
Global PIEZO1 Gain-of-Function Mutation Causes Cardiac Hypertrophy and Fibrosis in Mice

 , , , ,
, , , ,  and
and
Abstract
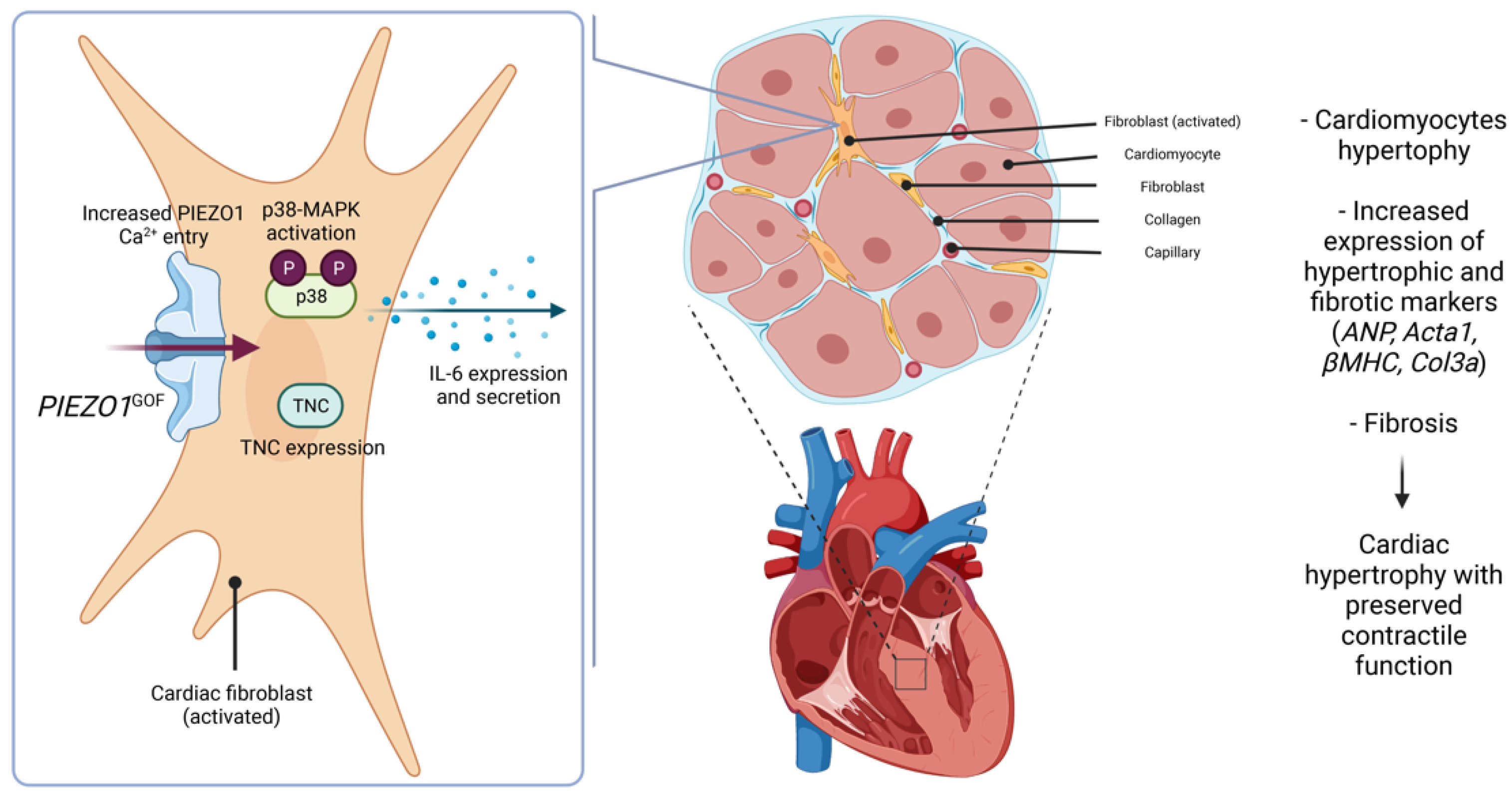
:1. Introduction
2. Materials and Methods
2.1. Genetically Modified Mice
2.2. Echocardiography
2.3. Blood Pressure Measurements
2.4. Animals and Tissue Harvest
2.5. Tissue Processing and Immunohistochemistry
2.5.1. Sectioning of Heart Muscle
2.5.2. In Situ Determination of Heart Cross-Sectional Area and Fibrosis
2.6. Slide Imaging and Quantification
2.7. Murine Cardiac Fibroblast Isolation and Culture
2.8. Quantitative RT-PCR of Cardiac Fibroblasts
2.9. Western Blotting
2.10. Intracellular Ca2+ Measurements
2.11. RNA Extraction and Quantitative RT-PCR of Heart Samples
2.12. Statistics
3. Results
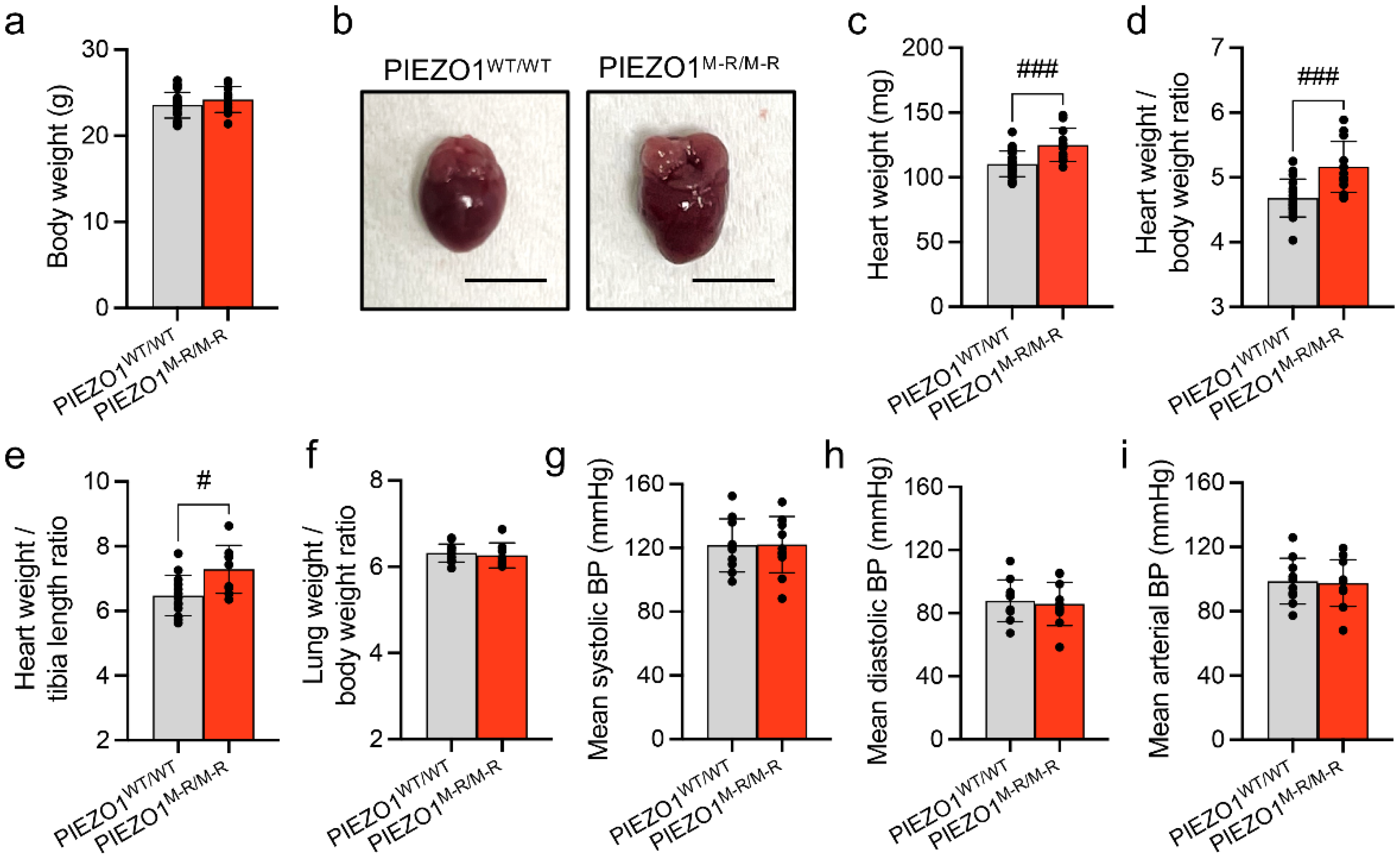
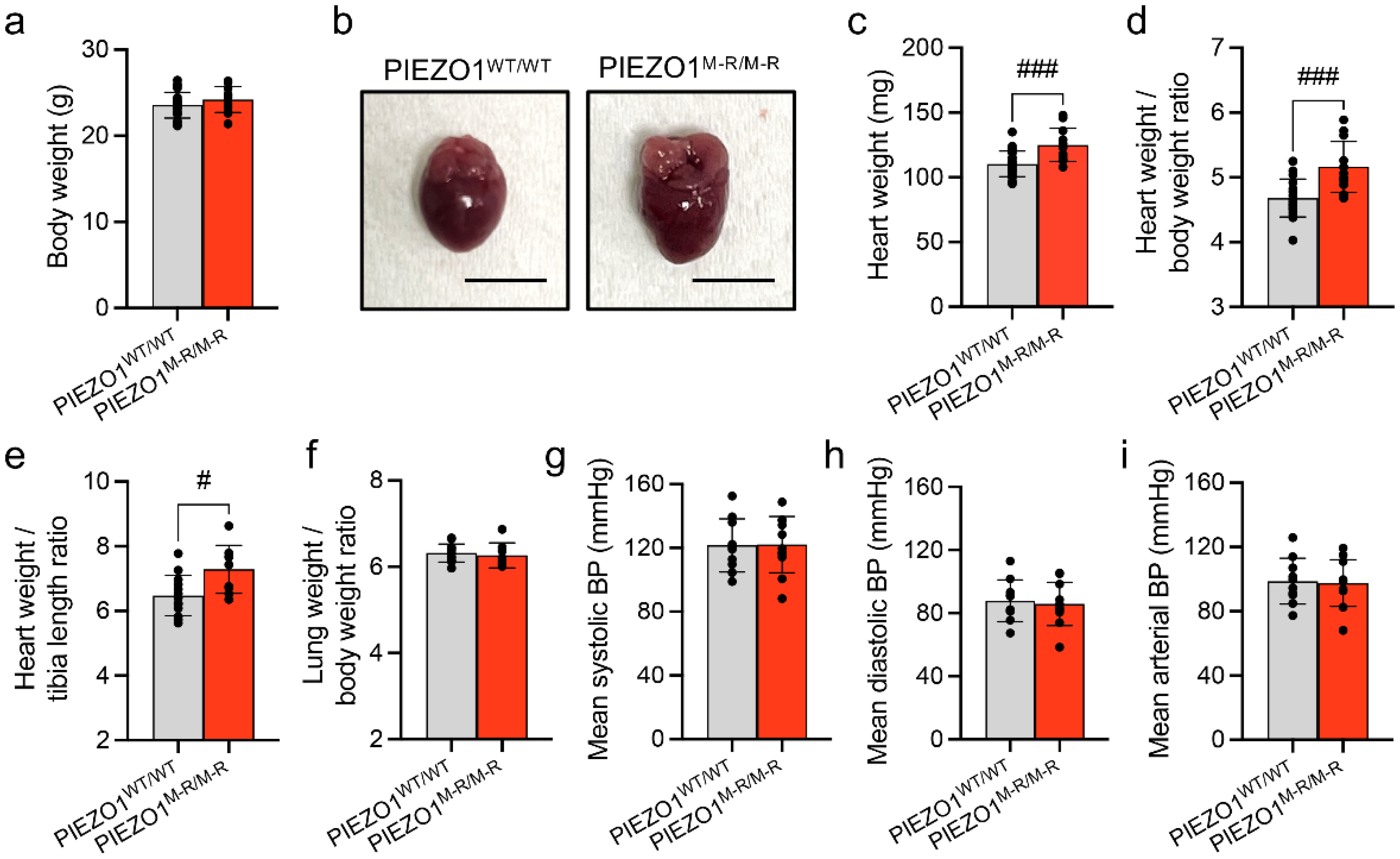
3.1. Increased Heart Size
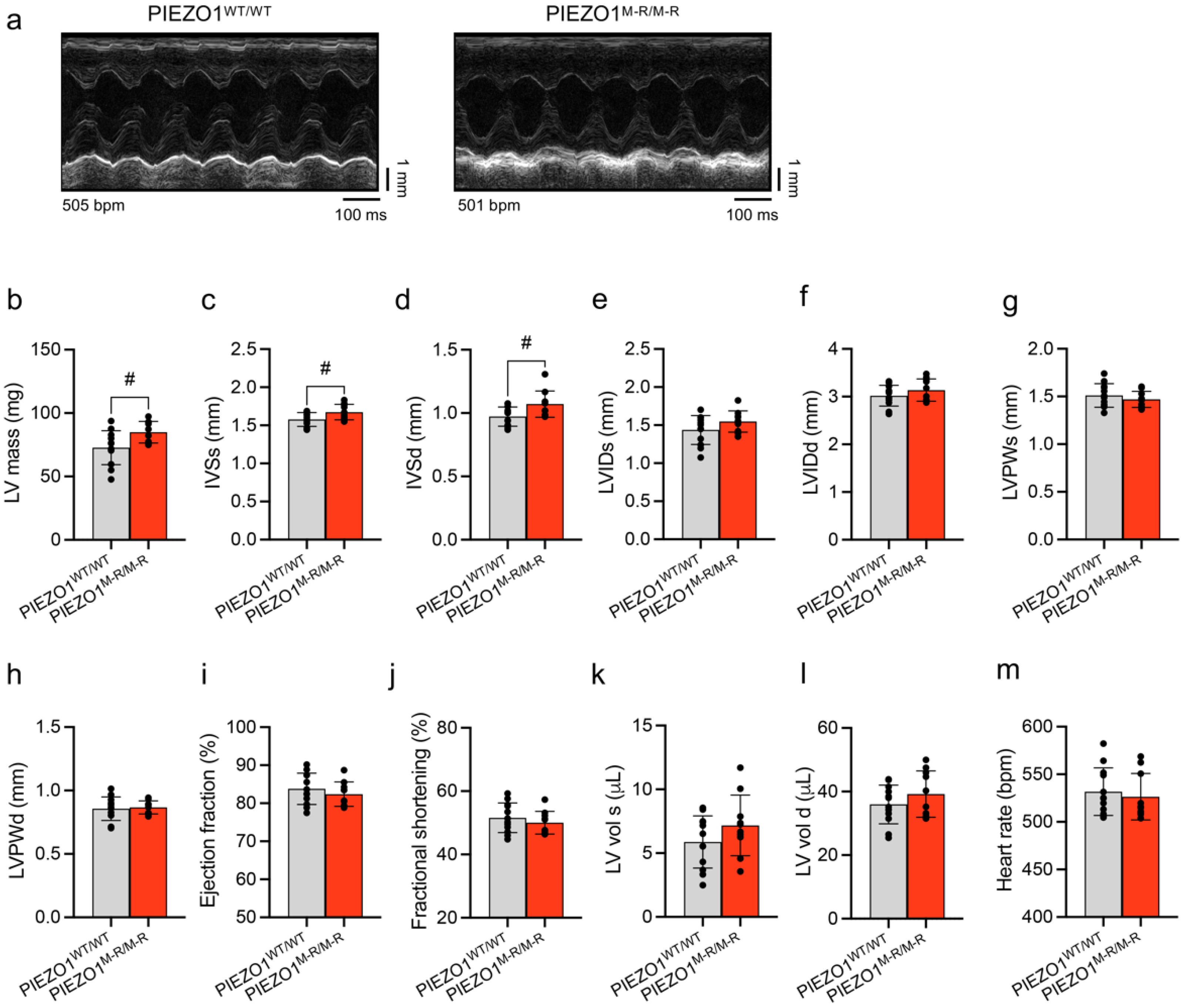
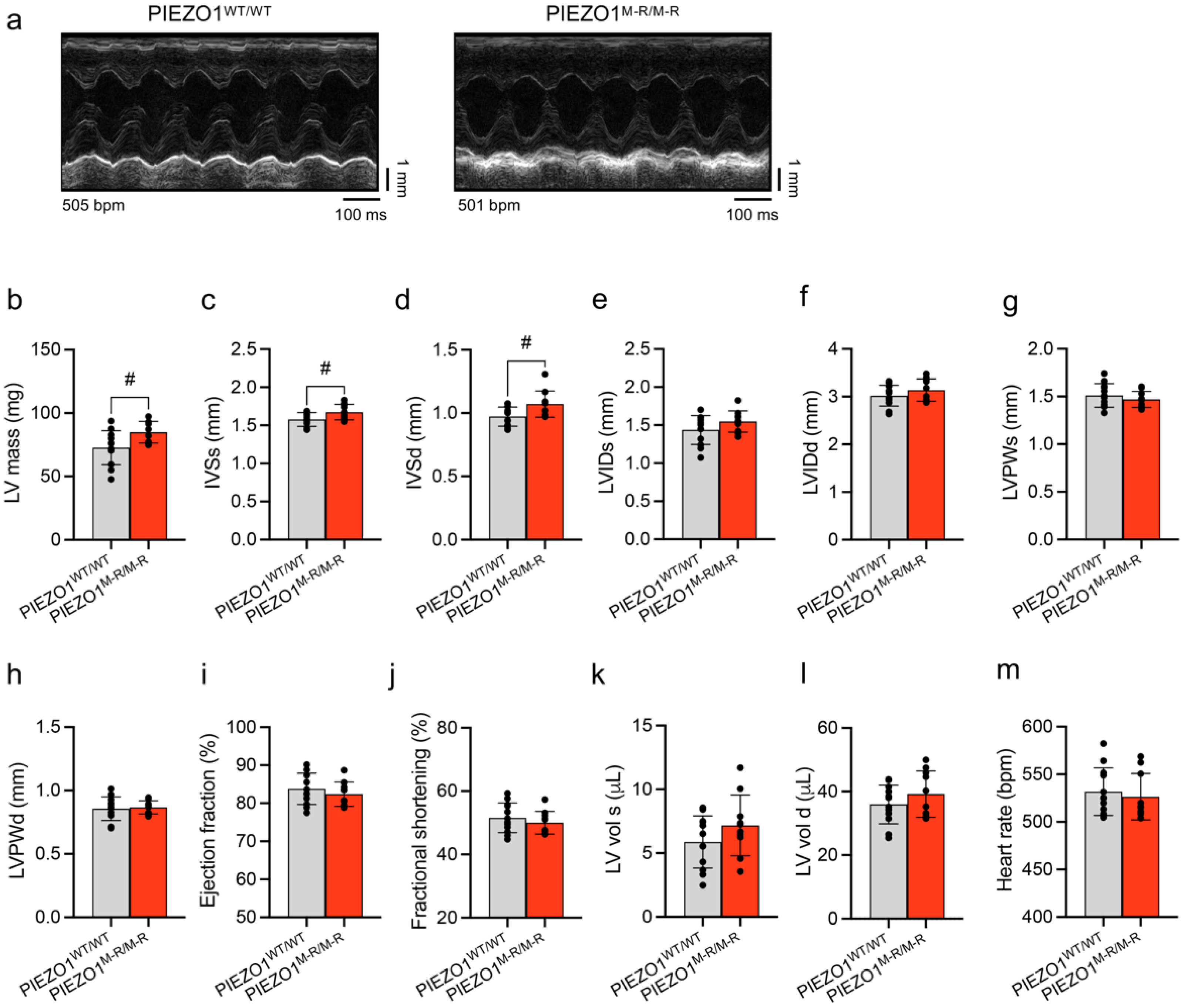
3.2. Unaffected In Vivo Cardiac Contractility despite Hypertrophy
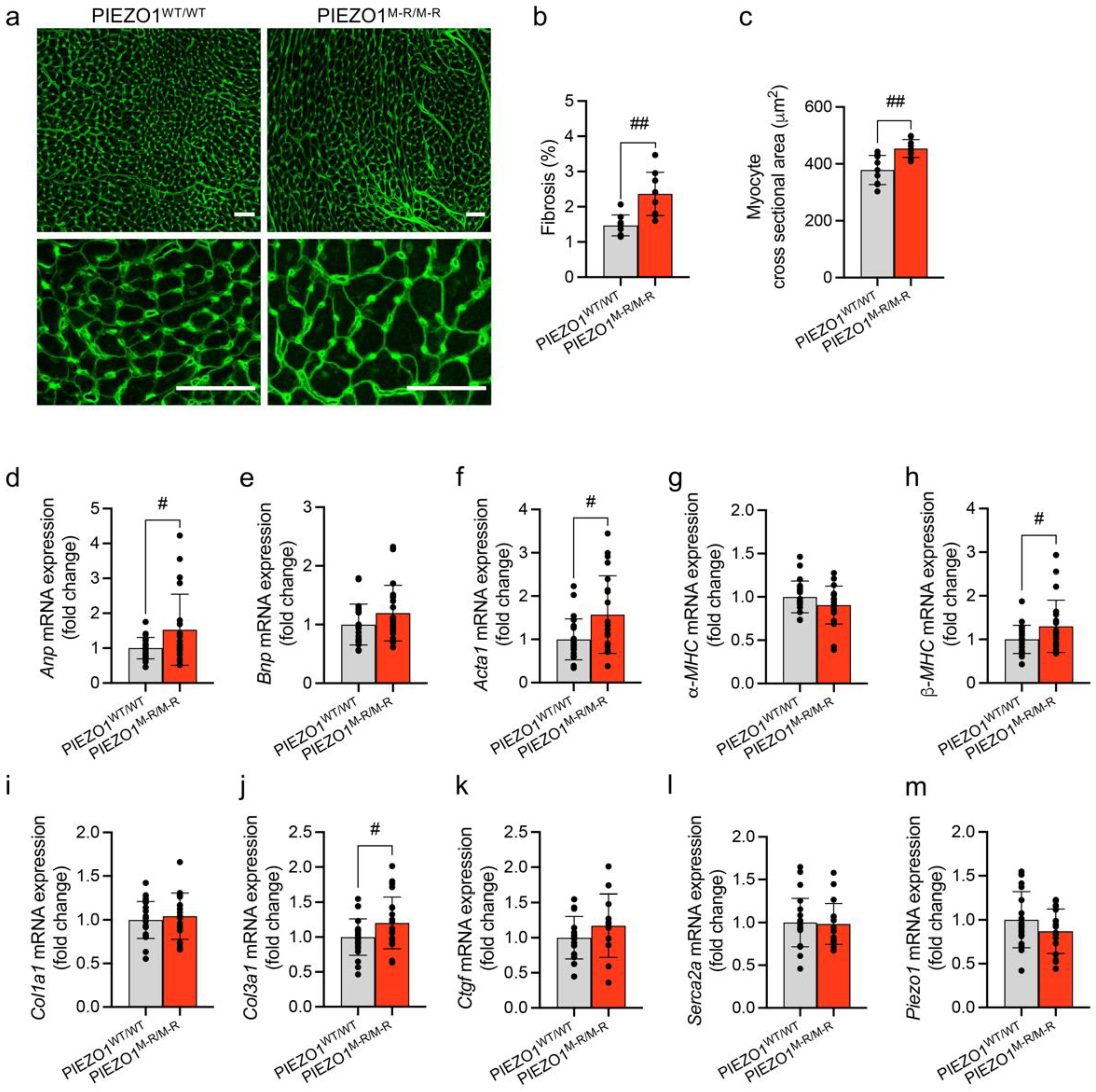
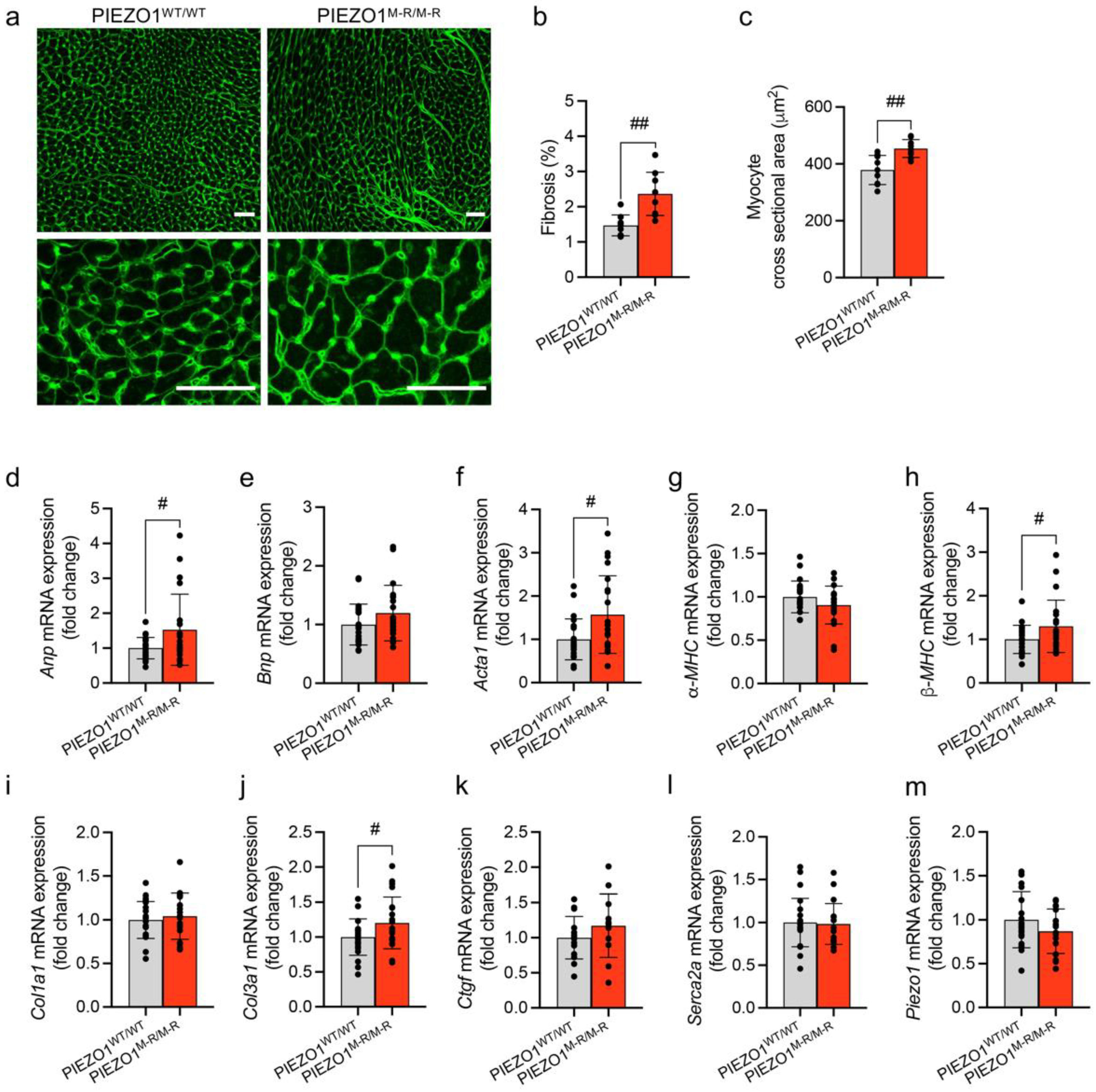
3.3. Cellular Hypertrophy, Altered Gene Expression and Fibrosis
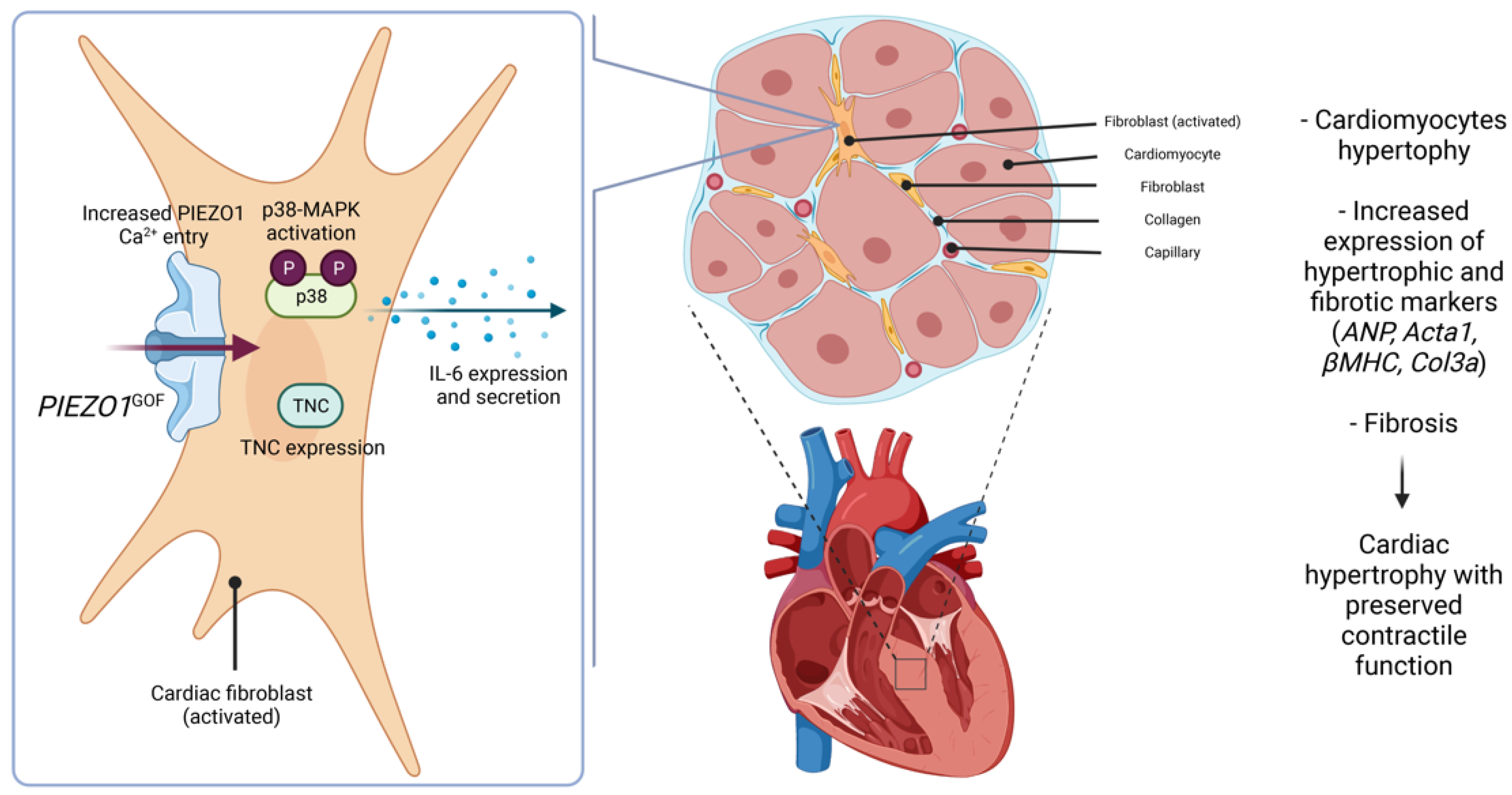
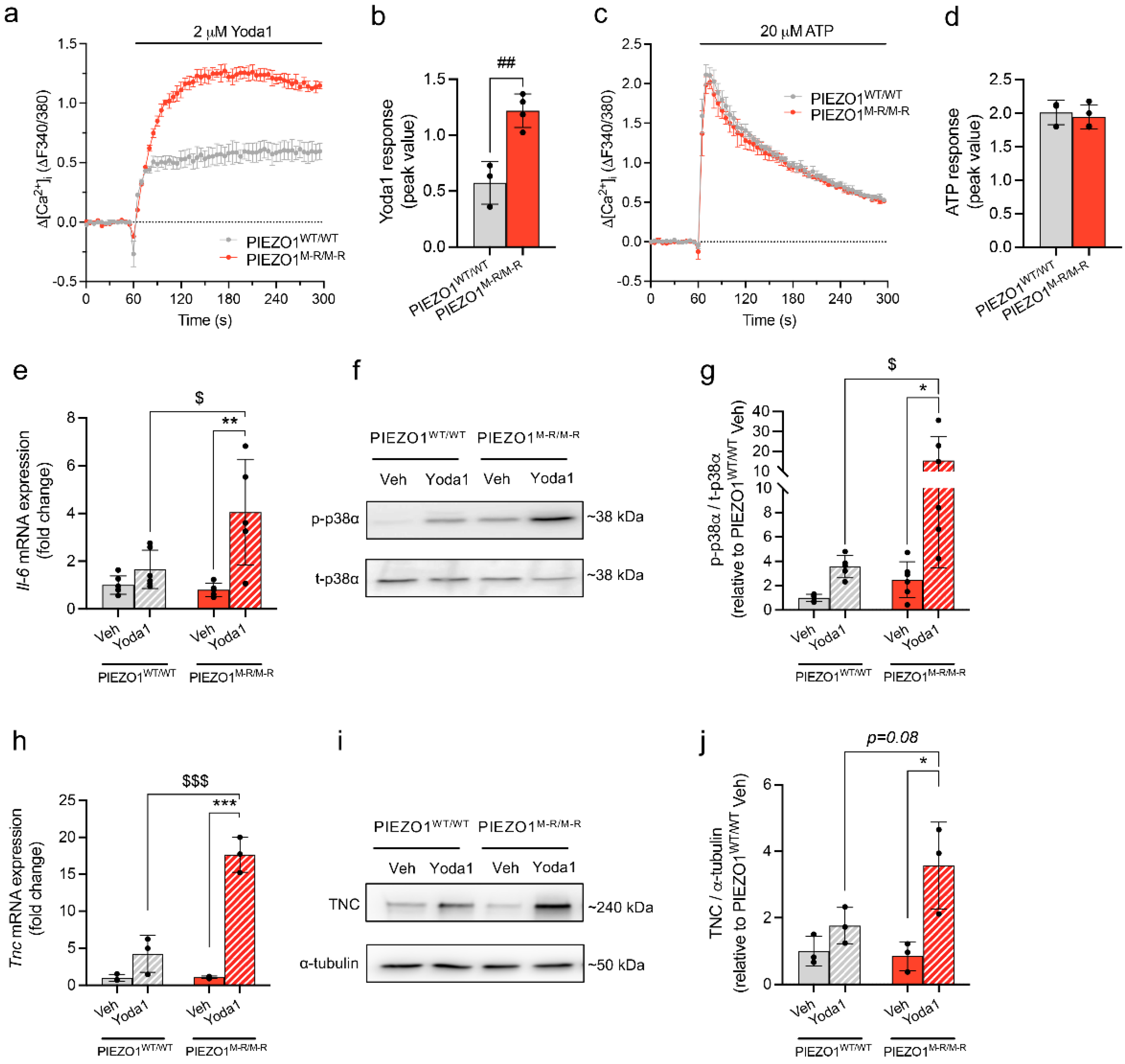
3.4. Increased PIEZO1 Function and Downstream Signalling in Cardiac Fibroblasts
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lyon, R.C.; Zanella, F.; Omens, J.H.; Sheikh, F. Mechanotransduction in Cardiac Hypertrophy and Failure. Circ. Res. 2015, 116, 1462–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manyari, D.E. Prognostic Implications of Echocardiographically Determined Left Ventricular Mass in the Framingham Heart Study. N. Engl. J. Med. 1990, 323, 1706–1707. [Google Scholar] [CrossRef] [PubMed]
- Ruwhof, C. Mechanical Stress-Induced Cardiac Hypertrophy: Mechanisms and Signal Transduction Pathways. Cardiovasc. Res. 2000, 47, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Herum, K.; Lunde, I.; McCulloch, A.; Christensen, G. The Soft- and Hard-Heartedness of Cardiac Fibroblasts: Mechanotransduction Signaling Pathways in Fibrosis of the Heart. J. Clin. Med. 2017, 6, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, A.; Kohl, P.; Peyronnet, R. Molecular Candidates for Cardiac Stretch-Activated Ion Channels. Glob. Cardiol. Sci. Pract. 2014, 2014, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyronnet, R.; Nerbonne, J.M.; Kohl, P. Cardiac Mechano-Gated Ion Channels and Arrhythmias. Circ. Res. 2016, 118, 311–329. [Google Scholar] [CrossRef] [Green Version]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 Are Essential Components of Distinct Mechanically Activated Cation Channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Coste, B.; Xiao, B.; Santos, J.S.; Syeda, R.; Grandl, J.; Spencer, K.S.; Kim, S.E.; Schmidt, M.; Mathur, J.; Dubin, A.E.; et al. Piezo Proteins Are Pore-Forming Subunits of Mechanically Activated Channels. Nature 2012, 483, 176–181. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Zhou, H.; Chi, S.; Wang, Y.; Wang, J.; Geng, J.; Wu, K.; Liu, W.; Zhang, T.; Dong, M.-Q.; et al. Structure and Mechanogating Mechanism of the Piezo1 Channel. Nature 2018, 554, 487–492. [Google Scholar] [CrossRef]
- Douguet, D.; Patel, A.; Xu, A.; Vanhoutte, P.M.; Honoré, E. Piezo Ion Channels in Cardiovascular Mechanobiology. Trends Pharmacol. Sci. 2019, 40, 956–970. [Google Scholar] [CrossRef] [Green Version]
