
Induction of Mitochondrial Fragmentation and Mitophagy after Neonatal Hypoxia–Ischemia

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals of In Vitro Experiments
2.2. Preparation of Primary Neurons
2.3. Sample Preparation for Live Cell Microscopy
2.4. Oxygen-Glucose Deprivation (OGD) of Primary Neurons
2.5. Quantification of Mitophagy and Mitochondrial Morphology Analysis in Primary Neurons
2.6. Measurement of Oxygen Consumption Rate (OCR)
2.7. Hypoxia–Ischemia Procedure
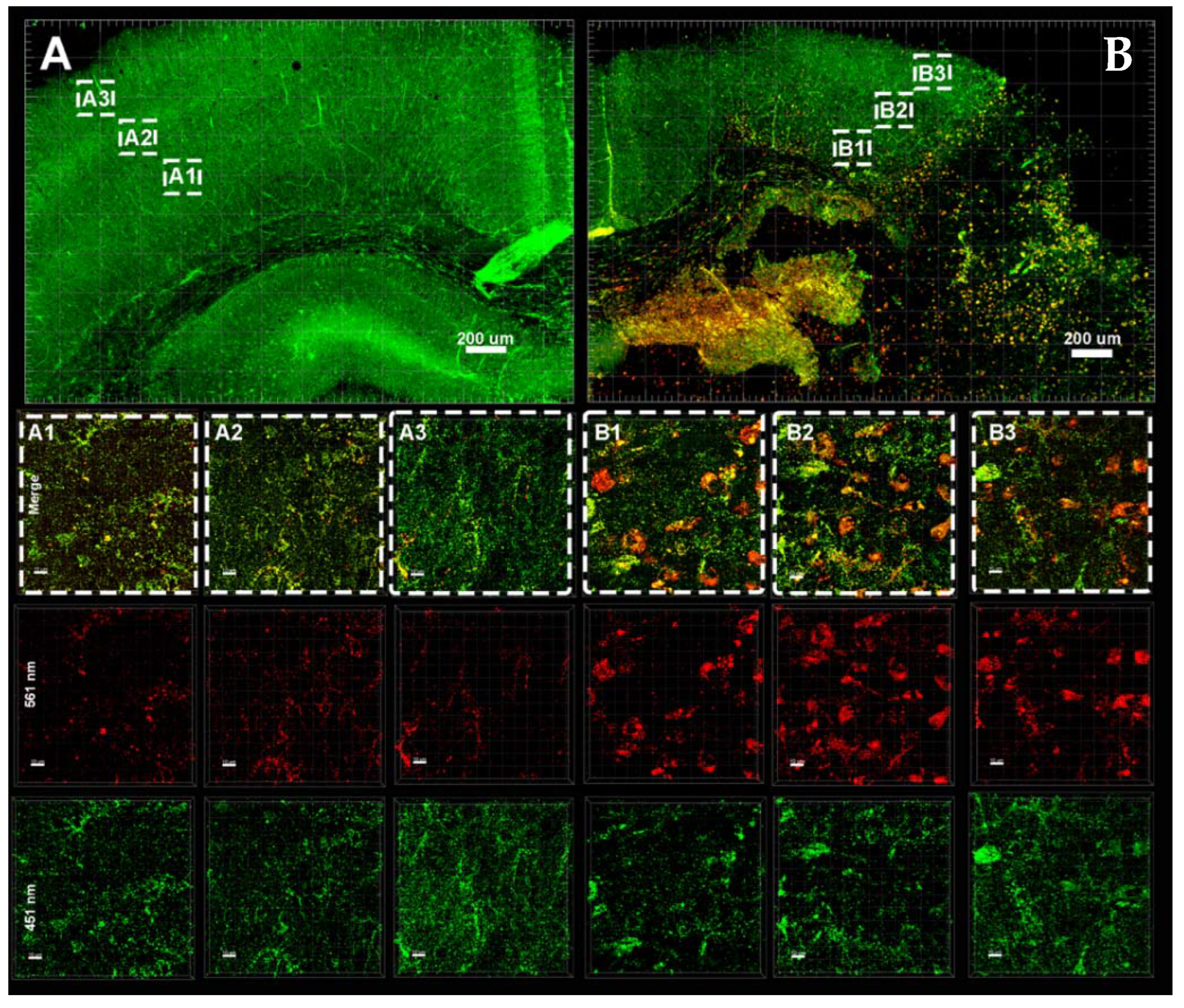
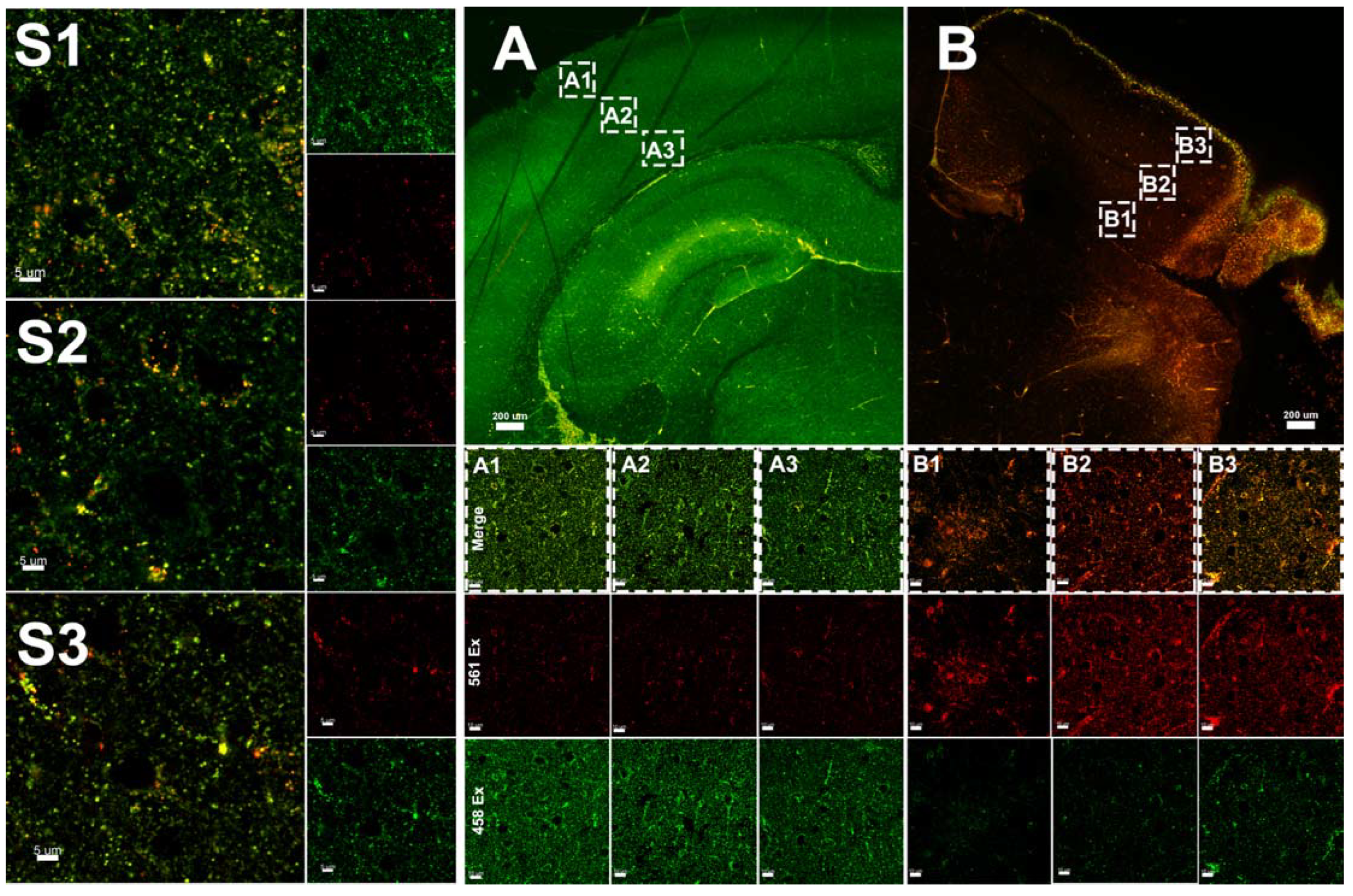
2.8. Assessment of Mitophagy in the Brain Tissue of Mt-Keima Mice
