. Alveolar macrophages rely on GM-CSF from alveolar epithelial type 2 cells before and after birth. J. Exp. Med. 2021, 218, e20210745. [Google Scholar] [CrossRef]
- Stanifer, M.L.; Guo, C.; Doldan, P.; Boulant, S. Importance of Type I and III Interferons at Respir.atory and Intestinal Barrier Surfaces. Front. Immunol. 2020, 11, 608645. [Google Scholar] [CrossRef] [PubMed]
- Nur Husna, S.M.; Tan, H.T.; Md Shukri, N.; Mohd Ashari, N.S.; Wong, K.K. Nasal Epithelial Barrier Integrity and Tight Junctions Disruption in Allergic Rhinitis: Overview and Pathogenic Insights. Front. Immunol. 2021, 12, 663626. [Google Scholar] [CrossRef]
- Hammad, H.; Lambrecht, B.N. Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity 2015, 43, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froidure, A.; Shen, C.; Pilette, C. Dendritic cells revisited in human allergic rhinitis and asthma. Allergy 2016, 71, 137–148. [Google Scholar] [CrossRef]
- Netea, M.G.; Dominguez-Andres, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front. Immunol. 2019, 10, 2759. [Google Scholar] [CrossRef] [Green Version]
- Shikotra, A.; Choy, D.F.; Ohri, C.M.; Doran, E.; Butler, C.; Hargadon, B.; Shelley, M.; Abbas, A.R.; Austin, C.D.; Jackman, J.; et al. Increased expression of immunoreactive thymic stromal lymphopoietin in patients with severe asthma. J. Allergy Clin. Immunol. 2012, 129, 104–111.e101–109. [Google Scholar] [CrossRef] [PubMed]
- Claudio, E.; Tassi, I.; Wang, H.; Tang, W.; Ha, H.L.; Siebenlist, U. Cutting Edge: IL-25 Targets Dendritic Cells To Attract IL-9-Producing T Cells in Acute Allergic Lung Inflammation. J. Immunol. 2015, 195, 3525–3529. [Google Scholar] [CrossRef] [Green Version]
- Musiol, S.; Alessandrini, F.; Schneider, E.; Jakwerth, C.A.; Chaker, A.M.; Guerth, F.; Ghiordanescu, I.; Schilling, J.T.; Kau, J.; Plaschke, M.; et al. TGF-β1 drives inflammatory T but not Treg cell compartment upon allergen exposure. Front. Immunol. 2022, 12, 763243. [Google Scholar] [CrossRef] [PubMed]
- Rate, A.; Upham, J.W.; Bosco, A.; McKenna, K.L.; Holt, P.G. Airway epithelial cells regulate the functional phenotype of locally differentiating dendritic cells: Implications for the pathogenesis of infectious and allergic airway disease. J. Immunol. 2009, 182, 72–83. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. Allergens and the airway epithelium response: Gateway to allergic sensitization. J. Allergy Clin. Immunol. 2014, 134, 499–507. [Google Scholar] [CrossRef]
- Komlosi, Z.I.; van de Veen, W.; Kovacs, N.; Szucs, G.; Sokolowska, M.; O’Mahony, L.; Akdis, M.; Akdis, C.A. Cellular and molecular mechanisms of allergic asthma. Mol. Asp. Med. 2022, 85, 100995. [Google Scholar] [CrossRef]
- Van der Velden, J.; Barker, D.; Barcham, G.; Koumoundouros, E.; Snibson, K. Increased mast cell density and airway responses to allergic and non-allergic stimuli in a sheep model of chronic asthma. PLoS ONE 2012, 7, e37161. [Google Scholar] [CrossRef]
- Shahabuddin, S.; Ji, R.; Wang, P.; Brailoiu, E.; Dun, N.; Yang, Y.; Aksoy, M.O.; Kelsen, S.G. CXCR3 chemokine receptor-induced chemotaxis in human airway epithelial cells: Role of p38 MAPK and PI3K signaling pathways. Am. J. Physiol. Cell Physiol. 2006, 291, C34–C39. [Google Scholar] [CrossRef] [PubMed]
- Weller, C.L.; Collington, S.J.; Hartnell, A.; Conroy, D.M.; Kaise, T.; Barker, J.E.; Wilson, M.S.; Taylor, G.W.; Jose, P.J.; Williams, T.J. Chemotactic action of prostaglandin E2 on mouse mast cells acting via the PGE2 receptor 3. Proc. Natl. Acad. Sci. USA 2007, 104, 11712–11717. [Google Scholar] [CrossRef] [Green Version]
