
GBA Variants and Parkinson Disease: Mechanisms and Treatments


Abstract
1. Introduction
2. Parkinson Disease
3. The GBA Gene
4. PD and the GBA Gene
4.1. The Link between GBA Mutations and PD
4.2. Presentation of GBA-PD
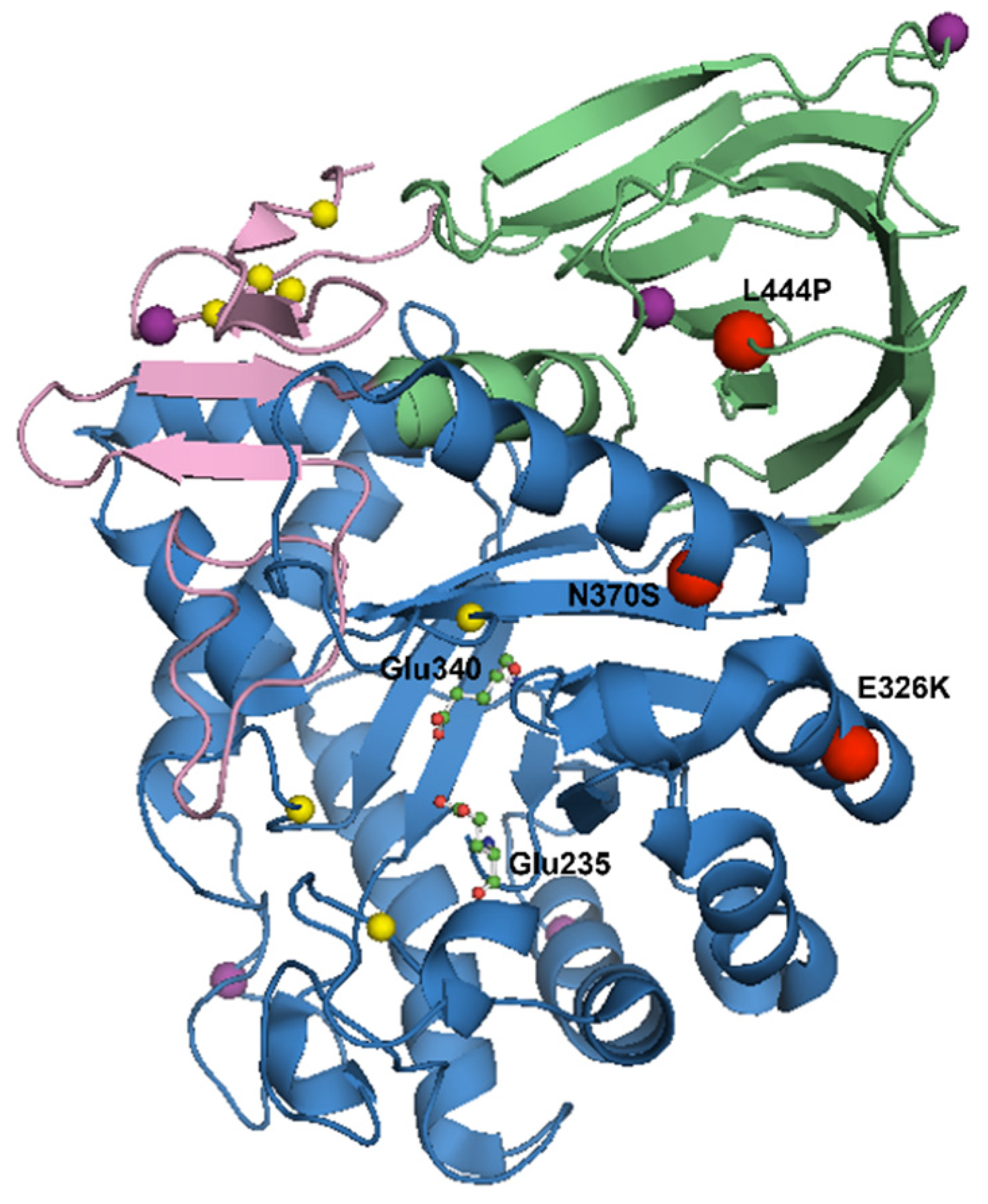
5. Mutations in the GBA Gene
6. GBA Activity and PD
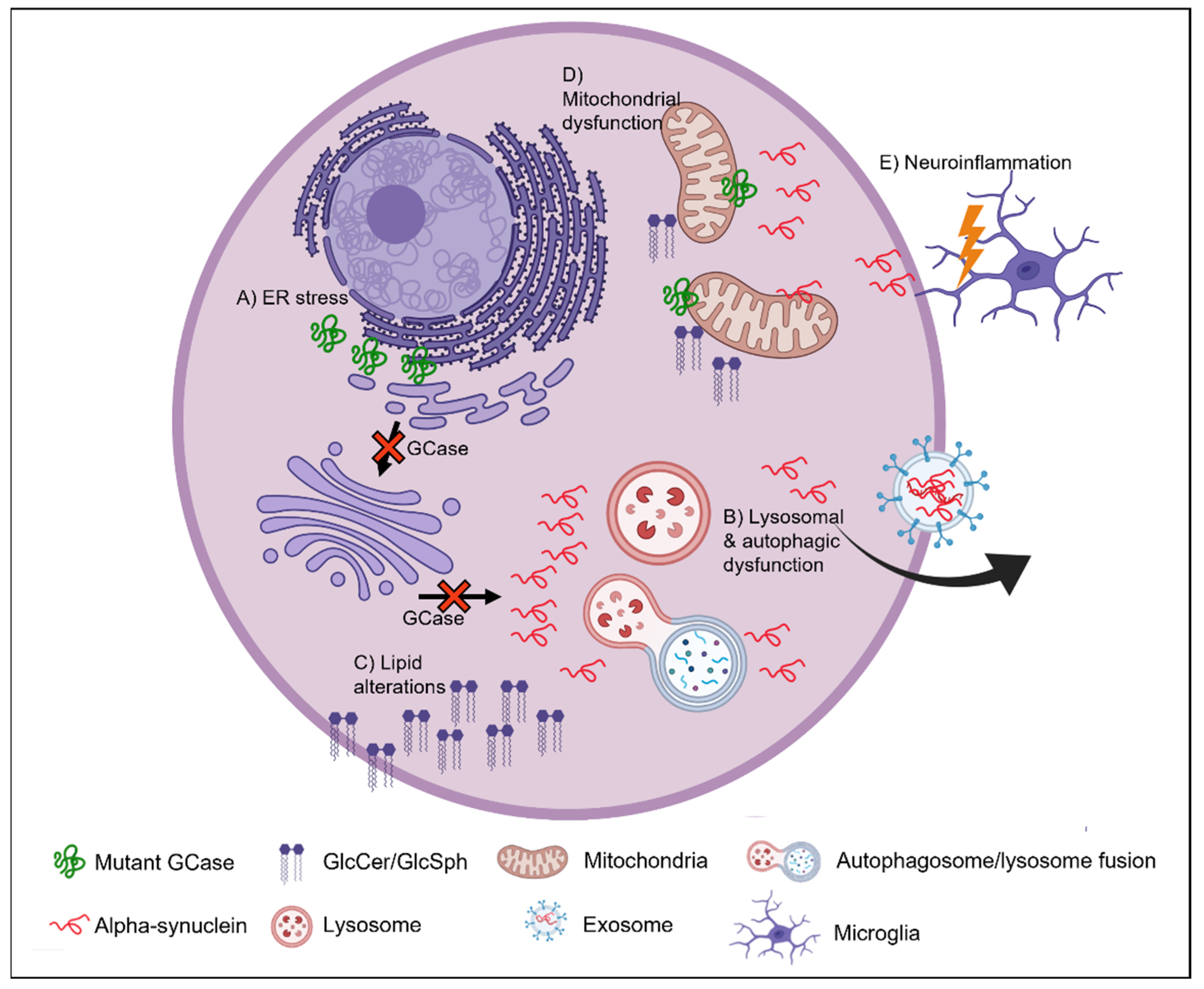
7. Mechanism Underlying GBA-PD
8. ER Stress
9. Autophagic-Lysosomal Pathway
10. Lipid Homeostasis
11. Mitochondrial Dysfunction
12. Neuroinflammation
13. GCase as a Therapeutic Target

{kind=link}
{kind=link}
| Treatment | Therapeutic Strategy | Drug Name | Phase in Drug Development | Reference |
|---|---|---|---|---|
| Substrate reduction | Reduce glycosphingolipid accumulation in the CNS | GZ667161 Venglustat Miglustat | Phase II completed for venglustat | [160,206,207] |
| Small molecule chaperones | Refold mutant GCase in the ER to improve trafficking to the lysosome and increase activity and stability while reducing ER stress | Ambroxol Isofagomine | Phase II completed for ambroxol | [39,90,138,211,212,213,214,215,216,217,218,219,220,221,222,223,224] |
| Gene therapy | Replace GCase activity and protein levels in the CNS | AAV-mediated delivery of recombinant GCase | Preclinical research ongoing Phase I/II ongoing for PR001 gene therapy (Prevail Therapeutics) | [111,120,205,209,210,225] |
| GCase activator | Increase GCase activity in the brain | BIA 28-6156/LTI-291 | Phase I completed | [205,226] |
| Transport vehicle modified recombinant GCase | Replace GCase activity and protein levels in the CNS | ETV:GBA | Preclinical research ongoing | [205] |
| Histone deacetylase inhibitors | Replace GCase activity and protein levels | LB-205 | Preclinical research ongoing | [227,228] |
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Braak, H. Neuropathological Staging of Brain Pathology in Sporadic Parkinson’s disease: Separating the Wheat from the Chaff. J. Park. Dis. 2017, 7, S71–S85. [Google Scholar] [CrossRef] [PubMed]
- Turpin, J.C.; Dubois, G.; Brice, A.; Masson, M.; Nadaud, M.C.; Boutry, J.M.; Schram, A.W.; Tager, J.M.; Baumann, N. Parkinsonian Symptomatology in a Patient with Type I (Adult) Gaucher’s Disease; Springer US: Boston, MA, USA, 1988; pp. 103–105. [Google Scholar]
- McKeran, R.O.; Bradbury, P.; Taylor, D.; Stern, G. Neurological involvement in type 1 (adult) Gaucher’s disease. J. Neurol. Neurosurg. Psychiatry 1985, 48, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Lopez, G. The link between the GBA gene and parkinsonism. Lancet Neurol. 2012, 11, 986–998. [Google Scholar] [CrossRef]
- Bultron, G.; Kacena, K.; Pearson, D.; Boxer, M.; Yang, R.; Sathe, S.; Pastores, G.; Mistry, P.K. The risk of Parkinson’s disease in type 1 Gaucher disease. J. Inherit. Metab. Dis. 2010, 33, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Gan-Or, Z.; Amshalom, I.; Kilarski, L.L.; Bar-Shira, A.; Gana-Weisz, M.; Mirelman, A.; Marder, K.; Bressman, S.; Giladi, N.; Orr-Urtreger, A. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 2015, 84, 880–887. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multi-center analysis of glucocerebrosidase mutations in Parkinson disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Tayebi, N.; Callahan, M.; Madike, V.; Stubblefield, B.K.; Orvisky, E.; Krasnewich, D.; Fillano, J.J.; Sidransky, E. Gaucher disease and parkinsonism: A phenotypic and genotypic characterization. Mol. Genet. Metab. 2001, 73, 313–321. [Google Scholar] [CrossRef]
- Hruska, K.S.; LaMarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef]
- Beutler, E.; Beutler, L.; West, C. Mutations in the gene encoding cytosolic beta-glucosidase in Gaucher disease. J. Lab. Clin. Med. 2004, 144, 65–68. [Google Scholar] [CrossRef]
- Gegg, M.E.; Schapira, A.H.V. The role of glucocerebrosidase in Parkinson disease pathogenesis. FEBS J. 2018, 285, 3591–3603. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Marsden, C.D. Parkinson’s disease. Lancet 1990, 335, 948–952. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Odagiri, S.; Miki, Y.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease and related neurodegenerative disorders. Mol. Neurobiol. 2013, 47, 495–508. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castano-Diez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef]
- Braak, H.; Tredici, K.D.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Ono, K. The Oligomer Hypothesis in α-Synucleinopathy. Neurochem. Res. 2017, 42, 3362–3371. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Alam, P.; Bousset, L.; Melki, R.; Otzen, D.E. α-Synuclein oligomers and fibrils: A spectrum of species, a spectrum of toxicities. J. Neurochem. 2019, 150, 522–534. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Cuervo, A.M. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol. 2007, 6, 352–361. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bove, J.; Rodriguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef]
- Reczek, D.; Schwake, M.; Schröder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 Is a Receptor for Lysosomal Mannose-6-Phosphate-Independent Targeting of β-Glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef]
- Grabowski, G.A.; Gatt, S.; Horowitz, M. Acid beta-glucosidase: Enzymology and molecular biology of Gaucher disease. Crit Rev. Biochem. Mol. Biol. 1990, 25, 385–414. [Google Scholar] [CrossRef]
- Bergmann, J.E.; Grabowski, G.A. Posttranslational processing of human lysosomal acid beta-glucosidase: A continuum of defects in Gaucher disease type 1 and type 2 fibroblasts. Am. J. Hum. Genet. 1989, 44, 741–750. [Google Scholar]
- Lieberman, R.L. A Guided Tour of the Structural Biology of Gaucher Disease: Acid-beta-Glucosidase and Saposin C. Enzym. Res. 2011, 2011, 973231. [Google Scholar] [CrossRef]
- Dvir, H.; Harel, M.; McCarthy, A.A.; Toker, L.; Silman, I.; Futerman, A.H.; Sussman, J.L. X-ray structure of human acid-beta-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 2003, 4, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.; Pasmanik-Chor, M.; Borochowitz, Z.; Falik-Zaccai, T.; Heldmann, K.; Carmi, R.; Parvari, R.; Beit-Or, H.; Goldman, B.; Peleg, L.; et al. Prevalence of glucocerebrosidase mutations in the Israeli Ashkenazi Jewish population. Hum. Mutat. 1998, 12, 240–244. [Google Scholar] [CrossRef]
- Charrow, J.; Andersson, H.C.; Kaplan, P.; Kolodny, E.H.; Mistry, P.; Pastores, G.; Rosenbloom, B.E.; Scott, C.R.; Wappner, R.S.; Weinreb, N.J.; et al. The gaucher registry: Demographics and disease characteristics of 1698 patients with gaucher disease. Arch. Intern. Med. 2000, 160, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef]
- Beavan, M.; McNeill, A.; Proukakis, C.; Hughes, D.A.; Mehta, A.; Schapira, A.H. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation-positive cohort. JAMA Neurol. 2015, 72, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Sidransky, E.; Verma, A.; Mixon, T.; Sandberg, G.D.; Wakefield, L.K.; Morrison, A.; Lwin, A.; Colegial, C.; Allman, J.M.; et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol. Genet. Metab. 2004, 82, 192–207. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, E.; Stubblefield, B.K.; McGlinchey, R.P.; McMahon, B.; Ory, D.S.; Sidransky, E. A characterization of Gaucher iPS-derived astrocytes: Potential implications for Parkinson’s disease. Neurobiol. Dis. 2020, 134, 104647. [Google Scholar] [CrossRef]