- Beech, D.J.; Kalli, A.C. Force Sensing by Piezo Channels in Cardiovascular Health and Disease. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 2228–2239. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yang, X.; Jiang, J.; Xiao, B. Structural Designs and Mechanogating Mechanisms of the Mechanosensitive Piezo Channels. Trends Biochem. Sci. 2021, 46, 472–488. [Google Scholar] [CrossRef] [PubMed]
- Blythe, N.M.; Muraki, K.; Ludlow, M.J.; Stylianidis, V.; Gilbert, H.T.J.; Evans, E.L.; Cuthbertson, K.; Foster, R.; Swift, J.; Li, J.; et al. Mechanically Activated Piezo1 Channels of Cardiac Fibroblasts Stimulate P38 Mitogen-Activated Protein Kinase Activity and Interleukin-6 Secretion. J. Biol. Chem. 2019, 294, 17395–17408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emig, R.; Knodt, W.; Krussig, M.J.; Zgierski-Johnston, C.M.; Gorka, O.; Groß, O.; Kohl, P.; Ravens, U.; Peyronnet, R. Piezo1 Channels Contribute to the Regulation of Human Atrial Fibroblast Mechanical Properties and Matrix Stiffness Sensing. Cells 2021, 10, 663. [Google Scholar] [CrossRef]
- Ploeg, M.C.; Munts, C.; Prinzen, F.W.; Turner, N.A.; van Bilsen, M.; van Nieuwenhoven, F.A. Piezo1 Mechanosensitive Ion Channel Mediates Stretch-Induced Nppb Expression in Adult Rat Cardiac Fibroblasts. Cells 2021, 10, 1745. [Google Scholar] [CrossRef]
- Jakob, D.; Klesen, A.; Allegrini, B.; Darkow, E.; Aria, D.; Emig, R.; Chica, A.S.; Rog-Zielinska, E.A.; Guth, T.; Beyersdorf, F.; et al. Piezo1 and BKCa Channels in Human Atrial Fibroblasts: Interplay and Remodelling in Atrial Fibrillation. J. Mol. Cell. Cardiol. 2021, 158, 49–62. [Google Scholar] [CrossRef]
- Liu, H.; Hu, J.; Zheng, Q.; Feng, X.; Zhan, F.; Wang, X.; Xu, G.; Hua, F. Piezo1 Channels as Force Sensors in Mechanical Force-Related Chronic Inflammation. Front. Immunol. 2022, 13, 816149. [Google Scholar] [CrossRef]
- Lindner, D.; Zietsch, C.; Tank, J.; Sossalla, S.; Fluschnik, N.; Hinrichs, S.; Maier, L.; Poller, W.; Blankenberg, S.; Schultheiss, H.-P.; et al. Cardiac Fibroblasts Support Cardiac Inflammation in Heart Failure. Basic Res. Cardiol. 2014, 109, 428. [Google Scholar] [CrossRef]
- Shinge, S.A.U.; Zhang, D.; Din, A.U.; Yu, F.; Nie, Y. Emerging Piezo1 Signaling in Inflammation and Atherosclerosis; a Potential Therapeutic Target. Int. J. Biol. Sci. 2022, 18, 923–941. [Google Scholar] [CrossRef]
- Atcha, H.; Jairaman, A.; Holt, J.R.; Meli, V.S.; Nagalla, R.R.; Veerasubramanian, P.K.; Brumm, K.T.; Lim, H.E.; Othy, S.; Cahalan, M.D.; et al. Mechanically Activated Ion Channel Piezo1 Modulates Macrophage Polarization and Stiffness Sensing. Nat. Commun. 2021, 12, 3256. [Google Scholar] [CrossRef]
- Lee, W.; Nims, R.J.; Savadipour, A.; Zhang, Q.; Leddy, H.A.; Liu, F.; McNulty, A.L.; Chen, Y.; Guilak, F.; Liedtke, W.B. Inflammatory Signaling Sensitizes Piezo1 Mechanotransduction in Articular Chondrocytes as a Pathogenic Feed-Forward Mechanism in Osteoarthritis. Proc. Natl. Acad. Sci. USA 2021, 118, e2001611118. [Google Scholar] [CrossRef] [PubMed]
- Albarrán-Juárez, J.; Iring, A.; Wang, S.; Joseph, S.; Grimm, M.; Strilic, B.; Wettschureck, N.; Althoff, T.F.; Offermanns, S. Piezo1 and Gq/G11 Promote Endothelial Inflammation Depending on Flow Pattern and Integrin Activation. J. Exp. Med. 2018, 215, 2655–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F.; Yin, K.; Wu, K.; Zhang, M.; Wang, S.; Cheng, H.; Zhou, Z.; Xiao, B. The Mechanosensitive Piezo1 Channel Mediates Heart Mechano-Chemo Transduction. Nat. Commun. 2021, 12, 869. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, S.; Li, W.; Ma, Y.; Shen, J.; Wang, Y.; Shen, Y.; Chen, J.; Ji, Y.; Xie, Y.; et al. Piezo1-Mediated Mechanotransduction Promotes Cardiac Hypertrophy by Impairing Calcium Homeostasis to Activate Calpain/Calcineurin Signaling. Hypertension 2021, 78, 647–660. [Google Scholar] [CrossRef]