- Girodet, P.O.; Ozier, A.; Trian, T.; Begueret, H.; Ousova, O.; Vernejoux, J.M.; Chanez, P.; Marthan, R.; Berger, P.; Tunon de Lara, J.M. Mast cell adhesion to bronchial smooth muscle in asthma specifically depends on CD51 and CD44 variant 6. Allergy 2010, 65, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Atiakshin, D.; Buchwalow, I.; Tiemann, M. Mast cell chymase: Morphofunctional characteristics. Histochem. Cell Biol. 2019, 152, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, A. Characterization of Innate Immune Responses to House Dust Mite Allergens: Pitfalls and Limitations. Front. Allergy 2021, 2, 662378. [Google Scholar] [CrossRef] [PubMed]
- Carter, R.J.; Bradding, P. The role of mast cells in the structural alterations of the airways as a potential mechanism in the pathogenesis of severe asthma. Curr. Pharm. Des. 2011, 17, 685–698. [Google Scholar] [CrossRef]
- Mootz, M.; Jakwerth, C.A.; Schmidt-Weber, C.B.; Zissler, U.M. Secretoglobins in the big picture of immunoregulation in airway diseases. Allergy 2022, 77, 767–777. [Google Scholar] [CrossRef]
- Seibold, M.A. Interleukin-13 Stimulation Reveals the Cellular and Functional Plasticity of the Airway Epithelium. Ann. Am. Thorac. Soc. 2018, 15 (Suppl. 2), S98–S102. [Google Scholar] [CrossRef]
- Lillehoj, E.P.; Kato, K.; Lu, W.; Kim, K.C. Cellular and molecular biology of airway mucins. Int. Rev. Cell Mol. Biol. 2013, 303, 139–202. [Google Scholar] [CrossRef]
- Bartel, S.; La Grutta, S.; Cilluffo, G.; Perconti, G.; Bongiovanni, A.; Giallongo, A.; Behrends, J.; Kruppa, J.; Hermann, S.; Chiang, D.; et al. Human airway epithelial extracellular vesicle miRNA signature is altered upon asthma development. Allergy 2020, 75, 346–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pua, H.H.; Ansel, K.M. MicroRNA regulation of allergic inflammation and asthma. Curr. Opin. Immunol. 2015, 36, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Simpson, L.J.; Patel, S.; Bhakta, N.R.; Choy, D.F.; Brightbill, H.D.; Ren, X.; Wang, Y.; Pua, H.H.; Baumjohann, D.; Montoya, M.M.; et al. A microRNA upregulated in asthma airway T cells promotes TH2 cytokine production. Nat. Immunol. 2014, 15, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Baumann, R.; Untersmayr, E.; Zissler, U.M.; Eyerich, S.; Adcock, I.M.; Brockow, K.; Biedermann, T.; Ollert, M.; Chaker, A.M.; Pfaar, O.; et al. Noninvasive and minimally invasive techniques for the diagnosis and management of allergic diseases. Allergy 2021, 76, 1010–1023. [Google Scholar] [CrossRef] [PubMed]
- Weidner, J.; Bartel, S.; Kilic, A.; Zissler, U.M.; Renz, H.; Schwarze, J.; Schmidt-Weber, C.B.; Maes, T.; Rebane, A.; Krauss-Etschmann, S.; et al. Spotlight on microRNAs in allergy and asthma. Allergy 2021, 76, 1661–1678. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.B.; Pua, H.H.; Happ, H.C.; Schneider, C.; von Moltke, J.; Locksley, R.M.; Baumjohann, D.; Ansel, K.M. MicroRNA regulation of type 2 innate lymphoid cell homeostasis and function in allergic inflammation. J. Exp. Med. 2017, 214, 3627–3643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Liu, L.; Wang, H.; Mandal, J.; Khan, P.; Hostettler, K.E.; Stolz, D.; Tamm, M.; Molino, A.; Lardinois, D.; et al. Constitutive high expression of protein arginine methyltransferase 1 in asthmatic airway smooth muscle cells is caused by reduced microRNA-19a expression and leads to enhanced remodeling. J. Allergy Clin. Immunol. 