- Maegawa, G.H.B.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.I.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.R.; et al. Identification and Characterization of Ambroxol as an Enzyme Enhancement Agent for Gaucher Disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef]
- Mata, I.F.; Samii, A.; Schneer, S.H.; Roberts, J.W.; Griffith, A.; Leis, B.C.; Schellenberg, G.D.; Sidransky, E.; Bird, T.D.; Leverenz, J.B.; et al. Glucocerebrosidase gene mutations: A risk factor for Lewy body disorders. Arch. Neurol. 2008, 65, 379–382. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Dinur, T.; Quinn, T.; Sakanaka, K.; Levy, O.; Waters, C.; Fahn, S.; Dorovski, T.; Chung, W.K.; Pauciulo, M.; et al. Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol. 2014, 71, 752–757. [Google Scholar] [CrossRef]
- Anheim, M.; Elbaz, A.; Lesage, S.; Durr, A.; Condroyer, C.; Viallet, F.; Pollak, P.; Bonaiti, B.; Bonaiti-Pellie, C.; Brice, A.; et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 2012, 78, 417–420. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Duran, R.; Hughes, D.A.; Mehta, A.; Schapira, A.H. A clinical and family history study of Parkinson’s disease in heterozygous glucocerebrosidase mutation carriers. J. Neurol. Neurosurg. Psychiatry 2012, 83, 853–854. [Google Scholar] [CrossRef] [PubMed]
- Lwin, A.; Orvisky, E.; Goker-Alpan, O.; LaMarca, M.E.; Sidransky, E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab. 2004, 81, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Schapira, A.H.V. The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem. 2016, 139, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Condroyer, C.; Pollak, P.; Durif, F.; Dupuits, C.; Viallet, F.; Lohmann, E.; Corvol, J.C.; Honore, A.; et al. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. 2011, 20, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Higgins, A.L.; Toffoli, M.; Mullin, S.; Lee, C.Y.; Koletsi, S.; Avenali, M.; Blandini, F.; Schapira, A.H. The remote assessment of parkinsonism supporting the ongoing development of interventions in Gaucher disease. Neurodegener. Dis. Manag. 2021, 11, 451–458. [Google Scholar] [CrossRef]
- Goker-Alpan, O.; Giasson, B.I.; Eblan, M.J.; Nguyen, J.; Hurtig, H.I.; Lee, V.M.Y.; Trojanowski, J.Q.; Sidransky, E. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology 2006, 67, 908–910. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Q.Y.; Zhao, Y.W.; Shu, L.; Guo, J.F.; Xu, Q.; Yan, X.X.; Tang, B.S. Effect of GBA Mutations on Phenotype of Parkinson’s Disease: A Study on Chinese Population and a Meta-Analysis. Parkinson’s Dis. 2015, 2015, 916971. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Bar-Shira, A.; Mirelman, A.; Gurevich, T.; Kedmi, M.; Giladi, N.; Orr-Urtreger, A. LRRK2 and GBA mutations differentially affect the initial presentation of Parkinson disease. Neurogenetics 2010, 11, 121–125. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Giladi, N.; Rozovski, U.; Shifrin, C.; Rosner, S.; Gurevich, T.; Bar-Shira, A.; Orr-Urtreger, A. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008, 70, 2277–2283. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Weil, R.S.; Bresner, C.; Lawton, M.A.; Grosset, K.A.; Tan, M.; Bajaj, N.; Barker, R.A.; Burn, D.J.; Foltynie, T.; et al. Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J. Neurol. Neurosurg. Psychiatry 2018, 89, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Goker-Alpan, O.; Lopez, G.; Vithayathil, J.; Davis, J.; Hallett, M.; Sidransky, E. The spectrum of parkinsonian manifestations associated with glucocerebrosidase mutations. Arch. Neurol. 2008, 65, 1353–1357. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Srulijes, K.; Pflederer, S.; Hauser, A.K.; Schulte, C.; Maetzler, W.; Gasser, T.; Berg, D. GBA-associated Parkinson’s disease: Reduced survival and more rapid progression in a prospective longitudinal study. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 407–411. [Google Scholar] [CrossRef]
- Winder-Rhodes, S.E.; Evans, J.R.; Ban, M.; Mason, S.L.; Williams-Gray, C.H.; Foltynie, T.; Duran, R.; Mencacci, N.E.; Sawcer, S.J.; Barker, R.A. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 2013, 136, 392–399. [Google Scholar] [CrossRef]
- Petrucci, S.; Ginevrino, M.; Trezzi, I.; Monfrini, E.; Ricciardi, L.; Albanese, A.; Avenali, M.; Barone, P.; Bentivoglio, A.R.; Bonifati, V.; et al. GBA-Related Parkinson’s Disease: Dissection of Genotype-Phenotype Correlates in a Large Italian Cohort. Mov. Disord. 2020, 35, 2106–2111. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Caccappolo, E.; Mejia-Santana, H.; Tang, M.; Rosado, L.; Orbe Reilly, M.; Ruiz, D.; Ross, B.; Verbitsky, M.; Kisselev, S.; et al. Cognitive performance of GBA mutation carriers with early-onset PD: The CORE-PD study. Neurology 2012, 78, 1434–1440. [Google Scholar] [CrossRef]
- Brockmann, K.; Srulijes, K.; Hauser, A.K.; Schulte, C.; Csoti, I.; Gasser, T.; Berg, D. GBA-associated PD presents with nonmotor characteristics. Neurology 2011, 77, 276–280. [Google Scholar] [CrossRef]
- Westbroek, W.; Gustafson, A.M.; Sidransky, E. Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol. Med. 2011, 17, 485–493. [Google Scholar] [CrossRef]
- Choi, J.H.; Stubblefield, B.; Cookson, M.R.; Goldin, E.; Velayati, A.; Tayebi, N.; Sidransky, E. Aggregation of α-synuclein in brain samples from subjects with glucocerebrosidase mutations. Mol. Genet. Metab. 2011, 104, 185–188. [Google Scholar] [CrossRef]
- Tayebi, N.; Walker, J.; Stubblefield, B.; Orvisky, E.; LaMarca, M.E.; Wong, K.; Rosenbaum, H.; Schiffmann, R.; Bembi, B.; Sidransky, E. Gaucher disease with parkinsonian manifestations: Does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol. Genet. Metab. 2003, 79, 104–109. [Google Scholar] [CrossRef]
- Nishioka, K.; Ross, O.A.; Vilarino-Guell, C.; Cobb, S.A.; Kachergus, J.M.; Mann, D.M.; Snowden, J.; Richardson, A.M.; Neary, D.; Robinson, C.A.; et al. Glucocerebrosidase mutations in diffuse Lewy body disease. Parkinsonism Relat. Disord. 2011, 17, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Parkkinen, L.; Neumann, J.; O’Sullivan, S.S.; Holton, J.L.; Revesz, T.; Hardy, J.; Lees, A.J. Glucocerebrosidase mutations do not cause increased Lewy body pathology in Parkinson’s disease. Mol. Genet. Metab. 2011, 103, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Reed, X.; Krohn, L.; Heilbron, K.; Bandres-Ciga, S.; Tan, M.; Gibbs, J.R.; Hernandez, D.G.; Kumaran, R.; Langston, R.; et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 2020, 143, 234–248. [Google Scholar] [CrossRef]