2.9. Calculation of Mitophagy
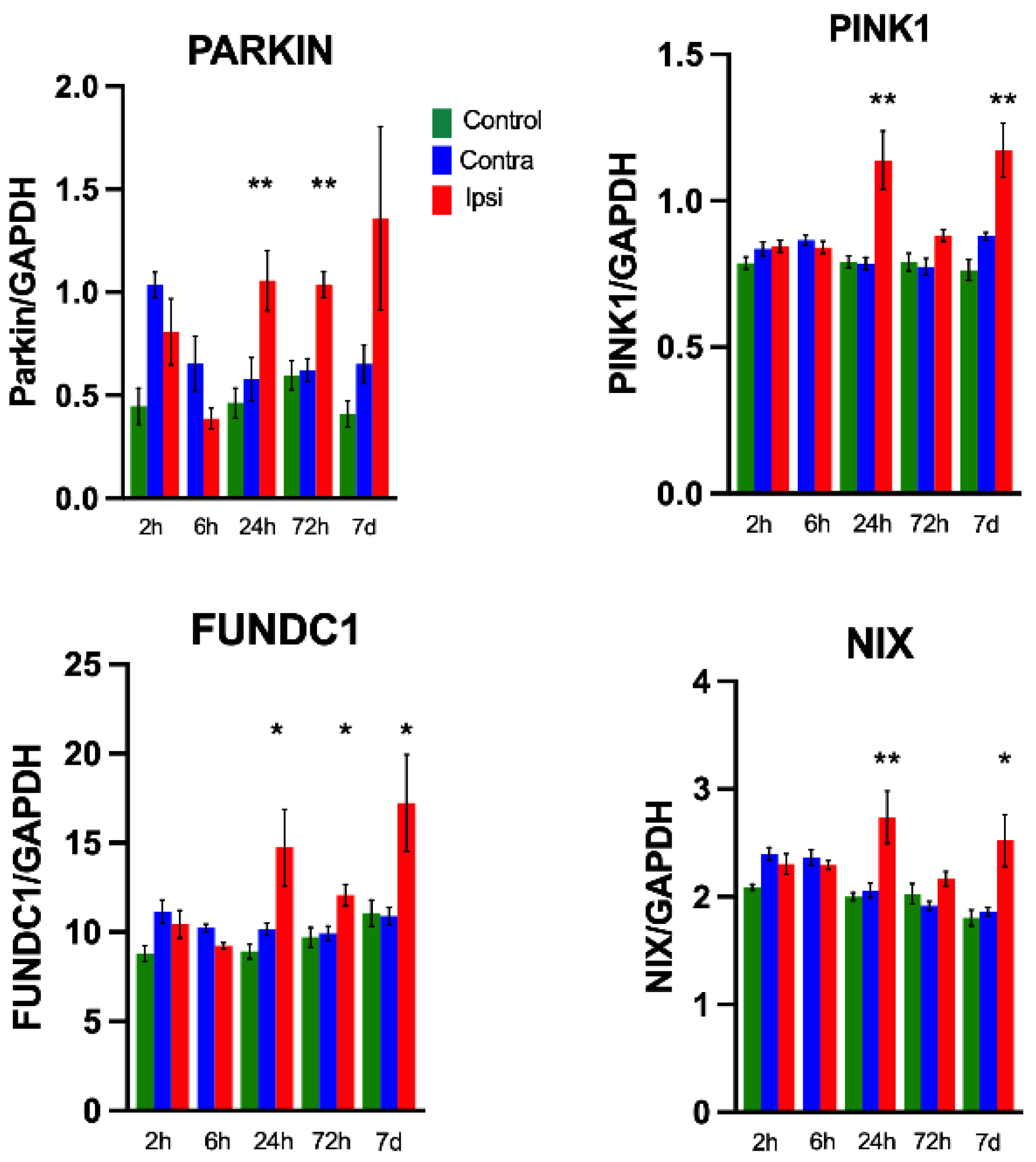
2.10. Quantitative Real-Time PCR
2.11. Preparation of Protein Extracts, Electrophoresis, and Western Blotting
2.12. Statistical Analysis
3. Results
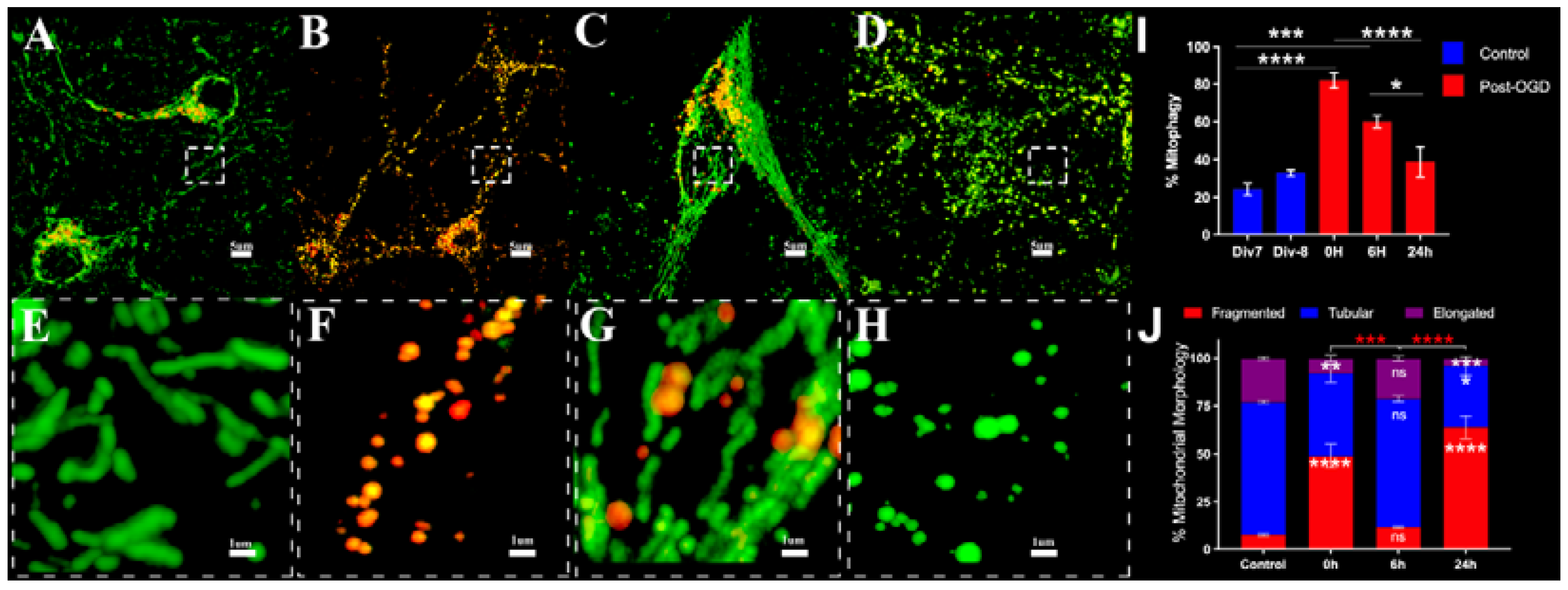
3.1. Oxygen Glucose Deprivation (OGD) Alters Mitophagy and