2017, 140, 510–524.e513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonkoly, E.; Janson, P.; Majuri, M.L.; Savinko, T.; Fyhrquist, N.; Eidsmo, L.; Xu, N.; Meisgen, F.; Wei, T.; Bradley, M.; et al. MiR-155 is overexpressed in patients with atopic dermatitis and modulates T-cell proliferative responses by targeting cytotoxic T lymphocyte-associated antigen 4. J. Allergy Clin. Immunol. 2010, 126, 581–589.e.581–520. [Google Scholar] [CrossRef] [PubMed]
- Johansson, K.; Malmhall, C.; Ramos-Ramirez, P.; Radinger, M. MicroRNA-155 is a critical regulator of type 2 innate lymphoid cells and IL-33 signaling in experimental models of allergic airway inflammation. J. Allergy Clin. Immunol. 2017, 139, 1007–1016.e1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polikepahad, S.; Knight, J.M.; Naghavi, A.O.; Oplt, T.; Creighton, C.J.; Shaw, C.; Benham, A.L.; Kim, J.; Soibam, B.; Harris, R.A.; et al. Proinflammatory role for let-7 microRNAS in experimental asthma. J. Biol. Chem. 2010, 285, 30139–30149. [Google Scholar] [CrossRef] [Green Version]
- Frey, A.; Lunding, L.P.; Ehlers, J.C.; Weckmann, M.; Zissler, U.M.; Wegmann, M. More Than Just a Barrier: The Immune Functions of the Airway Epithelium in Asthma Pathogenesis. Front. Immunol. 2020, 11, 761. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.; Homer, R.J.; Niu, N.; Bottomly, K. T helper 1 cells and interferon gamma regulate allergic airway inflammation and mucus production. J. Exp. Med. 1999, 190, 1309–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.J.; MacAry, P.A.; Eynott, P.; Moussavi, A.; Daniel, K.C.; Askenase, P.W.; Kemeny, D.M.; Chung, K.F. Allergen-specific Th1 cells counteract efferent Th2 cell-dependent bronchial hyperresponsiveness and eosinophilic inflammation partly via IFN-gam.mma. J. Immunol. 2001, 166, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, C.; Provost, K.; Niu, N.; Homer, R.; Cohn, L. IFN-gamma acts on the airway epithelium to inhibit local and systemic pathology in allergic airway disease. J. Immunol. 2011, 187, 3815–3820. [Google Scholar] [CrossRef] [Green Version]
- Sel, S.; Wegmann, M.; Dicke, T.; Sel, S.; Henke, W.; Yildirim, A.O.; Renz, H.; Garn, H. Effective prevention and therapy of experimental allergic asthma using a GATA-3-specific DNAzyme. J. Allergy Clin. Immunol. 2008, 121, 910–916.e915. [Google Scholar] [CrossRef]
- Brand, S.; Kesper, D.A.; Teich, R.; Kilic-Niebergall, E.; Pinkenburg, O.; Bothur, E.; Lohoff, M.; Garn, H.; Pfefferle, P.I.; Renz, H. DNA methylation of TH1/TH2 cytokine genes affects sensitization and progress of experimental asthma. J. Allergy Clin. Immunol. 2012, 129, 1602–1610.e1606. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Sad, S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol. Today 1996, 17, 138–146. [Google Scholar] [CrossRef]
- Steelant, B.; Farre, R.; Wawrzyniak, P.; Belmans, J.; Dekimpe, E.; Vanheel, H.; Van Gerven, L.; Kortekaas Krohn, I.; Bullens, D.M.A.; Ceuppens, J.L.; et al. Impaired barrier function in patients with house dust mite-induced allergic rhinitis is accompanied by decreased occludin and zonula occludens-1 expression. J. Allergy Clin. Immunol. 2016, 137, 1043–1053.e1045. [Google Scholar] [CrossRef] [Green Version]