- Stoker, T.B.; Camacho, M.; Winder-Rhodes, S.; Liu, G.; Scherzer, C.R.; Foltynie, T.; Barker, R.A.; Williams-Gray, C.H. A common polymorphism in SNCA is associated with accelerated motor decline in GBA-Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2020, 91, 673–674. [Google Scholar] [CrossRef] [PubMed]
- Goker-Alpan, O.; Stubblefield, B.K.; Giasson, B.I.; Sidransky, E. Glucocerebrosidase is present in alpha-synuclein inclusions in Lewy body disorders. Acta Neuropathol. 2010, 120, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Dandana, A.; Ben Khelifa, S.; Chahed, H.; Miled, A.; Ferchichi, S. Gaucher Disease: Clinical, Biological and Therapeutic Aspects. Pathobiology 2016, 83, 13–23. [Google Scholar] [CrossRef]
- Winfield, S.L.; Tayebi, N.; Martin, B.M.; Ginns, E.I.; Sidransky, E. Identification of three additional genes contiguous to the glucocerebrosidase locus on chromosome 1q21: Implications for Gaucher disease. Genome Res. 1997, 7, 1020–1026. [Google Scholar] [CrossRef][Green Version]
- Huh, Y.E.; Chiang, M.S.R.; Locascio, J.J.; Liao, Z.; Liu, G.; Choudhury, K.; Kuras, Y.I.; Tuncali, I.; Videnovic, A.; Hunt, A.L.; et al. Beta-Glucocerebrosidase activity in GBA-linked Parkinson disease: The type of mutation matters. Neurology 2020, 95, e685–e696. [Google Scholar] [CrossRef]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef]
- Smith, L.; Mullin, S.; Schapira, A.H.V. Insights into the structural biology of Gaucher disease. Exp. Neurol. 2017, 298, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Duran, R.; Mencacci, N.E.; Angeli, A.V.; Shoai, M.; Deas, E.; Houlden, H.; Mehta, A.; Hughes, D.; Cox, T.M.; Deegan, P.; et al. The glucocerobrosidase E326K variant predisposes to Parkinson’s disease, but does not cause Gaucher’s disease. Mov. Disord. 2013, 28, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Chabas, A.; Gort, L.; Diaz-Font, A.; Montfort, M.; Santamaria, R.; Cidras, M.; Grinberg, D.; Vilageliu, L. Perinatal lethal phenotype with generalized ichthyosis in a type 2 Gaucher disease patient with the [L444P;E326K]/P182L genotype: Effect of the E326K change in neonatal and classic forms of the disease. Blood Cells Mol. Dis. 2005, 35, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Liou, B.; Grabowski, G.A. Is E326K glucocerebrosidase a polymorphic or pathological variant? Mol. Genet. Metab. 2012, 105, 528–529. [Google Scholar] [CrossRef]
- Berge-Seidl, V.; Pihlstrøm, L.; Maple-Grødem, J.; Forsgren, L.; Linder, J.; Larsen, J.P.; Tysnes, O.-B.; Toft, M. The GBA variant E326K is associated with Parkinson’s disease and explains a genome-wide association signal. Neurosci. Lett. 2017, 658, 48–52. [Google Scholar] [CrossRef]
- den Heijer, J.M.; Cullen, V.C.; Quadri, M.; Schmitz, A.; Hilt, D.C.; Lansbury, P.; Berendse, H.W.; van de Berg, W.D.J.; de Bie, R.M.A.; Boertien, J.M.; et al. A Large-Scale Full GBA1 Gene Screening in Parkinson’s Disease in the Netherlands. Mov. Disord. 2020, 35, 1667–1674. [Google Scholar] [CrossRef]
- Ruskey, J.A.; Greenbaum, L.; Roncière, L.; Alam, A.; Spiegelman, D.; Liong, C.; Levy, O.A.; Waters, C.; Fahn, S.; Marder, K.S.; et al. Increased yield of full GBA sequencing in Ashkenazi Jews with Parkinson’s disease. Eur. J. Med. Genet. 2019, 62, 65–69. [Google Scholar] [CrossRef]
- Davis, M.Y.; Johnson, C.O.; Leverenz, J.B.; Weintraub, D.; Trojanowski, J.Q.; Chen-Plotkin, A.; Van Deerlin, V.M.; Quinn, J.F.; Chung, K.A.; Peterson-Hiller, A.L.; et al. Association of GBA Mutations and the E326K Polymorphism With Motor and Cognitive Progression in Parkinson Disease. JAMA Neurol. 2016, 73, 1217–1224. [Google Scholar] [CrossRef]
- Stoker, T.B.; Camacho, M.; Winder-Rhodes, S.; Liu, G.; Scherzer, C.R.; Foltynie, T.; Evans, J.; Breen, D.P.; Barker, R.A.; Williams-Gray, C.H. Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2020, 91, 695–702. [Google Scholar] [CrossRef]
- Greuel, A.; Trezzi, J.-P.; Glaab, E.; Ruppert, M.C.; Maier, F.; Jäger, C.; Hodak, Z.; Lohmann, K.; Ma, Y.; Eidelberg, D.; et al. GBA Variants in Parkinson’s Disease: Clinical, Metabolomic, and Multimodal Neuroimaging Phenotypes. Mov. Disord. 2020, 35, 2201–2210. [Google Scholar] [CrossRef]
- Mallett, V.; Ross, J.P.; Alcalay, R.N.; Ambalavanan, A.; Sidransky, E.; Dion, P.A.; Rouleau, G.A.; Gan-Or, Z. GBA p.T369M substitution in Parkinson disease: Polymorphism or association? A meta-analysis. Neurology. Genet. 2016, 2, e104. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, B.; Balwani, M.; Bronstein, J.M.; Kolodny, E.; Sathe, S.; Gwosdow, A.R.; Taylor, J.S.; Cole, J.A.; Zimran, A.; Weinreb, N.J. The incidence of Parkinsonism in patients with type 1 Gaucher disease: Data from the ICGG Gaucher Registry. Blood Cells Mol. Dis. 2011, 46, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Aharon-Peretz, J.; Rosenbaum, H.; Gershoni-Baruch, R. Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 2004, 351, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Chetrit, E.B.; Alcalay, R.N.; Steiner-Birmanns, B.; Altarescu, G.; Phillips, M.; Elstein, D.; Zimran, A. Phenotype in patients with Gaucher disease and Parkinson disease. Blood Cells Mol. Dis. 2013, 50, 218–221. [Google Scholar] [CrossRef]
- Fernandes, H.J.R.; Hartfield, E.M.; Christian, H.C.; Emmanoulidou, E.; Zheng, Y.; Booth, H.; Bogetofte, H.; Lang, C.; Ryan, B.J.; Sardi, S.P.; et al. ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons. Stem Cell Rep. 2016, 6, 342–356. [Google Scholar] [CrossRef]
- Alfonso, P.; Rodriguez-Rey, J.C.; Ganan, A.; Perez-Calvo, J.I.; Giralt, M.; Giraldo, P.; Pocovi, M. Expression and functional characterization of mutated glucocerebrosidase alleles causing Gaucher disease in Spanish patients. Blood Cells Mol. Dis. 2004, 32, 218–225. [Google Scholar] [CrossRef]
- Grace, M.E.; Newman, K.M.; Scheinker, V.; Bergfussman, A.; Grabowski, G.A. Analysis of Human Acid Beta-Glucosidase by Site-Directed Mutagenesis and Heterologous Expression. J. Biol. Chem. 1994, 269, 2283–2291. [Google Scholar] [CrossRef]
- Ohashi, T.; Hong, C.M.; Weiler, S.; Tomich, J.M.; Aerts, J.M.; Tager, J.M.; Barranger, J.A. Characterization of human glucocerebrosidase from different mutant alleles. J. Biol. Chem. 1991, 266, 3661–3667. [Google Scholar] [CrossRef]
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.Y.; Whitworth, A.J.; Schapira, A.H. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016, 6, 31380. [Google Scholar] [CrossRef]
- Maor, G.; Rencus-Lazar, S.; Filocamo, M.; Steller, H.; Segal, D.; Horowitz, M. Unfolded protein response in Gaucher disease: From human to Drosophila. Orphanet J. Rare Dis. 2013, 8, 140. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef] [PubMed]
- Ron, I.; Horowitz, M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum. Mol. Genet. 2005, 14, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Bendikov-Bar, I.; Ron, I.; Filocamo, M.; Horowitz, M. Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol. Dis. 2011, 46, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Wegrzynowicz, M.; Rusconi, R.; Deangeli, G.; Di Monte, D.A.; Spillantini, M.G.; Schapira, A.H.V. The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain A J. Neurol. 2017, 140, 2706–2721. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kim, D.; Kim, S.; Kim, S.; Karuppagounder, S.S.; Kwon, S.-H.; Lee, S.; Kam, T.-I.; Lee, S.; Ham, S.; et al. α-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol. Neurodegener. 2018, 13, 1. [Google Scholar] [CrossRef]