- Liang, J.; Huang, B.; Yuan, G.; Chen, Y.; Liang, F.; Zeng, H.; Zheng, S.; Cao, L.; Geng, D.; Zhou, S. Stretch-Activated Channel Piezo1 Is up-Regulated in Failure Heart and Cardiomyocyte Stimulated by AngII. Am. J. Transl. Res. 2017, 9, 2945–2955. [Google Scholar]
- Yu, Z.-Y.; Gong, H.; Kesteven, S.; Guo, Y.; Wu, J.; Li, J.; Iismaa, S.; Kaidonis, X.; Graham, R.; Cox, C.; et al. Piezo1 Is the Cardiac Mechanosensor That Initiates the Hypertrophic Response to Pressure Overload. 2021; In Review. [Google Scholar] [CrossRef]
- Zarychanski, R.; Schulz, V.P.; Houston, B.L.; Maksimova, Y.; Houston, D.S.; Smith, B.; Rinehart, J.; Gallagher, P.G. Mutations in the Mechanotransduction Protein PIEZO1 Are Associated with Hereditary Xerocytosis. Blood 2012, 120, 1908–1915. [Google Scholar] [CrossRef] [Green Version]
- Bae, C.; Gnanasambandam, R.; Nicolai, C.; Sachs, F.; Gottlieb, P.A. Xerocytosis Is Caused by Mutations That Alter the Kinetics of the Mechanosensitive Channel PIEZO1. Proc. Natl. Acad. Sci. USA 2013, 110, E1162–E1168. [Google Scholar] [CrossRef] [Green Version]
- Demolombe, S.; Duprat, F.; Honoré, E.; Patel, A. Slower Piezo1 Inactivation in Dehydrated Hereditary Stomatocytosis (Xerocytosis). Biophys. J. 2013, 105, 833–834. [Google Scholar] [CrossRef] [Green Version]
- Albuisson, J.; Murthy, S.E.; Bandell, M.; Coste, B.; Louis-dit-Picard, H.; Mathur, J.; Fénéant-Thibault, M.; Tertian, G.; de Jaureguiberry, J.-P.; Syfuss, P.-Y.; et al. Dehydrated Hereditary Stomatocytosis Linked to Gain-of-Function Mutations in Mechanically Activated PIEZO1 Ion Channels. Nat. Commun. 2013, 4, 1884. [Google Scholar] [CrossRef]
- Evans, E.L.; Povstyan, O.V.; De Vecchis, D.; Macrae, F.; Lichtenstein, L.; Futers, T.S.; Parsonage, G.; Humphreys, N.E.; Adamson, A.; Kalli, A.C.; et al. RBCs Prevent Rapid PIEZO1 Inactivation and Expose Slow Deactivation as a Mechanism of Dehydrated Hereditary Stomatocytosis. Blood 2020, 136, 140–144. [Google Scholar] [CrossRef]
- Ma, S.; Cahalan, S.; LaMonte, G.; Grubaugh, N.D.; Zeng, W.; Murthy, S.E.; Paytas, E.; Gamini, R.; Lukacs, V.; Whitwam, T.; et al. Common PIEZO1 Allele in African Populations Causes RBC Dehydration and Attenuates Plasmodium Infection. Cell 2018, 173, 443–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartoli, F.; Debant, M.; Chuntharpursat-Bon, E.; Evans, E.L.; Musialowski, K.E.; Parsonage, G.; Morley, L.C.; Futers, T.S.; Sukumar, P.; Bowen, T.S.; et al. Endothelial Piezo1 Sustains Muscle Capillary Density and Contributes to Physical Activity. J. Clin. Investig. 2022, 132, e141775. [Google Scholar] [CrossRef] [PubMed]
- Emde, B.; Heinen, A.; Gödecke, A.; Bottermann, K. Wheat Germ Agglutinin Staining as a Suitable Method for Detection and Quantification of Fibrosis in Cardiac Tissue after Myocardial Infarction. Eur. J. Histochem. 2014, 58, 2448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bageghni, S.A.; Hemmings, K.E.; Zava, N.; Denton, C.P.; Porter, K.E.; Ainscough, J.F.X.; Drinkhill, M.J.; Turner, N.A. Cardiac Fibroblast-specific P38α MAP Kinase Promotes Cardiac Hypertrophy via a Putative Paracrine Interleukin-6 Signaling Mechanism. FASEB J. 2018, 32, 4941–4954. [Google Scholar] [CrossRef] [Green Version]