- Smallcombe, C.C.; Linfield, D.T.; Harford, T.J.; Bokun, V.; Ivanov, A.I.; Piedimonte, G.; Rezaee, F. Disruption of the airway epithelial barrier in a murine model of Respir.atory syncytial virus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L358–L368. [Google Scholar] [CrossRef]
- Lai, J.F.; Thompson, L.J.; Ziegler, S.F. TSLP drives acute TH2-cell differentiation in lungs. J. Allergy Clin. Immunol. 2020, 146, 1406–1418.e1407. [Google Scholar] [CrossRef]
- Roan, F.; Obata-Ninomiya, K.; Ziegler, S.F. Epithelial cell-derived cytokines: More than just signaling the alarm. J. Clin. Investig. 2019, 129, 1441–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.G.; Zhang, T.T.; Li, H.T.; Chen, F.H.; Zou, X.L.; Ji, J.Z.; Chen, H. Neutralization of TSLP inhibits airway remodeling in a murine model of allergic asthma induced by chronic exposure to house dust mite. PLoS ONE 2013, 8, e51268. [Google Scholar] [CrossRef]
- Zhang, F.Q.; Han, X.P.; Zhang, F.; Ma, X.; Xiang, D.; Yang, X.M.; Ou-Yang, H.F.; Li, Z. Therapeutic efficacy of a co-blockade of IL-13 and IL-25 on airway inflammation and remodeling in a mouse model of asthma. Int. Immunopharmacol. 2017, 46, 133–140. [Google Scholar] [CrossRef]
- Tanabe, T.; Shimokawaji, T.; Kanoh, S.; Rubin, B.K. IL-33 stimulates CXCL8/IL-8 secretion in goblet cells but not normally differentiated airway cells. Clin. Exp. Allergy 2014, 44, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNamee, E.N.; Biette, K.A.; Hammer, J.; Harris, R.; Miyazawa, H.; Lee, J.J.; Furuta, G.T.; Masterson, J.C. Targeting granulocyte-macrophage colony-stimulating factor in epithelial and vascular remodeling in experimental eosinophilic esophagitis. Allergy 2017, 72, 1232–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.A.; Bercik, P. Macrophages are related to goblet cell hyperplasia and induce MUC5B but not MUC5AC in human bronchus epithelial cells. Lab. Investig. 2012, 92, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Bankova, L.G.; Dwyer, D.F.; Yoshimoto, E.; Ualiyeva, S.; McGinty, J.W.; Raff, H.; von Moltke, J.; Kanaoka, Y.; Frank Austen, K.; Barrett, N.A. The cysteinyl leukotriene 3 receptor regulates expansion of IL-25-producing airway brush cells leading to type 2 inflammation. Sci. Immunol. 2018, 3, eaat9453. [Google Scholar] [CrossRef] [Green Version]
- Bell, S.M.; Zhang, L.; Xu, Y.; Besnard, V.; Wert, S.E.; Shroyer, N.; Whitsett, J.A. Kruppel-like factor 5 controls villus formation and initiation of cytodifferentiation in the embryonic intestinal epithelium. Dev. Biol. 2013, 375, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Denney, L.; Byrne, A.J.; Shea, T.J.; Buckley, J.S.; Pease, J.E.; Herledan, G.M.; Walker, S.A.; Gregory, L.G.; Lloyd, C.M. Pulmonary Epithelial Cell-Derived Cytokine TGF-beta1 Is a Critical Cofactor for Enhanced Innate Lymphoid Cell Function. Immunity 2015, 43, 945–958. [Google Scholar] [CrossRef] [Green Version]
- Gregory, L.G.; Jones, C.P.; Walker, S.A.; Sawant, D.; Gowers, K.H.; Campbell, G.A.; McKenzie, A.N.; Lloyd, C.M. IL-25 drives remodelling in allergic airways disease induced by house dust mite. Thorax 2013, 68, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Hammad, H.; Chieppa, M.; Perros, F.; Willart, M.A.; Germain, R.N.; Lambrecht, B.N. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat. Med. 2009, 15, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Iijima, K.; Kobayashi, T.; Hara, K.; Kephart, G.M.; Ziegler, S.F.; McKenzie, A.N.; Kita, H. IL-33 and thymic stromal lymphopoietin mediate immune pathology in response to chronic airborne allergen exposure. J. Immunol. 