- Huang, Y.; Deng, L.; Zhong, Y.; Yi, M. The Association between E326K of GBA and the Risk of Parkinson’s Disease. Parkinson’s Dis. 2018, 2018, 1048084. [Google Scholar] [CrossRef]
- Horowitz, M.; Pasmanik-Chor, M.; Ron, I.; Kolodny, E.H. The enigma of the E326K mutation in acid beta-glucocerebrosidase. Mol. Genet. Metab. 2011, 104, 35–38. [Google Scholar] [CrossRef]
- Montfort, M.; Chabás, A.; Vilageliu, L.; Grinberg, D. Functional analysis of 13 GBA mutant alleles identified in Gaucher disease patients: Pathogenic changes and “modifier” polymorphisms. Hum. Mutat. 2004, 23, 567–575. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Levy, O.A.; Waters, C.C.; Fahn, S.; Ford, B.; Kuo, S.H.; Mazzoni, P.; Pauciulo, M.W.; Nichols, W.C.; Gan-Or, Z.; et al. Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain A J. Neurol. 2015, 138, 2648–2658. [Google Scholar] [CrossRef]
- Grace, M.E.; Ashton-Prolla, P.; Pastores, G.M.; Soni, A.; Desnick, R.J. Non-pseudogene-derived complex acid beta-glucosidase mutations causing mild type 1 and severe type 2 gaucher disease. J. Clin. Investig. 1999, 103, 817–823. [Google Scholar] [CrossRef]
- Malini, E.; Grossi, S.; Deganuto, M.; Rosano, C.; Parini, R.; Dominisini, S.; Cariati, R.; Zampieri, S.; Bembi, B.; Filocamo, M.; et al. Functional analysis of 11 novel GBA alleles. Eur. J. Hum. Genet. EJHG 2014, 22, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Wolf, P.; Chiang, M.S.R.; Helesicova, K.; Zhang, X.K.; Merchant, K.; Hutten, S.J.; Scherzer, C.; Caspell-Garcia, C.; Blauwendraat, C.; et al. Longitudinal Measurements of Glucocerebrosidase activity in Parkinson’s patients. Ann. Clin. Transl. Neurol. 2020, 7, 1816–1830. [Google Scholar] [CrossRef] [PubMed]
- Torralba, M.A.; Pérez-Calvo, J.I.; Pastores, G.M.; Cenarro, A.; Giraldo, P.; Pocoví, M. Identification and Characterization of a Novel Mutation c.1090G>T (G325W) and Nine Common Mutant Alleles Leading to Gaucher Disease in Spanish Patients. Blood Cells Mol. Dis. 2001, 27, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Liou, B.; Kazimierczuk, A.; Zhang, M.; Scott, C.R.; Hegde, R.S.; Grabowski, G.A. Analyses of variant acid beta-glucosidases: Effects of Gaucher disease mutations. J. Biol. Chem. 2006, 281, 4242–4253. [Google Scholar] [CrossRef]
- Gegg, M.E.; Burke, D.; Heales, S.J.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef]
- Gundner, A.L.; Duran-Pacheco, G.; Zimmermann, S.; Ruf, I.; Moors, T.; Baumann, K.; Jagasia, R.; van de Berg, W.D.J.; Kremer, T. Path mediation analysis reveals GBA impacts Lewy body disease status by increasing alpha-synuclein levels. Neurobiol. Dis. 2019, 121, 205–213. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain A J. Neurol. 2014, 137, 834–848. [Google Scholar] [CrossRef]
- Cullen, V.; Sardi, P.; Ng, J.; Xu, Y.H.; Sun, Y.; Tomlinson, J.J.; Kolodziej, P.; Kahn, I.; Saftig, P.; Woulfe, J.; et al. Acid beta-Glucosidase Mutants Linked to Gaucher Disease, Parkinson Disease, and Lewy Body Dementia Alter alpha-Synuclein Processing. Ann. Neurol. 2011, 69, 940–953. [Google Scholar] [CrossRef]
- Fishbein, I.; Kuo, Y.M.; Giasson, B.I.; Nussbaum, R.L. Augmentation of phenotype in a transgenic Parkinson mouse heterozygous for a Gaucher mutation. Brain A J. Neurol. 2014, 137, 3235–3247. [Google Scholar] [CrossRef]
- Sardi, S.P.; Clarke, J.; Kinnecom, C.; Tamsett, T.J.; Li, L.; Stanek, L.M.; Passini, M.A.; Grabowski, G.A.; Schlossmacher, M.G.; Sidman, R.L.; et al. CNS expression of glucocerebrosidase corrects alpha-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc. Natl. Acad. Sci. USA 2011, 108, 12101–12106. [Google Scholar] [CrossRef]
- Xu, Y.H.; Xu, K.; Sun, Y.; Liou, B.; Quinn, B.; Li, R.H.; Xue, L.; Zhang, W.; Setchell, K.D.; Witte, D.; et al. Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Hum. Mol. Genet. 2014, 23, 3943–3957. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.H.; Sun, Y.; Ran, H.; Quinn, B.; Witte, D.; Grabowski, G.A. Accumulation and distribution of alpha-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol. Genet. Metab. 2011, 102, 436–447. [Google Scholar] [CrossRef]
- Ginns, E.I.; Mak, S.K.; Ko, N.; Karlgren, J.; Akbarian, S.; Chou, V.P.; Guo, Y.; Lim, A.; Samuelsson, S.; LaMarca, M.L.; et al. Neuroinflammation and alpha-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol. Genet. Metab. 2014, 111, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Manning-Boğ, A.B.; Schüle, B.; Langston, J.W. Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: A biological link between Gaucher disease and parkinsonism. NeuroToxicology 2009, 30, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Cleeter, M.W.; Chau, K.Y.; Gluck, C.; Mehta, A.; Hughes, D.A.; Duchen, M.; Wood, N.W.; Hardy, J.; Mark Cooper, J.; Schapira, A.H. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 2013, 62, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Abul Khair, S.B.; Dhanushkodi, N.R.; Ardah, M.T.; Chen, W.; Yang, Y.; Haque, M.E. Silencing of Glucocerebrosidase Gene in Drosophila Enhances the Aggregation of Parkinson’s Disease Associated alpha-Synuclein Mutant A53T and Affects Locomotor Activity. Front. Neurosci. 2018, 12, 81. [Google Scholar] [CrossRef]
- Bae, E.-J.; Yang, N.Y.; Lee, C.; Lee, H.-J.; Kim, S.; Sardi, S.P.; Lee, S.-J. Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and α-synuclein aggregation. Exp. Mol. Med. 2015, 47, e153. [Google Scholar] [CrossRef]
- Jo, J.; Yang, L.; Tran, H.D.; Yu, W.; Sun, A.X.; Chang, Y.Y.; Jung, B.C.; Lee, S.J.; Saw, T.Y.; Xiao, B.; et al. Lewy Body-like Inclusions in Human Midbrain Organoids Carrying Glucocerebrosidase and alpha-Synuclein Mutations. Ann. Neurol. 2021, 90, 490–505. [Google Scholar] [CrossRef]
- Sardi, P.S.; Shihabuddin, L.S.; Sidman, R.L.; Cheng, S.H. Augmenting CNS Glucocerebrosidase Activity as a Therapeutic Strategy for Parkinsonism and Other Gaucher-Related Synucleinopathies. Mol. Ther. 2013, 21, S14–S15. [Google Scholar] [CrossRef]
- Woodard, C.M.; Campos, B.A.; Kuo, S.H.; Nirenberg, M.J.; Nestor, M.W.; Zimmer, M.; Mosharov, E.V.; Sulzer, D.; Zhou, H.Y.; Paull, D.; et al. iPSC-Derived Dopamine Neurons Reveal Differences between Monozygotic Twins Discordant for Parkinson’s Disease. Cell Rep. 2014, 9, 1173–1182. [Google Scholar] [CrossRef]
- Yang, J.; Hertz, E.; Zhang, X.; Leinartaite, L.; Lundius, E.G.; Li, J.; Svenningsson, P. Overexpression of alpha-synuclein simultaneously increases glutamate NMDA receptor phosphorylation and reduces glucocerebrosidase activity. Neurosci. Lett. 2016, 611, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Sedor, S.; McGeary, I.; Cornblath, E.J.; Peng, C.; Riddle, D.M.; Li, H.L.; Zhang, B.; Brown, H.J.; Olufemi, M.F.; et al. Glucocerebrosidase Activity Modulates Neuronal Susceptibility to Pathological alpha-Synuclein Insult. Neuron 2020, 105, 822–836.e7. [Google Scholar] [CrossRef]