- Shimojo, N.; Hashizume, R.; Kanayama, K.; Hara, M.; Suzuki, Y.; Nishioka, T.; Hiroe, M.; Yoshida, T.; Imanaka-Yoshida, K. Tenascin-C May Accelerate Cardiac Fibrosis by Activating Macrophages via the Integrin AVβ3/Nuclear Factor–ΚB/Interleukin-6 Axis. Hypertension 2015, 66, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Carrier, L. Asymmetric Septal Hypertrophy in Heterozygous CMyBP-C Null Mice. Cardiovasc. Res. 2004, 63, 293–304. [Google Scholar] [CrossRef]
- Antunes, M.D.O.; Scudeler, T.L. Hypertrophic Cardiomyopathy. Int. J. Cardiol. Heart Vasc. 2020, 27, 100503. [Google Scholar] [CrossRef]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Olivotto, I.; Cecchi, F.; Poggesi, C.; Yacoub, M.H. Patterns of Disease Progression in Hypertrophic Cardiomyopathy: An Individualized Approach to Clinical Staging. Circ. Heart Fail. 2012, 5, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Yalçin, F.; Kucukler, N.; Cingolani, O.; Mbiyangandu, B.; Sorensen, L.; Pinherio, A.; Abraham, M.R.; Abraham, T.P. Evolution of Ventricular Hypertrophy and Myocardial Mechanics in Physiological and Pathological Hypertrophy. J. Appl. Physiol. 2019, 126, 354–362. [Google Scholar] [CrossRef]
- Dirkx, E.; da Costa Martins, P.A.; De Windt, L.J. Regulation of Fetal Gene Expression in Heart Failure. Biochim. Biophys. Acta 2013, 1832, 2414–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergeeva, I.A.; Christoffels, V.M. Regulation of Expression of Atrial and Brain Natriuretic Peptide, Biomarkers for Heart Development and Disease. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2013, 1832, 2403–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suo, M.; Hautala, N.; Földes, G.; Szokodi, I.; Tóth, M.; Leskinen, H.; Uusimaa, P.; Vuolteenaho, O.; Nemer, M.; Ruskoaho, H. Posttranscriptional Control of BNP Gene Expression in Angiotensin II–Induced Hypertension. Hypertension 2002, 39, 803–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, O.; Ogawa, Y.; Itoh, H.; Suga, S.; Komatsu, Y.; Kishimoto, I.; Nishino, K.; Yoshimasa, T.; Nakao, K. Rapid Transcriptional Activation and Early MRNA Turnover of Brain Natriuretic Peptide in Cardiocyte Hypertrophy. Evidence for Brain Natriuretic Peptide as an “Emergency” Cardiac Hormone against Ventricular Overload. J. Clin. Investig. 1995, 96, 1280–1287. [Google Scholar] [CrossRef] [Green Version]
- Sakata, Y.; Yamamoto, K.; Masuyama, T.; Mano, T.; Nishikawa, N.; Kuzuya, T.; Miwa, T.; Hori, M. Ventricular Production of Natriuretic Peptides and Ventricular Structural Remodeling in Hypertensive Heart Failure. J. Hypertens. 2001, 19, 1905–1912. [Google Scholar] [CrossRef]
- Doust, J.; Lehman, R.; Glasziou, P. The Role of BNP Testing in Heart Failure. Am. Fam. Physician 2006, 74, 1893–1898. [Google Scholar]
- Taegtmeyer, H.; Sen, S.; Vela, D. Return to the Fetal Gene Program: A Suggested Metabolic Link to Gene Expression in the Heart. Ann. N. Y. Acad. Sci. 2010, 1188, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Hinderer, S.; Schenke-Layland, K. Cardiac Fibrosis—A Short Review of Causes and Therapeutic Strategies. Adv. Drug Deliv. Rev. 2019, 146, 77–82. [Google Scholar] [CrossRef]