2014, 193, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Millien, V.O.; Lu, W.; Shaw, J.; Yuan, X.; Mak, G.; Roberts, L.; Song, L.Z.; Knight, J.M.; Creighton, C.J.; Luong, A.; et al. Cleavage of fibrinogen by proteinases elicits allergic responses through Toll-like receptor 4. Science 2013, 341, 792–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Kleer, I.M.; Kool, M.; de Bruijn, M.J.; Willart, M.; van Moorleghem, J.; Schuijs, M.J.; Plantinga, M.; Beyaert, R.; Hams, E.; Fallon, P.G.; et al. Perinatal Activation of the Interleukin-33 Pathway Promotes Type 2 Immunity in the Developing Lung. Immunity 2016, 45, 1285–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, S.; Custovic, A.; Ghazal, P.; Grigg, J.; Gore, M.; Henderson, J.; Lloyd, C.M.; Marsland, B.; Power, U.F.; Roberts, G.; et al. Pulmonary epithelial barrier and immunological functions at birth and in early life-key determinants of the development of asthma? A description of the protocol for the Breathing Together study. Wellcome Open Res. 2018, 3, 60. [Google Scholar] [CrossRef] [Green Version]
- Weckmann, M.; Thiele, D.; Liboschik, L.; Bahmer, T.; Pech, M.; Dittrich, A.M.; Fuchs, O.; Happle, C.; Schaub, B.; Ricklefs, I.; et al. Cytokine levels in children and adults with wheezing and asthma show specific patterns of variability over time. Clin. Exp. Immunol. 2021, 204, 152–164. [Google Scholar] [CrossRef]
- Stein, M.M.; Hrusch, C.L.; Gozdz, J.; Igartua, C.; Pivniouk, V.; Murray, S.E.; Ledford, J.G.; Marques Dos Santos, M.; Anderson, R.L.; Metwali, N.; et al. Innate Immunity and Asthma Risk in Amish and Hutterite Farm Children. N. Engl. J. Med. 2016, 375, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Schuijs, M.J.; Willart, M.A.; Vergote, K.; Gras, D.; Deswarte, K.; Ege, M.J.; Madeira, F.B.; Beyaert, R.; van Loo, G.; Bracher, F.; et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science 2015, 349, 1106–1110. [Google Scholar] [CrossRef]
- Ruysseveldt, E.; Martens, K.; Steelant, B. Airway Basal Cells, Protectors of Epithelial Walls in Health and Respir.atory Diseases. Front. Allergy 2022, 2, 787128. [Google Scholar] [CrossRef]
- Van Dyken, S.J.; Nussbaum, J.C.; Lee, J.; Molofsky, A.B.; Liang, H.E.; Pollack, J.L.; Gate, R.E.; Haliburton, G.E.; Ye, C.J.; Marson, A.; et al. A tissue checkpoint regulates type 2 immunity. Nat. Immunol. 2016, 17, 1381–1387. [Google Scholar] [CrossRef] [Green Version]
- Maggi, L.; Mazzoni, A.; Capone, M.; Liotta, F.; Annunziato, F.; Cosmi, L. The dual function of ILC2: From host protection to pathogenic players in type 2 asthma. Mol. Asp. Med. 2021, 80, 100981. [Google Scholar] [CrossRef]
- Halwani, R.; Al-Muhsen, S.; Al-Jahdali, H.; Hamid, Q. Role of transforming growth factor-beta in airway remodeling in asthma. Am. J. Respir. Cell Mol. Biol. 2011, 44, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Ojiaku, C.A.; Yoo, E.J.; Panettieri, R.A., Jr. Transforming Growth Factor beta1 Function in Airway Remodeling and Hyperresponsiveness. The Missing Link? Am. J. Respir. Cell Mol. Biol. 2017, 56, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Ricardo-Gonzalez, R.R.; Van Dyken, S.J.; Schneider, C.; Lee, J.; Nussbaum, J.C.; Liang, H.E.; Vaka, D.; Eckalbar, W.L.; Molofsky, A.B.; Erle, D.J.; et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat. Immunol. 2018, 19, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Lechner, A.; Henkel, F.; Hartung, F.; Bohnacker, S.; Alessandrini, F.; Gubernatorova, E.O.; Drutskaya, M.S.; Angioni, C.; Schreiber, Y.; Haimerl, P.; et al. Macrophages acquire a TNF-dependent inflammatory memory in allergic asthma. J. Allergy Clin. Immunol. 2021. [Google Scholar] [CrossRef]