- Dermentzaki, G.; Dimitriou, E.; Xilouri, M.; Michelakakis, H.; Stefanis, L. Loss of β-Glucocerebrosidase Activity Does Not Affect Alpha-Synuclein Levels or Lysosomal Function in Neuronal Cells. PLoS ONE 2013, 8, e60674. [Google Scholar] [CrossRef]
- Huebecker, M.; Moloney, E.B.; van der Spoel, A.C.; Priestman, D.A.; Isacson, O.; Hallett, P.J.; Platt, F.M. Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 40. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hallett, P.; Isacson, O. Progressive decline of glucocerebrosidase in aging and Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 433–438. [Google Scholar] [CrossRef]
- Moors, T.E.; Paciotti, S.; Ingrassia, A.; Quadri, M.; Breedveld, G.; Tasegian, A.; Chiasserini, D.; Eusebi, P.; Duran-Pacheco, G.; Kremer, T.; et al. Characterization of Brain Lysosomal Activities in GBA-Related and Sporadic Parkinson’s Disease and Dementia with Lewy Bodies. Mol. Neurobiol. 2019, 56, 1344–1355. [Google Scholar] [CrossRef]
- Chiasserini, D.; Paciotti, S.; Eusebi, P.; Persichetti, E.; Tasegian, A.; Kurzawa-Akanbi, M.; Chinnery, P.F.; Morris, C.M.; Calabresi, P.; Parnetti, L.; et al. Selective loss of glucocerebrosidase activity in sporadic Parkinson’s disease and dementia with Lewy bodies. Mol. Neurodegener. 2015, 10, 15. [Google Scholar] [CrossRef]
- Parnetti, L.; Chiasserini, D.; Persichetti, E.; Eusebi, P.; Varghese, S.; Qureshi, M.M.; Dardis, A.; Deganuto, M.; De Carlo, C.; Castrioto, A.; et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1019–1027. [Google Scholar] [CrossRef]
- Atashrazm, F.; Hammond, D.; Perera, G.; Dobson-Stone, C.; Mueller, N.; Pickford, R.; Kim, W.S.; Kwok, J.B.; Lewis, S.J.G.; Halliday, G.M.; et al. Reduced glucocerebrosidase activity in monocytes from patients with Parkinson’s disease. Sci. Rep. 2018, 8, 15446. [Google Scholar] [CrossRef]
- Wang, F.; Song, W.; Brancati, G.; Segatori, L. Inhibition of endoplasmic reticulum-associated degradation rescues native folding in loss of function protein misfolding diseases. J. Biol. Chem. 2011, 286, 43454–43464. [Google Scholar] [CrossRef] [PubMed]
- Braunstein, H.; Maor, G.; Chicco, G.; Filocamo, M.; Zimran, A.; Horowitz, M. UPR activation and CHOP mediated induction of GBA1 transcription in Gaucher disease. Blood Cells Mol. Dis. 2018, 68, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Kurzawa-Akanbi, M.; Hanson, P.S.; Blain, P.G.; Lett, D.J.; McKeith, I.G.; Chinnery, P.F.; Morris, C.M. Glucocerebrosidase Mutations alter the endoplasmic reticulum and lysosomes in Lewy body disease. J. Neurochem. 2012, 123, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Korkotian, E.; Schwarz, A.; Pelled, D.; Schwarzmann, G.; Segal, M.; Futerman, A.H. Elevation of intracellular glucosylceramide levels results in an increase in endoplasmic reticulum density and in functional calcium stores in cultured neurons. J. Biol. Chem. 1999, 274, 21673–21678. [Google Scholar] [CrossRef]
- Stojkovska, I.; Wani, W.Y.; Zunke, F.; Belur, N.R.; Pavlenko, E.A.; Mwenda, N.; Sharma, K.; Francelle, L.; Mazzulli, J.R. Rescue of α-synuclein aggregation in Parkinson’s patient neurons by synergistic enhancement of ER proteostasis and protein trafficking. Neuron 2022, 110, 436–451.e411. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Yang, S.Y.; Beavan, M.; Chau, K.Y.; Taanman, J.W.; Schapira, A.H. A Human Neural Crest Stem Cell-Derived Dopaminergic Neuronal Model Recapitulates Biochemical Abnormalities in GBA1 Mutation Carriers. Stem Cell Rep. 2017, 8, 728–742. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Graham, A.-R.; Brown, E.; McLean, J.R.; Hayes, M.A.; Beagan, J.; Izen, S.C.; et al. Sustained Systemic Glucocerebrosidase Inhibition Induces Brain α-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice. Antioxid. Redox Signal. 2015, 23, 550–564. [Google Scholar] [CrossRef]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.; Duchen, M.R. Mitochondria and quality control defects in a mouse model of Gaucher disease—Links to Parkinson’s disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef]
- Du, T.T.; Wang, L.; Duan, C.L.; Lu, L.L.; Zhang, J.L.; Gao, G.; Qiu, X.B.; Wang, X.M.; Yang, H. GBA deficiency promotes SNCA/alpha-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015, 11, 1803–1820. [Google Scholar] [CrossRef]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Doherty, M.K.; Whitfield, P.D.; Schapira, A.H.V. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: Relevance to Parkinson disease. Hum. Mol. Genet. 2016, 25, 3432–3445. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Gegg, M.; Chau, D.; Schapira, A. Glucocerebrosidase activity, cathepsin D and monomeric alpha-synuclein interactions in a stem cell derived neuronal model of a PD associated GBA1 mutation. Neurobiol. Dis. 2020, 134, 104620. [Google Scholar] [CrossRef] [PubMed]
- Klucken, J.; Poehler, A.-M.; Ebrahimi-Fakhari, D.; Schneider, J.; Nuber, S.; Rockenstein, E.; Schlötzer-Schrehardt, U.; Hyman, B.T.; McLean, P.J.; Masliah, E.; et al. Alpha-synuclein aggregation involves a bafilomycin A(1)-sensitive autophagy pathway. Autophagy 2012, 8, 754–766. [Google Scholar] [CrossRef]
- Kuo, S.H.; Tasset, I.; Cheng, M.M.; Diaz, A.; Pan, M.K.; Lieberman, O.J.; Hutten, S.J.; Alcalay, R.N.; Kim, S.; Ximénez-Embún, P.; et al. Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci. Adv. 2022, 8, eabm6393. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.J.; Yang, N.Y.; Song, M.; Lee, C.S.; Lee, J.S.; Jung, B.C.; Lee, H.J.; Kim, S.; Masliah, E.; Sardi, S.P.; et al. Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat. Commun. 2014, 5, 4755. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Verona, G.; Schapira, A.H.V. Glucocerebrosidase deficiency promotes release of alpha-synuclein fibrils from cultured neurons. Hum. Mol. Genet. 2020, 29, 1716–1728. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.A.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef]
- Cerri, S.; Ghezzi, C.; Ongari, G.; Croce, S.; Avenali, M.; Zangaglia, R.; Di Monte, D.A.; Valente, E.M.; Blandini, F. GBA Mutations Influence the Release and Pathological Effects of Small Extracellular Vesicles from Fibroblasts of Patients with Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 2215. [Google Scholar] [CrossRef]
- Jewett, K.A.; Thomas, R.E.; Phan, C.Q.; Lin, B.; Milstein, G.; Yu, S.; Bettcher, L.F.; Neto, F.C.; Djukovic, D.; Raftery, D.; et al. Glucocerebrosidase reduces the spread of protein aggregation in a Drosophila melanogaster model of neurodegeneration by regulating proteins trafficked by extracellular vesicles. PLoS Genet. 2021, 17, e1008859. [Google Scholar] [CrossRef]