- Carver, W.; Nagpal, M.L.; Nachtigal, M.; Borg, T.K.; Terracio, L. Collagen Expression in Mechanically Stimulated Cardiac Fibroblasts. Circ. Res. 1991, 69, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Chow, L.T.C.; Shum, I.O.L.; Qin, L.; Sanderson, J.E. Left and Right Ventricular Collagen Type I/III Ratios and Remodeling Post-Myocardial Infarction. J. Card. Fail. 1999, 5, 117–126. [Google Scholar] [CrossRef]
- Tayebjee, M.H.; MacFadyen, R.J.; Lip, G.Y. Extracellular Matrix Biology: A New Frontier in Linking the Pathology and Therapy of Hypertension? J. Hypertens. 2003, 21, 2211–2218. [Google Scholar] [CrossRef] [PubMed]
- Soufen, H.N.; Salemi, V.M.C.; Aneas, I.M.S.; Ramires, F.J.A.; Benício, A.M.D.; Benvenuti, L.A.; Krieger, J.E.; Mady, C. Collagen Content, but Not the Ratios of Collagen Type III/I MRNAs, Differs among Hypertensive, Alcoholic, and Idiopathic Dilated Cardiomyopathy. Braz. J. Med. Biol. Res. 2008, 41, 1098–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigil-Garcia, M.; Demkes, C.J.; Eding, J.E.C.; Versteeg, D.; de Ruiter, H.; Perini, I.; Kooijman, L.; Gladka, M.M.; Asselbergs, F.W.; Vink, A.; et al. Gene Expression Profiling of Hypertrophic Cardiomyocytes Identifies New Players in Pathological Remodelling. Cardiovasc. Res. 2021, 117, 1532–1545. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.; Corden, B.; Cook, S.A. Targeting Cardiac Fibrosis in Heart Failure with Preserved Ejection Fraction: Mirage or Miracle? EMBO Mol. Med. 2020, 12, e10865. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, M.; Shimizu, M.; Ino, H.; Okeie, K.; Yamaguchi, M.; Fujino, N.; Mabuchi, H.; Nakanishi, I. Collagen Remodeling and Cardiac Dysfunction in Patients with Hypertrophic Cardiomyopathy: The Significance of Type III and VI Collagens. Clin. Cardiol. 2001, 24, 325–329. [Google Scholar] [CrossRef]
- Krenning, G.; Zeisberg, E.M.; Kalluri, R. The Origin of Fibroblasts and Mechanism of Cardiac Fibrosis. J. Cell. Physiol. 2010, 225, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Kuivaniemi, H.; Tromp, G. Type III Collagen (COL3A1): Gene and Protein Structure, Tissue Distribution, and Associated Diseases. Gene 2019, 707, 151–171. [Google Scholar] [CrossRef]
- Garg, P.; Assadi, H.; Jones, R.; Chan, W.B.; Metherall, P.; Thomas, R.; van der Geest, R.; Swift, A.J.; Al-Mohammad, A. Left Ventricular Fibrosis and Hypertrophy Are Associated with Mortality in Heart Failure with Preserved Ejection Fraction. Sci. Rep. 2021, 11, 617. [Google Scholar] [CrossRef]
- González, A.; Schelbert, E.B.; Díez, J.; Butler, J. Myocardial Interstitial Fibrosis in Heart Failure. J. Am. Coll. Cardiol. 2018, 71, 1696–1706. [Google Scholar] [CrossRef]
- Furihata, T.; Kinugawa, S.; Takada, S.; Fukushima, A.; Takahashi, M.; Homma, T.; Masaki, Y.; Tsuda, M.; Matsumoto, J.; Mizushima, W.; et al. The Experimental Model of Transition from Compensated Cardiac Hypertrophy to Failure Created by Transverse Aortic Constriction in Mice. IJC Heart Vasc. 2016, 11, 24–28. [Google Scholar] [CrossRef]
- Bartoli, F.; Bailey, M.A.; Rode, B.; Mateo, P.; Antigny, F.; Bedouet, K.; Gerbaud, P.; Gosain, R.; Plante, J.; Norman, K.; et al. Orai1 Channel Inhibition Preserves Left Ventricular Systolic Function and Normal Ca 2+ Handling After Pressure Overload. Circulation 2020, 141, 199–216. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Niu, T.; Wang, W.; Li, J.; Li, S.; Janicki, J.S.; Ruiz, S.; Meyer, C.J.; Wang, X.L.; Tang, D.; et al. Triterpenoid Dihydro-CDDO-Trifluoroethyl Amide Protects against Maladaptive Cardiac Remodeling and Dysfunction in Mice: A Critical Role of Nrf2. PLoS ONE 2012, 7, e44899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohberger, B.; Kaltenegger, H.; Weigl, L.; Mann, A.; Kullich, W.; Stuendl, N.; Leithner, A.; Steinecker-Frohnwieser, B. Mechanical Exposure and Diacerein Treatment Modulates Integrin-FAK-MAPKs Mechanotransduction in Human Osteoarthritis Chondrocytes. Cell. Signal. 2019, 56, 23–30. [Google Scholar] [CrossRef]