- Endo, Y.; Hirahara, K.; Iinuma, T.; Shinoda, K.; Tumes, D.J.; Asou, H.K.; Matsugae, N.; Obata-Ninomiya, K.; Yamamoto, H.; Motohashi, S.; et al. The interleukin-33-p38 kinase axis confers memory T helper 2 cell pathogenicity in the airway. Immunity 2015, 42, 294–308. [Google Scholar] [CrossRef] [Green Version]
- Coquet, J.M.; Schuijs, M.J.; Smyth, M.J.; Deswarte, K.; Beyaert, R.; Braun, H.; Boon, L.; Karlsson Hedestam, G.B.; Nutt, S.L.; Hammad, H.; et al. Interleukin-21-Producing CD4(+) T Cells Promote Type 2 Immunity to House Dust Mites. Immunity 2015, 43, 318–330. [Google Scholar] [CrossRef] [Green Version]
- Wythe, S.E.; Dodd, J.S.; Openshaw, P.J.; Schwarze, J. OX40 ligand and programmed cell death 1 ligand 2 expression on inflammatory dendritic cells regulates CD4 T cell cytokine production in the lung during viral disease. J. Immunol. 2012, 188, 1647–1655. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Wang, Y.H.; Duramad, O.; Hori, T.; Delespesse, G.J.; Watanabe, N.; Qin, F.X.; Yao, Z.; Cao, W.; Liu, Y.J. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J. Exp. Med. 2005, 202, 1213–1223. [Google Scholar] [CrossRef] [Green Version]
- Seo, G.Y.; Giles, D.A.; Kronenberg, M. The role of innate lymphoid cells in response to microbes at mucosal surfaces. Mucosal Immunol. 2020, 13, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Ualiyeva, S.; Lemire, E.; Aviles, E.C.; Wong, C.; Boyd, A.A.; Lai, J.; Liu, T.; Matsumoto, I.; Barrett, N.A.; Boyce, J.A.; et al. Tuft cell-produced cysteinyl leukotrienes and IL-25 synergistically initiate lung type 2 inflammation. Sci. Immunol. 2021, 6, eabj0474. [Google Scholar] [CrossRef] [PubMed]
- Mirchandani, A.S.; Besnard, A.G.; Yip, E.; Scott, C.; Bain, C.C.; Cerovic, V.; Salmond, R.J.; Liew, F.Y. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J. Immunol. 2014, 192, 2442–2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Moltke, J.; Ji, M.; Liang, H.E.; Locksley, R.M. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 2016, 529, 221–225. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, M.; Furukawa, K.T.; Morimoto, M. Pulmonary neuroendocrine cells: Physiology, tissue homeostasis and disease. Dis. Model. Mech. 2020, 13, dmm046920. [Google Scholar] [CrossRef]
- Sui, P.; Wiesner, D.L.; Xu, J.; Zhang, Y.; Lee, J.; Van Dyken, S.; Lashua, A.; Yu, C.; Klein, B.S.; Locksley, R.M.; et al. Pulmonary neuroendocrine cells amplify allergic asthma responses. Science 2018, 360, eaan8546. [Google Scholar] [CrossRef] [Green Version]
- Kiniwa, T.; Moro, K. Localization and site-specific cell-cell interactions of group 2 innate lymphoid cells. Int. Immunol. 2021, 33, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Barrios, J.; Kho, A.T.; Aven, L.; Mitchel, J.A.; Park, J.A.; Randell, S.H.; Miller, L.A.; Tantisira, K.G.; Ai, X. Pulmonary Neuroendocrine Cells Secrete gamma-Aminobutyric Acid to Induce Goblet Cell Hyperplasia in Primate Models. Am. J. Respir. Cell Mol. Biol. 2019, 60, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; Ural, B.B.; Farber, D.L. Tissue-specific immunity for a changing world. Cell 2021, 184, 1517–1529. [Google Scholar] [CrossRef]