- Thomas, R.E.; Vincow, E.S.; Merrihew, G.E.; MacCoss, M.J.; Davis, M.Y.; Pallanck, L.J. Glucocerebrosidase deficiency promotes protein aggregation through dysregulation of extracellular vesicles. PLoS Genet. 2018, 14, e1007694. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Wegrzynowicz, M.; Harrison, I.F.; Verona, G.; Bellotti, V.; Spillantini, M.G.; Schapira, A.H.V. L444P Gba1 mutation increases formation and spread of α-synuclein deposits in mice injected with mouse α-synuclein pre-formed fibrils. PLoS ONE 2020, 15, e0238075. [Google Scholar] [CrossRef] [PubMed]
- Farmer, B.C.; Walsh, A.E.; Kluemper, J.C.; Johnson, L.A. Lipid Droplets in Neurodegenerative Disorders. Front. Neurosci. 2020, 14, 742. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.; Baldwin, A.; Gilbert, R.; Muller, H. Glucocerebrosidase rescues alpha-synuclein from amyloid formation. bioRxiv 2018. [Google Scholar] [CrossRef]
- Galvagnion, C.; Brown, J.W.P.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070. [Google Scholar] [CrossRef]
- Farfel-Becker, T.; Vitner, E.B.; Kelly, S.L.; Bame, J.R.; Duan, J.; Shinder, V.; Merrill, A.H., Jr.; Dobrenis, K.; Futerman, A.H. Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration. Hum. Mol. Genet. 2014, 23, 843–854. [Google Scholar] [CrossRef]
- Melo, F.; Caballero, L.; Zamorano, E.; Ventura, N.; Navarro, C.; Doll, I.; Zamorano, P.; Cornejo, A. The Cytotoxic Effect of alpha-Synuclein Aggregates. Chemphyschem 2021, 22, 526–532. [Google Scholar] [CrossRef]
- Fabelo, N.; Martin, V.; Santpere, G.; Marin, R.; Torrent, L.; Ferrer, I.; Diaz, M. Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol. Med. 2011, 17, 1107–1118. [Google Scholar] [CrossRef]
- Sardi, S.P.; Viel, C.; Clarke, J.; Treleaven, C.M.; Richards, A.M.; Park, H.; Olszewski, M.A.; Dodge, J.C.; Marshall, J.; Makino, E.; et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. USA 2017, 114, 2699–2704. [Google Scholar] [CrossRef]
- Nagata, M.; Izumi, Y.; Ishikawa, E.; Kiyotake, R.; Doi, R.; Iwai, S.; Omahdi, Z.; Yamaji, T.; Miyamoto, T.; Bamba, T.; et al. Intracellular metabolite β-glucosylceramide is an endogenous Mincle ligand possessing immunostimulatory activity. Proc. Natl. Acad. Sci. USA 2017, 114, E3285–E3294. [Google Scholar] [CrossRef]
- Orvisky, E.; Park, J.K.; LaMarca, M.E.; Ginns, E.I.; Martin, B.M.; Tayebi, N.; Sidransky, E. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: Correlation with phenotype and genotype. Mol. Genet. Metab. 2002, 76, 262–270. [Google Scholar] [CrossRef]
- Hallett, P.J.; Huebecker, M.; Brekk, O.R.; Moloney, E.B.; Rocha, E.M.; Priestman, D.A.; Platt, F.M.; Isacson, O. Glycosphingolipid levels and glucocerebrosidase activity are altered in normal aging of the mouse brain. Neurobiol. Aging 2018, 67, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Galvagnion, C.; Cerri, S.; Schapira, A.H.V.; Blandini, F.; Di Monte, D.A. Sphingolipid changes in Parkinson L444P GBA mutation fibroblasts promote α-synuclein aggregation. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Hein, L.K.; Duplock, S.; Hopwood, J.J.; Fuller, M. Lipid composition of microdomains is altered in a cell model of Gaucher disease. J. Lipid Res. 2008, 49, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Varela, A.R.; Ventura, A.E.; Carreira, A.C.; Fedorov, A.; Futerman, A.H.; Prieto, M.; Silva, L.C. Pathological levels of glucosylceramide change the biophysical properties of artificial and cell membranes. Phys. Chem. Chem. Phys. 2016, 19, 340–346. [Google Scholar] [CrossRef]
- Gegg, M.E.; Sweet, L.; Wang, B.H.; Shihabuddin, L.S.; Sardi, S.P.; Schapira, A.H. No evidence for substrate accumulation in Parkinson brains with GBA mutations. Mov. Disord. 2015, 30, 1085–1089. [Google Scholar] [CrossRef]
- Clark, L.N.; Chan, R.; Cheng, R.; Liu, X.; Park, N.; Parmalee, N.; Kisselev, S.; Cortes, E.; Torres, P.A.; Pastores, G.M.; et al. Gene-wise association of variants in four lysosomal storage disorder genes in neuropathologically confirmed Lewy body disease. PLoS ONE 2015, 10, e0125204. [Google Scholar] [CrossRef]
- Nilsson, O.; Svennerholm, L. Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease. J. Neurochem. 1982, 39, 709–718. [Google Scholar] [CrossRef]
- Taguchi, Y.V.; Liu, J.; Ruan, J.; Pacheco, J.; Zhang, X.; Abbasi, J.; Keutzer, J.; Mistry, P.K.; Chandra, S.S. Glucosylsphingosine Promotes alpha-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J. Neurosci. 2017, 37, 9617–9631. [Google Scholar] [CrossRef]
- Paul, A.; Jacoby, G.; Laor Bar-Yosef, D.; Beck, R.; Gazit, E.; Segal, D. Glucosylceramide Associated with Gaucher Disease Forms Amyloid-like Twisted Ribbon Fibrils That Induce alpha-Synuclein Aggregation. ACS Nano 2021, 15, 11854–11868. [Google Scholar] [CrossRef]
- Zunke, F.; Moise, A.C.; Belur, N.R.; Gelyana, E.; Stojkovska, I.; Dzaferbegovic, H.; Toker, N.J.; Jeon, S.; Fredriksen, K.; Mazzulli, J.R. Reversible Conformational Conversion of α-Synuclein into Toxic Assemblies by Glucosylceramide. Neuron 2018, 97, 92–107.e10. [Google Scholar] [CrossRef] [PubMed]
- Glajch, K.E.; Moors, T.E.; Chen, Y.; Bechade, P.A.; Nam, A.Y.; Rajsombath, M.M.; McCaffery, T.D.; Dettmer, U.; Weihofen, A.; Hirst, W.D.; et al. Wild-type GBA1 increases the alpha-synuclein tetramer-monomer ratio, reduces lipid-rich aggregates, and attenuates motor and cognitive deficits in mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2103425118. [Google Scholar] [CrossRef] [PubMed]
- Guedes, L.C.; Chan, R.B.; Gomes, M.A.; Conceicao, V.A.; Machado, R.B.; Soares, T.; Xu, Y.; Gaspar, P.; Carrico, J.A.; Alcalay, R.N.; et al. Serum lipid alterations in GBA-associated Parkinson’s disease. Parkinsonism Relat. Disord. 2017, 44, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Moloney, E.B.; Moskites, A.; Ferrari, E.J.; Isacson, O.; Hallett, P.J. The glycoprotein GPNMB is selectively elevated in the substantia nigra of Parkinson’s disease patients and increases after lysosomal stress. Neurobiol. Dis. 2018, 120, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, V.; Liu, J.; Yang, R.; Lin, H.; Lischuk, A.; Pastores, G.; Zhang, X.; Chuang, W.L.; Mistry, P.K. Validating glycoprotein non-metastatic melanoma B (gpNMB, osteoactivin), a new biomarker of Gaucher disease. Blood Cells Mol. Dis. 2018, 68, 47–53. [Google Scholar] [CrossRef]
- Schapira, A.H.; Gegg, M. Mitochondrial contribution to Parkinson’s disease pathogenesis. Parkinsons Dis. 2011, 2011, 159160. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Kinghorn, K.J.; Grönke, S.; Castillo-Quan, J.I.; Woodling, N.S.; Li, L.; Sirka, E.; Gegg, M.; Mills, K.; Hardy, J.; Bjedov, I.; et al. A Drosophila Model of Neuronopathic Gaucher Disease Demonstrates Lysosomal-Autophagic Defects and Altered mTOR Signalling and Is Functionally Rescued by Rapamycin. J. Neurosci. 2016, 36, 11654–11670. [Google Scholar] [CrossRef]
- Schöndorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.J.; Myer, B.J.; Khokher, A.M.; Rushton, N.; Cox, T.M. Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: Increased release of interleukin-6 and interleukin-10. QJM Mon. J. Assoc. Physicians 1997, 90, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Mistry, P.K.; Liu, J.; Yang, M.; Nottoli, T.; McGrath, J.; Jain, D.; Zhang, K.; Keutzer, J.; Chuang, W.L.; Mehal, W.Z.; et al. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc. Natl. Acad. Sci. USA 2010, 107, 19473–19478. [Google Scholar] [CrossRef]