- Liu, S.; Xu, X.; Fang, Z.; Ning, Y.; Deng, B.; Pan, X.; He, Y.; Yang, Z.; Huang, K.; Li, J. Piezo1 Impairs Hepatocellular Tumor Growth via Deregulation of the MAPK-Mediated YAP Signaling Pathway. Cell Calcium 2021, 95, 102367. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wan, P.; Zhang, J.; Li, X.; Xing, J.; Zou, Y.; Wang, K.; Peng, H.; Zhu, Q.; Cao, L.; et al. Targeting Mechanosensitive Piezo1 Alleviated Renal Fibrosis Through P38MAPK-YAP Pathway. Front. Cell Dev. Biol. 2021, 9, 741060. [Google Scholar] [CrossRef]
- He, J.; Fang, B.; Shan, S.; Xie, Y.; Wang, C.; Zhang, Y.; Zhang, X.; Li, Q. Mechanical Stretch Promotes Hypertrophic Scar Formation through Mechanically Activated Cation Channel Piezo1. Cell Death Dis. 2021, 12, 226. [Google Scholar] [CrossRef]
- Banerjee, I.; Fuseler, J.W.; Intwala, A.R.; Baudino, T.A. IL-6 Loss Causes Ventricular Dysfunction, Fibrosis, Reduced Capillary Density, and Dramatically Alters the Cell Populations of the Developing and Adult Heart. Am. J. Physiol.-Heart Circ. Physiol. 2009, 296, H1694–H1704. [Google Scholar] [CrossRef]
- Takeuchi, T.; Yoshida, H.; Tanaka, S. Role of Interleukin-6 in Bone Destruction and Bone Repair in Rheumatoid Arthritis. Autoimmun. Rev. 2021, 20, 102884. [Google Scholar] [CrossRef]
- Hall, C.; Gehmlich, K.; Denning, C.; Pavlovic, D. Complex Relationship Between Cardiac Fibroblasts and Cardiomyocytes in Health and Disease. J. Am. Heart Assoc. 2021, 10, e019338. [Google Scholar] [CrossRef]
- Fontes, J.A.; Rose, N.R.; Čiháková, D. The Varying Faces of IL-6: From Cardiac Protection to Cardiac Failure. Cytokine 2015, 74, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Meléndez, G.C.; McLarty, J.L.; Levick, S.P.; Du, Y.; Janicki, J.S.; Brower, G.L. Interleukin 6 Mediates Myocardial Fibrosis, Concentric Hypertrophy, and Diastolic Dysfunction in Rats. Hypertension 2010, 56, 225–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danesh, J.; Kaptoge, S.; Mann, A.G.; Sarwar, N.; Wood, A.; Angleman, S.B.; Wensley, F.; Higgins, J.P.T.; Lennon, L.; Eiriksdottir, G.; et al. Long-Term Interleukin-6 Levels and Subsequent Risk of Coronary Heart Disease: Two New Prospective Studies and a Systematic Review. PLoS Med. 2008, 5, e78. [Google Scholar] [CrossRef] [PubMed]
- Su, J.-H.; Luo, M.-Y.; Liang, N.-; Gong, S.-X.; Chen, W.; Huang, W.-Q.; Tian, Y.; Wang, A.-P. Interleukin-6: A Novel Target for Cardio-Cerebrovascular Diseases. Front. Pharmacol. 2021, 12, 745061. [Google Scholar] [CrossRef] [PubMed]
- Bürger, A.; Benicke, M.; Deten, A.; Zimmer, H.-G. Catecholamines Stimulate Interleukin-6 Synthesis in Rat Cardiac Fibroblasts. Am. J. Physiol.-Heart Circ. Physiol. 2001, 281, H14–H21. [Google Scholar] [CrossRef] [PubMed]
- Camelliti, P.; Borg, T.; Kohl, P. Structural and Functional Characterisation of Cardiac Fibroblasts. Cardiovasc. Res. 2005, 65, 40–51. [Google Scholar] [CrossRef]