- Clark, H.R.; McKenney, C.; Livingston, N.M.; Gershman, A.; Sajjan, S.; Chan, I.S.; Ewald, A.J.; Timp, W.; Wu, B.; Singh, A.; et al. Epigenetically regulated digital signaling defines epithelial innate immunity at the tissue level. Nat. Commun. 2021, 12, 1836. [Google Scholar] [CrossRef]
- Fanucchi, S.; Dominguez-Andres, J.; Joosten, L.A.B.; Netea, M.G.; Mhlanga, M.M. The Intersection of Epigenetics and Metabolism in Trained Immunity. Immunity 2021, 54, 32–43. [Google Scholar] [CrossRef]
- Natoli, G.; Ostuni, R. Adaptation and memory in immune responses. Nat. Immunol. 2019, 20, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Ordovas-Montanes, J.; Beyaz, S.; Rakoff-Nahoum, S.; Shalek, A.K. Distribution and storage of inflammatory memory in barrier tissues. Nat. Rev. Immunol. 2020, 20, 308–320. [Google Scholar] [CrossRef]
- Ordovas-Montanes, J.; Dwyer, D.F.; Nyquist, S.K.; Buchheit, K.M.; Vukovic, M.; Deb, C.; Wadsworth, M.H., 2nd; Hughes, T.K.; Kazer, S.W.; Yoshimoto, E.; et al. Allergic inflammatory memory in human Respir.atory epithelial progenitor cells. Nature 2018, 560, 649–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlier, F.M.; Detry, B.; Lecocq, B.; Collin, A.M.; Planté-Bordeneuve, T.; Verleden, S.E.; Stanciu-Pop, C.M.; Rondelet, B.; Janssens, W.; Ambroise, J.; et al. The memory of airway epithelium damage in smokers and COPD patients. bioRxiv 2021. [Google Scholar] [CrossRef]
- Boothby, I.C.; Kinet, M.J.; Boda, D.P.; Kwan, E.Y.; Clancy, S.; Cohen, J.N.; Habrylo, I.; Lowe, M.M.; Pauli, M.; Yates, A.E.; et al. Early-life inflammation primes a T helper 2 cell-fibroblast niche in skin. Nature 2021, 599, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, D.F.; Ordovas-Montanes, J.; Allon, S.J.; Buchheit, K.M.; Vukovic, M.; Derakhshan, T.; Feng, C.; Lai, J.; Hughes, T.K.; Nyquist, S.K.; et al. Human airway mast cells proliferate and acquire distinct inflammation-driven phenotypes during type 2 inflammation. Sci. Immunol. 2021, 6, eabb7221. [Google Scholar] [CrossRef]
- Dougherty, R.H.; Sidhu, S.S.; Raman, K.; Solon, M.; Solberg, O.D.; Caughey, G.H.; Woodruff, P.G.; Fahy, J.V. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. J. Allergy Clin. Immunol. 2010, 125, 1046–1053.e1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, M.C.; Lai, Y.; Nolin, J.D.; Long, S.; Chen, C.C.; Piliponsky, A.M.; Altemeier, W.A.; Larmore, M.; Frevert, C.W.; Mulligan, M.S.; et al. Airway epithelium-shifted mast cell infiltration regulates asthmatic inflammation via IL-33 signaling. J. Clin. Investig. 2019, 129, 4979–4991. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, C.G.K.; Miao, V.N.; Owings, A.H.; Navia, A.W.; Tang, Y.; Bromley, J.D.; Lotfy, P.; Sloan, M.; Laird, H.; Williams, H.B.; et al. Impaired local intrinsic i.immunity to SARS-CoV-2 infection in severe COVID-19. Cell 2021, 184, 4713–4733.e4722. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thurmann, L.; Kurth, F.; Volker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Fanucchi, S.; Fok, E.T.; Dalla, E.; Shibayama, Y.; Borner, K.; Chang, E.Y.; Stoychev, S.; Imakaev, M.; Grimm, D.; Wang, K.C.; et al. Immune genes are primed for robust transcription by proximal long noncoding RNAs located in nuclear compartments. Nat. Genet. 2019, 51, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.B.; Cowley, C.J.; Sajjath, S.M.; Barrows, D.; Yang, Y.; Carroll, T.S.; Fuchs, E. Establishment, maintenance, and recall of inflammatory memory. Cell Stem Cell 2021, 28, 1758–1774.e1758. [Google Scholar] [CrossRef] [PubMed]