- Vitner, E.B.; Salomon, R.; Farfel-Becker, T.; Meshcheriakova, A.; Ali, M.; Klein, A.D.; Platt, F.M.; Cox, T.M.; Futerman, A.H. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat. Med. 2014, 20, 204–208. [Google Scholar] [CrossRef]
- Keatinge, M.; Bui, H.; Menke, A.; Chen, Y.C.; Sokol, A.M.; Bai, Q.; Ellett, F.; Da Costa, M.; Burke, D.; Gegg, M.; et al. Glucocerebrosidase 1 deficient Danio rerio mirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum. Mol. Genet. 2015, 24, 6640–6652. [Google Scholar] [CrossRef]
- Vitner, E.B.; Farfel-Becker, T.; Eilam, R.; Biton, I.; Futerman, A.H. Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain A J. Neurol. 2012, 135, 1724–1735. [Google Scholar] [CrossRef]
- Enquist, I.B.; Lo Bianco, C.; Ooka, A.; Nilsson, E.; Mansson, J.E.; Ehinger, M.; Richter, J.; Brady, R.O.; Kirik, D.; Karlsson, S. Murine models of acute neuronopathic Gaucher disease. Proc. Natl. Acad. Sci. USA 2007, 104, 17483–17488. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Couch, Y.; Richardson, J.; Cooper, J.M.; Wood, M.J. Alpha-synuclein release by neurons activates the inflammatory response in a microglial cell line. Neurosci. Res. 2011, 69, 337–342. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Lee, E.J.; Woo, M.S.; Moon, P.G.; Baek, M.C.; Choi, I.Y.; Kim, W.K.; Junn, E.; Kim, H.S. Alpha-synuclein activates microglia by inducing the expressions of matrix metalloproteinases and the subsequent activation of protease-activated receptor-1. J. Immunol. 2010, 185, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef]
- Thome, A.D.; Harms, A.S.; Volpicelli-Daley, L.A.; Standaert, D.G. microRNA-155 Regulates Alpha-Synuclein-Induced Inflammatory Responses in Models of Parkinson Disease. J. Neurosci. 2016, 36, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, F.; Cerf, L.; Dehay, B.; Ramos-Gonzalez, P.; De Giorgi, F.; Bourdenx, M.; Bessede, A.; Obeso, J.A.; Matute, C.; Ichas, F.; et al. In vitro alpha-synuclein neurotoxicity and spreading among neurons and astrocytes using Lewy body extracts from Parkinson disease brains. Neurobiol. Dis. 2017, 103, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Loria, F.; Vargas, J.Y.; Bousset, L.; Syan, S.; Salles, A.; Melki, R.; Zurzolo, C. Alpha-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol. 2017, 134, 789–808. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.G.; Eblan, M.J.; Gutti, U.; Hruska, K.S.; Stubblefield, B.K.; Goker-Alpan, O.; LaMarca, M.E.; Sidransky, E. Glucocerebrosidase mutations in Chinese subjects from Taiwan with sporadic Parkinson disease. Mol. Genet. Metab. 2007, 91, 195–200. [Google Scholar] [CrossRef]
- Angeli, A.; Mencacci, N.E.; Duran, R.; Aviles-Olmos, I.; Kefalopoulou, Z.; Candelario, J.; Rusbridge, S.; Foley, J.; Pradhan, P.; Jahanshahi, M.; et al. Genotype and phenotype in Parkinson’s disease: Lessons in heterogeneity from deep brain stimulation. Mov. Disord. 2013, 28, 1370–1375. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, E.; Deroma, L.; Bembi, B.; Deegan, P.; Hollak, C.; Weinreb, N.J.; Cox, T.M. Enzyme replacement and substrate reduction therapy for Gaucher disease. Cochrane Database Syst. Rev. 2015, 2015, CD010324. [Google Scholar] [CrossRef]
- Chauhan, A.; Tikoo, A.; Kapur, A.K.; Singh, M. The taming of the cell penetrating domain of the HIV Tat: Myths and realities. J. Control. Release Off. J. Control. Release Soc. 2007, 117, 148–162. [Google Scholar] [CrossRef]
- Gillmeister, M.P.; Betenbaugh, M.J.; Fishman, P.S. Cellular trafficking and photochemical internalization of cell penetrating peptide linked cargo proteins: A dual fluorescent labeling study. Bioconjug. Chem. 2011, 22, 556–566. [Google Scholar] [CrossRef]
- Fu, A.; Wang, Y.; Zhan, L.; Zhou, R. Targeted delivery of proteins into the central nervous system mediated by rabies virus glycoprotein-derived peptide. Pharm. Res. 2012, 29, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Zhang, M.; Gao, F.; Xu, X.; Chen, Z. A novel peptide delivers plasmids across blood-brain barrier into neuronal cells as a single-component transfer vector. PLoS ONE 2013, 8, e59642. [Google Scholar] [CrossRef] [PubMed]
- Gramlich, P.A.; Westbroek, W.; Feldman, R.A.; Awad, O.; Mello, N.; Remington, M.P.; Sun, Y.; Zhang, W.; Sidransky, E.; Betenbaugh, M.J.; et al. A peptide-linked recombinant glucocerebrosidase for targeted neuronal delivery: Design, production, and assessment. J. Biotechnol. 2016, 221, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ysselstein, D.; Young, T.J.; Nguyen, M.; Padmanabhan, S.; Hirst, W.D.; Dzamko, N.; Krainc, D. Evaluation of Strategies for Measuring Lysosomal Glucocerebrosidase Activity. Mov. Disord. 2021, 36, 2719–2730. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Yun, S.P.; Lee, S.; Umanah, G.E.; Bandaru, V.V.R.; Yin, X.; Rhee, P.; Karuppagounder, S.S.; Kwon, S.-H.; Lee, H.; et al. GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers. Proc. Natl. Acad. Sci. USA 2018, 115, 798–803. [Google Scholar] [CrossRef]
- Peterschmitt, M.J.; Saiki, H.; Hatano, T.; Gasser, T.; Isaacson, S.H.; Gaemers, S.J.M.; Minini, P.; Saubadu, S.; Sharma, J.; Walbillic, S.; et al. Safety, Pharmacokinetics, and Pharmacodynamics of Oral Venglustat in Patients with Parkinson’s Disease and a GBA Mutation: Results from Part 1 of the Randomized, Double-Blinded, Placebo-Controlled MOVES-PD Trial. J. Parkinson’s Dis. 2022, 12, 557–570. [Google Scholar] [CrossRef]
- Hudry, E.; Vandenberghe, L.H. Therapeutic AAV Gene Transfer to the Nervous System: A Clinical Reality. Neuron 2019, 101, 839–862. [Google Scholar] [CrossRef]
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Brown, E.; Hayes, M.A.; Beagan, J.; McLean, J.R.; Izen, S.C.; Perez-Torres, E.; et al. Glucocerebrosidase gene therapy prevents alpha-synucleinopathy of midbrain dopamine neurons. Neurobiol. Dis. 2015, 82, 495–503. [Google Scholar] [CrossRef]
- Sucunza, D.; Rico, A.J.; Roda, E.; Collantes, M.; González-Aseguinolaza, G.; Rodríguez-Pérez, A.I.; Peñuelas, I.; Vázquez, A.; Labandeira-García, J.L.; Broccoli, V.; et al. Glucocerebrosidase Gene Therapy Induces Alpha-Synuclein Clearance and Neuroprotection of Midbrain Dopaminergic Neurons in Mice and Macaques. Int. J. Mol. Sci. 2021, 22, 4825. [Google Scholar] [CrossRef]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hallqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the Treatment of Patients with Parkinson Disease with and without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial. JAMA Neurol. 2020, 77, 427–434. [Google Scholar] [CrossRef]