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J. Fibroblast-Specific Genetic Manipulation of P38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 2017, 136, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Sinfield, J.K.; Das, A.; O’Regan, D.J.; Ball, S.G.; Porter, K.E.; Turner, N.A. P38 MAPK Alpha Mediates Cytokine-Induced IL-6 and MMP-3 Expression in Human Cardiac Fibroblasts. Biochem. Biophys. Res. Commun. 2013, 430, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Imanaka-Yoshida, K.; Hiroe, M.; Yoshida, T. Interaction between Cell and Extracellular Matrix in Heart Disease: Multiple Roles of Tenascin-C in Tissue Remodeling. Histol. Histopathol. 2004, 19, 517–525. [Google Scholar] [CrossRef]
- Imanaka-Yoshida, K.; Tawara, I.; Yoshida, T. Tenascin-C in Cardiac Disease: A Sophisticated Controller of Inflammation, Repair, and Fibrosis. Am. J. Physiol. Cell Physiol. 2020, 319, C781–C796. [Google Scholar] [CrossRef]
- Matsumoto, K.-I.; Aoki, H. The Roles of Tenascins in Cardiovascular, Inflammatory, and Heritable Connective Tissue Diseases. Front. Immunol. 2020, 11, 609752. [Google Scholar] [CrossRef]
- Imanaka-Yoshida, K.; Hiroe, M.; Nishikawa, T.; Ishiyama, S.; Shimojo, T.; Ohta, Y.; Sakakura, T.; Yoshida, T. Tenascin-C Modulates Adhesion of Cardiomyocytes to Extracellular Matrix during Tissue Remodeling after Myocardial Infarction. Lab. Investig. J. Tech. Methods Pathol. 2001, 81, 1015–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, D.; Kozuka, Y.; Noro, A.; Ogawa, T.; Imanaka-Yoshida, K.; Yoshida, T. Tenascin-C Induces Phenotypic Changes in Fibroblasts to Myofibroblasts with High Contractility through the Integrin Avβ1/Transforming Growth Factor β/SMAD Signaling Axis in Human Breast Cancer. Am. J. Pathol. 2020, 190, 2123–2135. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Wang, W.; Morales-Nebreda, L.; Feng, G.; Wu, M.; Zhou, X.; Lafyatis, R.; Lee, J.; Hinchcliff, M.; Feghali-Bostwick, C.; et al. Tenascin-C Drives Persistence of Organ Fibrosis. Nat. Commun. 2016, 7, 11703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podesser, B.K.; Kreibich, M.; Dzilic, E.; Santer, D.; Förster, L.; Trojanek, S.; Abraham, D.; Krššák, M.; Klein, K.U.; Tretter, E.V.; et al. Tenascin-C Promotes Chronic Pressure Overload-Induced Cardiac Dysfunction, Hypertrophy and Myocardial Fibrosis. J. Hypertens. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Tong, X.; Zhang, J.; Shen, M.; Zhang, J. Silencing of Tenascin-C Inhibited Inflammation and Apoptosis Via PI3K/Akt/NF-ΚB Signaling Pathway in Subarachnoid Hemorrhage Cell Model. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2020, 29, 104485. [Google Scholar] [CrossRef] [Green Version]
- Maqbool, A.; Spary, E.J.; Manfield, I.W.; Ruhmann, M.; Zuliani-Alvarez, L.; Gamboa-Esteves, F.O.; Porter, K.E.; Drinkhill, M.J.; Midwood, K.S.; Turner, N.A. Tenascin C Upregulates Interleukin-6 Expression in Human Cardiac Myofibroblasts via Toll-like Receptor 4. World J. Cardiol. 2016, 8, 340–350. [Google Scholar] [CrossRef]
- Fujimoto, M.; Suzuki, H.; Shiba, M.; Shimojo, N.; Imanaka-Yoshida, K.; Yoshida, T.; Kanamaru, K.; Matsushima, S.; Taki, W. Tenascin-C Induces Prolonged Constriction of Cerebral Arteries in Rats. Neurobiol. Dis. 2013, 55, 104–109. [Google Scholar] [CrossRef]
- Suzuki, H.; Fujimoto, M.; Kawakita, F.; Liu, L.; Nakatsuka, Y.; Nakano, F.; Nishikawa, H.; Okada, T.; Kanamaru, H.; Imanaka-Yoshida, K.; et al. Tenascin-C in Brain Injuries and Edema after Subarachnoid Hemorrhage: Findings from Basic and Clinical Studies. J. Neurosci. Res. 2020, 98, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, J.-H.; Zhang, Y.-Y.; Wang, Y.-Z.; Wang, J.; Zhao, Y.; Jin, X.-X.; Xue, G.-L.; Li, P.-H.; Sun, Y.-L.; et al. Deletion of Interleukin-6 Alleviated Interstitial Fibrosis in Streptozotocin-Induced Diabetic Cardiomyopathy of Mice through Affecting TGFβ1 and MiR-29 Pathways. Sci. Rep. 2016, 6, 23010. [Google Scholar] [CrossRef]