- Bintu, L.; Yong, J.; Antebi, Y.E.; McCue, K.; Kazuki, Y.; Uno, N.; Oshimura, M.; Elowitz, M.B. Dynamics of epigenetic regulation at the single-cell level. Science 2016, 351, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Martins, A.G.; Mane, K.; Lindsey, B.B.; Ogava, R.L.T.; Castro, I.; Jagne, Y.J.; Sallah, H.J.; Armitage, E.P.; Jarju, S.; Ahadzie, B.; et al. Prior upregulation of interferon pathways in the nasopharynx impacts viral shedding following live attenuated influenza vaccine challenge in children. Cell Rep. Med. 2021, 2, 100465. [Google Scholar] [CrossRef] [PubMed]
- Singanayagam, A.; Glanville, N.; Girkin, J.L.; Ching, Y.M.; Marcellini, A.; Porter, J.D.; Toussaint, M.; Walton, R.P.; Finney, L.J.; Aniscenko, J.; et al. Corticosteroid suppression of antiviral immunity increases bacterial loads and mucus production in COPD exacerbations. Nat. Commun. 2018, 9, 2229. [Google Scholar] [CrossRef]
- Mostafa, M.M.; Rider, C.F.; Shah, S.; Traves, S.L.; Gordon, P.M.K.; Miller-Larsson, A.; Leigh, R.; Newton, R. Glucocorticoid-driven transcriptomes in human airway epithelial cells: Commonalities, differences and functional insight from cell lines and primary cells. BMC Med. Genom. 2019, 12, 29. [Google Scholar] [CrossRef]
- Wu, Q.; Chen, J.; Ren, Y.; Qiu, H.; Yuan, L.; Deng, H.; Zhang, Y.; Zheng, R.; Hong, H.; Sun, Y.; et al. Artificial intelligence for cellular phenotyping diagnosis of nasal polyps by whole-slide imaging. EBioMedicine 2021, 66, 103336. [Google Scholar] [CrossRef]
- Soyka, M.B.; Wawrzyniak, P.; Eiwegger, T.; Holzmann, D.; Treis, A.; Wanke, K.; Kast, J.I.; Akdis, C.A. Defective epithelial barrier in chronic rhinosinusitis: The regulation of tight junctions by IFN-gamma and IL-4. J. Allergy Clin. Immunol. 2012, 130, 1087–1096.e1010. [Google Scholar] [CrossRef]
- Hirsch, A.G.; Stewart, W.F.; Sundaresan, A.S.; Young, A.J.; Kennedy, T.L.; Scott Greene, J.; Feng, W.; Tan, B.K.; Schleimer, R.P.; Kern, R.C.; et al. Nasal and sinus symptoms and chronic rhinosinusitis in a population-based sample. Allergy 2017, 72, 274–281. [Google Scholar] [CrossRef]
- Hastan, D.; Fokkens, W.J.; Bachert, C.; Newson, R.B.; Bislimovska, J.; Bockelbrink, A.; Bousquet, P.J.; Brozek, G.; Bruno, A.; Dahlen, S.E.; et al. Chronic rhinosinusitis in Europe—An underestimated disease. A GA(2)LEN study. Allergy 2011, 66, 1216–1223. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakwerth, C.A.; Ordovas-Montanes, J.; Blank, S.; Schmidt-Weber, C.B.; Zissler, U.M. Role of Respiratory Epithelial Cells in Allergic Diseases. Cells 2022, 11, 1387. https://doi.org/10.3390/cells11091387
Jakwerth CA, Ordovas-Montanes J, Blank S, Schmidt-Weber CB, Zissler UM. Role of Respiratory Epithelial Cells in Allergic Diseases. Cells. 2022; 11(9):1387. https://doi.org/10.3390/cells11091387
Chicago/Turabian StyleJakwerth, Constanze A., Jose Ordovas-Montanes, Simon Blank, Carsten B. Schmidt-Weber, and Ulrich M. Zissler. 2022. "Role of Respiratory Epithelial Cells in Allergic Diseases" Cells 11, no. 9: 1387. https://doi.org/10.3390/cells11091387
APA StyleJakwerth, C. A., Ordovas-Montanes, J., Blank, S., Schmidt-Weber, C. B., & Zissler, U. M. (2022). Role of Respiratory Epithelial Cells in Allergic Diseases. Cells, 11(9), 1387. https://doi.org/10.3390/cells11091387