- Zimran, A.; Altarescu, G.; Elstein, D. Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood Cells Mol. Dis. 2013, 50, 134–137. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Magalhaes, J.; Shen, C.; Chau, K.Y.; Hughes, D.; Mehta, A.; Foltynie, T.; Cooper, J.M.; Abramov, A.Y.; Gegg, M.; et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 2014, 137, 1481–1495. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Schapira, A.H. Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci. Rep. 2018, 8, 1385. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Ko, W.K.; Li, Q.; Bezard, E.; Schapira, A.H. Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse 2017, 71, e21967. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Daly, L.; Bezard, E.; Schapira, A.H. Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann. Neurol. 2016, 80, 766–775. [Google Scholar] [CrossRef] [PubMed]
- Maor, G.; Cabasso, O.; Krivoruk, O.; Rodriguez, J.; Steller, H.; Segal, D.; Horowitz, M. The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum. Mol. Genet. 2016, 25, 2712–2727. [Google Scholar] [CrossRef]
- Aflaki, E.; Borger, D.K.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.J.; Westbroek, W.; Zheng, W.; Sullivan, P.; et al. A New Glucocerebrosidase Chaperone Reduces α-Synuclein and Glycolipid Levels in iPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef]
- Bendikov-Bar, I.; Maor, G.; Filocamo, M.; Horowitz, M. Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis. 2013, 50, 141–145. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Zunke, F.; Tsunemi, T.; Toker, N.J.; Jeon, S.; Burbulla, L.F.; Patnaik, S.; Sidransky, E.; Marugan, J.J.; Sue, C.M.; et al. Activation of β-Glucocerebrosidase Reduces Pathological α-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 2016, 36, 7693–7706. [Google Scholar] [CrossRef]
- Shanmuganathan, M.; Britz-McKibbin, P. Inhibitor screening of pharmacological chaperones for lysosomal beta-glucocerebrosidase by capillary electrophoresis. Anal. Bioanal. Chem. 2011, 399, 2843–2853. [Google Scholar] [CrossRef]
- Patnaik, S.; Zheng, W.; Choi, J.H.; Motabar, O.; Southall, N.; Westbroek, W.; Lea, W.A.; Velayati, A.; Goldin, E.; Sidransky, E.; et al. Discovery, structure-activity relationship, and biological evaluation of noninhibitory small molecule chaperones of glucocerebrosidase. J. Med. Chem. 2012, 55, 5734–5748. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Shimoda, M.; Ito, K.; Hanai, S.; Aizawa, H.; Kato, T.; Kawasaki, K.; Yamaguchi, T.; Ryoo, H.D.; Goto-Inoue, N.; et al. Expression of Human Gaucher Disease Gene GBA Generates Neurodevelopmental Defects and ER Stress in Drosophila Eye. PLoS ONE 2013, 8, e69147, Erratum in PLoS ONE 2015, 10, e0135619. [Google Scholar] [CrossRef] [PubMed]
- Richter, F.; Fleming, S.M.; Watson, M.; Lemesre, V.; Pellegrino, L.; Ranes, B.; Zhu, C.; Mortazavi, F.; Mulligan, C.K.; Sioshansi, P.C.; et al. A GCase chaperone improves motor function in a mouse model of synucleinopathy. Neurotherapeutics 2014, 11, 840–856. [Google Scholar] [CrossRef] [PubMed]
- Morabito, G.; Giannelli, S.G.; Ordazzo, G.; Bido, S.; Castoldi, V.; Indrigo, M.; Cabassi, T.; Cattaneo, S.; Luoni, M.; Cancellieri, C.; et al. AAV-PHP.B-Mediated Global-Scale Expression in the Mouse Nervous System Enables GBA1 Gene Therapy for Wide Protection from Synucleinopathy. Mol. Ther. 2017, 25, 2727–2742. [Google Scholar] [CrossRef]
- den Heijer, J.M.; Kruithof, A.C.; van Amerongen, G.; de Kam, M.L.; Thijssen, E.; Grievink, H.W.; Moerland, M.; Walker, M.; Been, K.; Skerlj, R.; et al. A randomized single and multiple ascending dose study in healthy volunteers of LTI-291, a centrally penetrant glucocerebrosidase activator. Br. J. Clin. Pharmacol. 2021, 87, 3561–3573. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yang, C.; Chen, M.; Ye, D.Y.; Lonser, R.R.; Brady, R.O.; Zhuang, Z. Histone deacetylase inhibitors prevent the degradation and restore the activity of glucocerebrosidase in Gaucher disease. Proc. Natl. Acad. Sci. USA 2011, 108, 21200–21205. [Google Scholar] [CrossRef]
- Yang, C.; Rahimpour, S.; Lu, J.; Pacak, K.; Ikejiri, B.; Brady, R.O.; Zhuang, Z. Histone deacetylase inhibitors increase glucocerebrosidase activity in Gaucher disease by modulation of molecular chaperones. Proc. Natl. Acad. Sci. USA 2013, 110, 966–971. [Google Scholar] [CrossRef]
- Jung, O.; Patnaik, S.; Marugan, J.; Sidransky, E.; Westbroek, W. Progress and potential of non-inhibitory small molecule chaperones for the treatment of Gaucher disease and its implications for Parkinson disease. Expert Rev. Proteom. 2016, 13, 471–479. [Google Scholar] [CrossRef]
- Bendikov-Bar, I.; Maor, G.; Horowitz, M. Processing and maturation of human glucocerebrosidase. In Advances in Gaucher Disease: Basic and Clinical Perspectives; Future Medicine Ltd.: London, UK, 2013; pp. 140–157. [Google Scholar] [CrossRef]
- Khanna, R.; Benjamin, E.R.; Pellegrino, L.; Schilling, A.; Rigat, B.A.; Soska, R.; Nafar, H.; Ranes, B.E.; Feng, J.; Lun, Y.; et al. The pharmacological chaperone isofagomine increases the activity of the Gaucher disease L444P mutant form of beta-glucosidase. FEBS J. 2010, 277, 1618–1638. [Google Scholar] [CrossRef]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef]
- Decressac, M.; Björklund, A. mTOR inhibition alleviates L-DOPA-induced dyskinesia in parkinsonian rats. J. Parkinson’s Dis. 2013, 3, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Fog, C.K.; Zago, P.; Malini, E.; Solanko, L.M.; Peruzzo, P.; Bornaes, C.; Magnoni, R.; Mehmedbasic, A.; Petersen, N.H.T.; Bembi, B.; et al. The heat shock protein amplifier arimoclomol improves refolding, maturation and lysosomal activity of glucocerebrosidase. EBioMedicine 2018, 38, 142–153. [Google Scholar] [CrossRef] [PubMed]


| Mutation | Penetrance of Mutation | Location of Mutation | Effect on GCase | GD | GBA-PD | References |
|---|---|---|---|---|---|---|
| N370S | 0.08–71.8% | Interface of domains II and III | Loss of GCase activity Activation of the UPR Alpha-synuclein pathology | Generally mild, non-neuronopathic GD | Lower disease penetrance and a milder clinical phenotype | [5,6,7,40,47,52,73,80,83,84,85,86,87,88,89,90,91,92,93] |
| L444P | 0.06–18.8% | Domain II | Loss of GCase activity Activation of the UPR Alpha-synuclein pathology | Generally severe, neuronopathic GD | Higher disease penetrance and a worse clinical phenotype | [5,6,7,40,47,52,73,80,83,87,88,89,90,91,92,94,95,96] |
| E326K | 2.8–3.88% | Surface of domain III | Reduces GCase activity to a lesser extent than GD-causing mutations | No clinical manifestation | Worse clinical phenotype | [44,73,74,75,79,80,93,97,98,99,100,101,102,103,104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, L.; Schapira, A.H.V. GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells 2022, 11, 1261. https://doi.org/10.3390/cells11081261
Smith L, Schapira AHV. GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells. 2022; 11(8):1261. https://doi.org/10.3390/cells11081261
Chicago/Turabian StyleSmith, Laura, and Anthony H. V. Schapira. 2022. "GBA Variants and Parkinson Disease: Mechanisms and Treatments" Cells 11, no. 8: 1261. https://doi.org/10.3390/cells11081261
APA StyleSmith, L., & Schapira, A. H. V. (2022). GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells, 11(8), 1261. https://doi.org/10.3390/cells11081261