- Wang, L.; You, X.; Lotinun, S.; Zhang, L.; Wu, N.; Zou, W. Mechanical Sensing Protein PIEZO1 Regulates Bone Homeostasis via Osteoblast-Osteoclast Crosstalk. Nat. Commun. 2020, 11, 282. [Google Scholar] [CrossRef] [Green Version]
- Kakkar, R.; Lee, R.T. Intramyocardial Fibroblast Myocyte Communication. Circ. Res. 2010, 106, 47–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiu, K.; Nagai, R. Fibroblast-Mediated Pathways in Cardiac Hypertrophy. J. Mol. Cell. Cardiol. 2014, 70, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, S.L.; Keenan, W.T.; Athanas, A.J.; Proudfoot, J.A.; Zangwill, L.M.; Ayyagari, R.; Liebmann, J.M.; Girkin, C.A.; Patapoutian, A.; Weinreb, R.N. Investigation of Associations between Piezo1 Mechanoreceptor Gain-of-Function Variants and Glaucoma-Related Phenotypes in Humans and Mice. Sci. Rep. 2020, 10, 19013. [Google Scholar] [CrossRef]
- Nguetse, C.N.; Purington, N.; Ebel, E.R.; Shakya, B.; Tetard, M.; Kremsner, P.G.; Velavan, T.P.; Egan, E.S. A Common Polymorphism in the Mechanosensitive Ion Channel PIEZO1 Is Associated with Protection from Severe Malaria in Humans. Proc. Natl. Acad. Sci. USA 2020, 117, 9074–9081. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Anp | AGGCCATATTGGAGCAAATC | CTCCTCCAGGTGGTCTAGCA |
| Bnp | ATGGATCTCCTGAAGGTGCTG | GTGCTGCCTTGAGACCGAA |
| Acta1 | CGTGAAGCCTCACTTCCTACC | AGAGCCGTTGTCACACACAA |
| α-MHC | GCCCAGTACCTCCGAAAGTC | GCCTTAACATACTCCTCCTTGTC |
| β-MHC | CTACAGGCCTGGGCTTACCT | TCTCCTTCTCAGACTTCCGC |
| Col1a1 | GCTCCTCTTAGGGGCCACT | CCACGTCTCACCATTGGGG |
| Col3a1 | CTGTAACATGGAAACTGGGGAAA | CCATAGCTGAACTGAAAACCACC |
| Ctgf | GGGCCTCTTCTGCGATTTC | ATCCAGGCAAGTGCATTGGTA |
| Serca2a | CACACCGCTGAATCTGAC | GGAAGCGGTTACTCCAGT |
| Piezo1 | TGAGCCCTTCCCCAACAATAC | CTGCAGGTGGTTCTGGATATAG |
| Rpl32 | GCTGCTGATGTGCAACAAA | GGGATTGGTGACTCTGATGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartoli, F.; Evans, E.L.; Blythe, N.M.; Stewart, L.; Chuntharpursat-Bon, E.; Debant, M.; Musialowski, K.E.; Lichtenstein, L.; Parsonage, G.; Futers, T.S.; et al. Global PIEZO1 Gain-of-Function Mutation Causes Cardiac Hypertrophy and Fibrosis in Mice. Cells 2022, 11, 1199. https://doi.org/10.3390/cells11071199
Bartoli F, Evans EL, Blythe NM, Stewart L, Chuntharpursat-Bon E, Debant M, Musialowski KE, Lichtenstein L, Parsonage G, Futers TS, et al. Global PIEZO1 Gain-of-Function Mutation Causes Cardiac Hypertrophy and Fibrosis in Mice. Cells. 2022; 11(7):1199. https://doi.org/10.3390/cells11071199
Chicago/Turabian StyleBartoli, Fiona, Elizabeth L. Evans, Nicola M. Blythe, Leander Stewart, Eulashini Chuntharpursat-Bon, Marjolaine Debant, Katie E. Musialowski, Laeticia Lichtenstein, Gregory Parsonage, T. Simon Futers, and et al. 2022. "Global PIEZO1 Gain-of-Function Mutation Causes Cardiac Hypertrophy and Fibrosis in Mice" Cells 11, no. 7: 1199. https://doi.org/10.3390/cells11071199
APA StyleBartoli, F., Evans, E. L., Blythe, N. M., Stewart, L., Chuntharpursat-Bon, E., Debant, M., Musialowski, K. E., Lichtenstein, L., Parsonage, G., Futers, T. S., Turner, N. A., & Beech, D. J. (2022). Global PIEZO1 Gain-of-Function Mutation Causes Cardiac Hypertrophy and Fibrosis in Mice. Cells, 11(7), 1199. https://doi.org/10.3390/cells11071199