Induces Mitochondrial Fragmentation in Primary Neurons
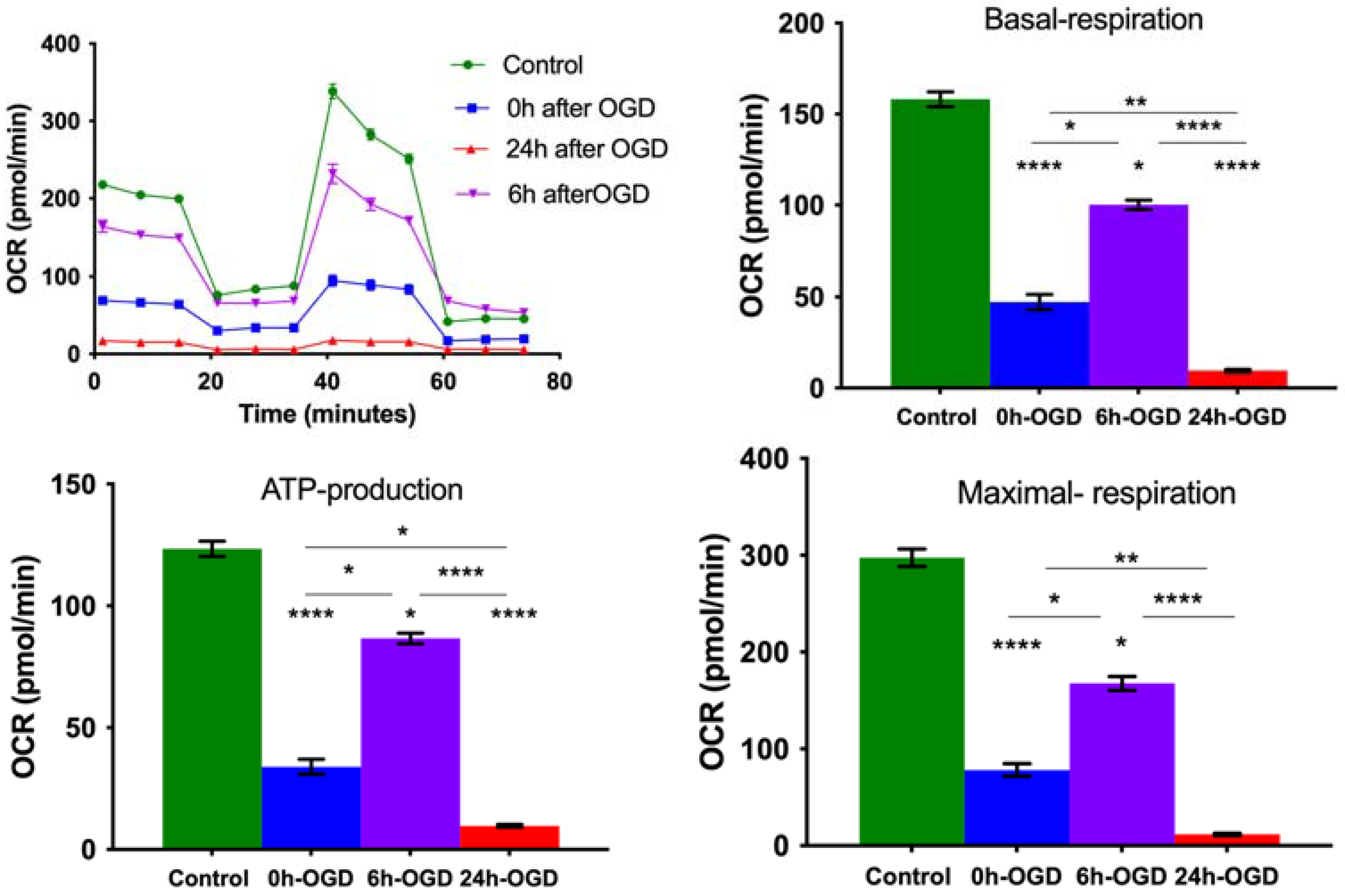
3.2. OGD Reduces Mitochondrial Oxygen Consumption Rate in Primary Neurons
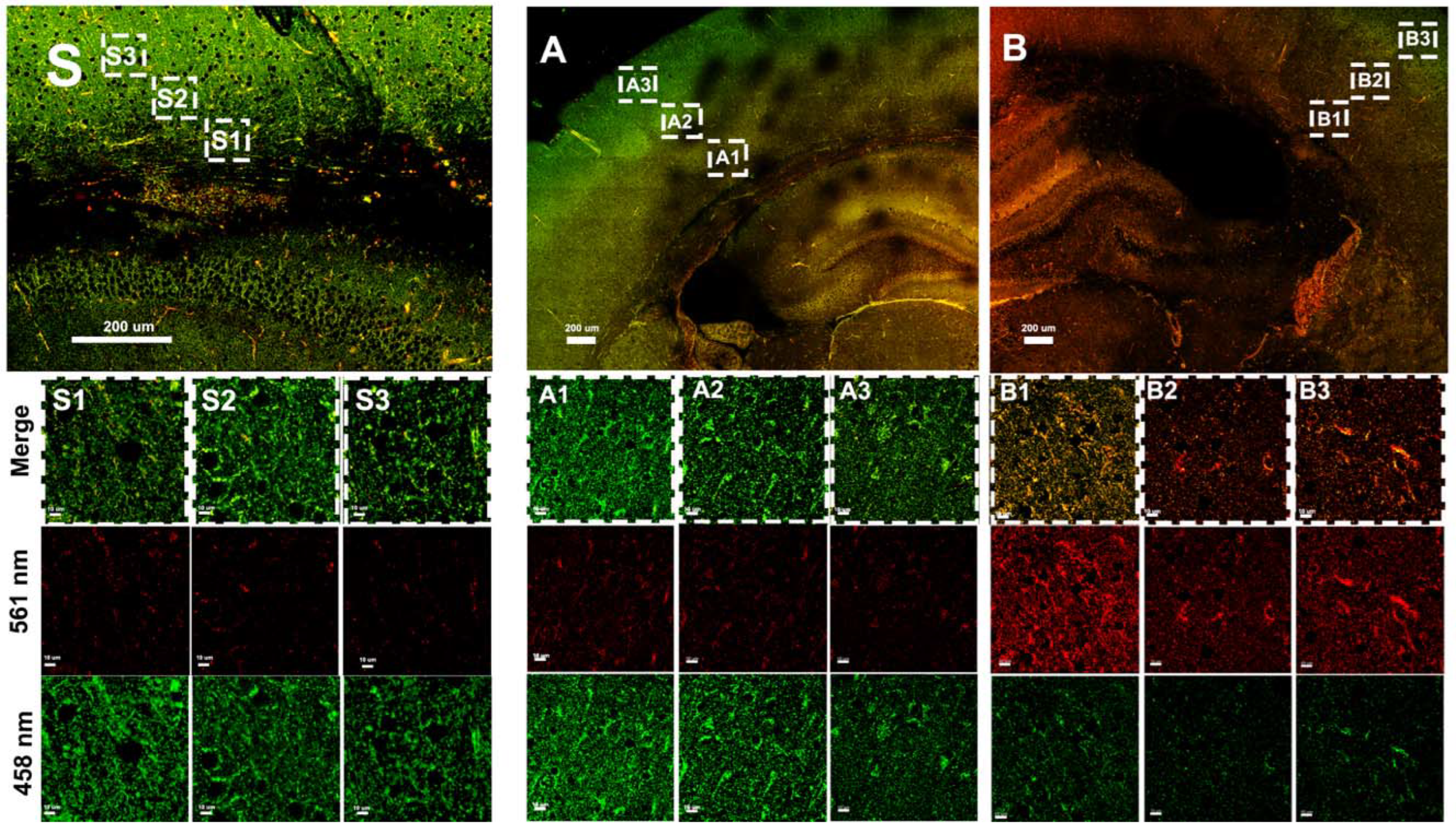
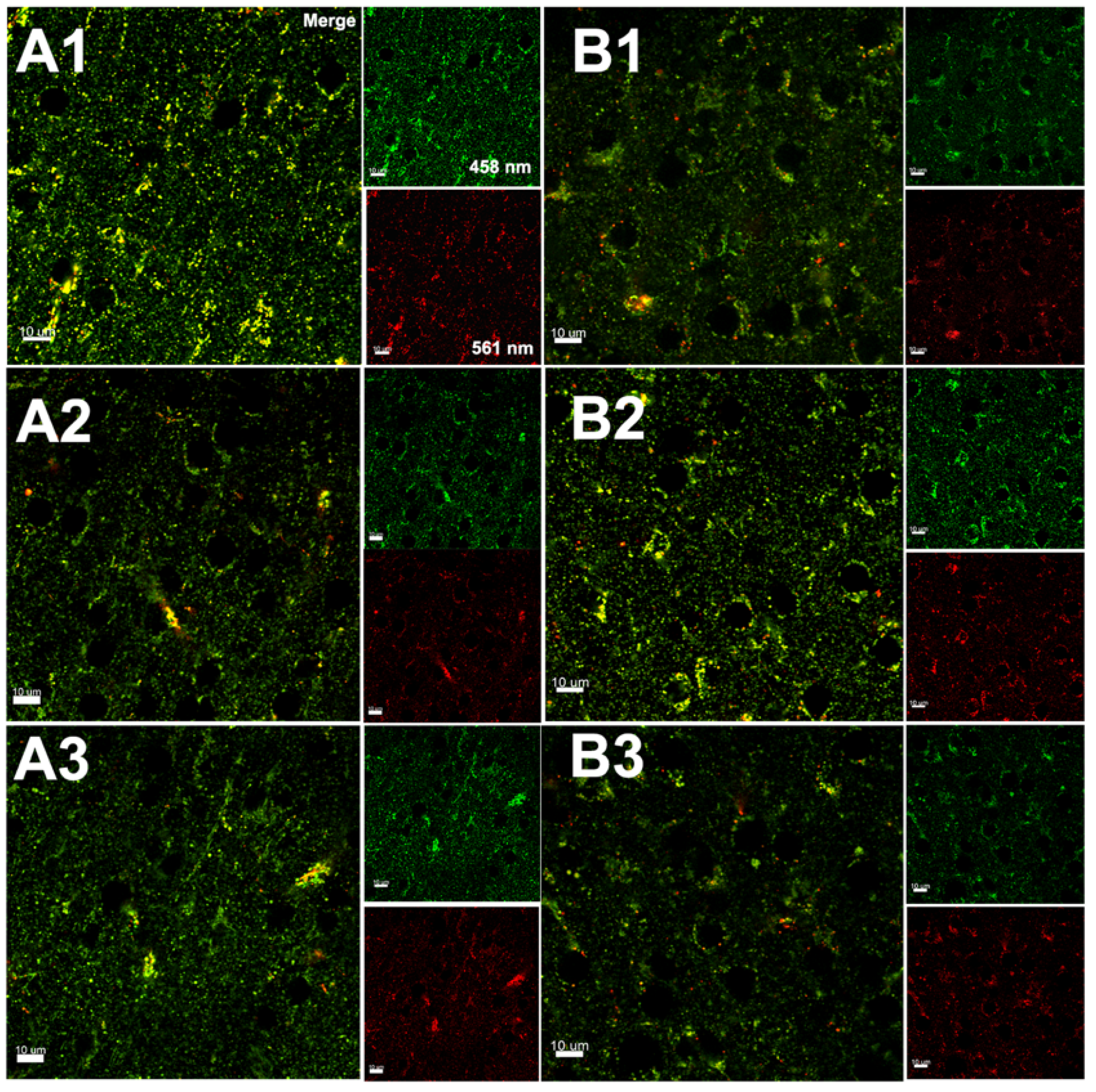
3.3. Hypoxia–Ischemia Alters Mitophagy in the Neonatal Brain
3.4. Hypoxia–Ischemia Upregulates Mitophagy Gene Expression
3.5. Mitophagy Protein Expression Is Altered In Vitro in Primary Neurons after OGD and after HI in the Neonatal Brain
3.5.1. Expression of Mitophagy-Related Proteins in Primary Neurons after OGD
3.5.2. Expression of Mitophagy-Related Proteins in Brain Homogenates after HI
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blomgren, K.; Hagberg, H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic. Biol. Med. 2006, 40, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Niatsetskaya, Z.V.; Sosunov, S.A.; Matsiukevich, D.; Utkina-Sosunova, I.V.; Ratner, V.I.; Starkov, A.A.; Ten, V.S. The Oxygen Free Radicals Originating from Mitochondrial Complex I Contribute to Oxidative Brain Injury Following Hypoxia-Ischemia in Neonatal Mice. J. Neurosci. 2012, 32, 3235–3244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ten, V.S.; Starkov, A. Hypoxic-Ischemic Injury in the Developing Brain: The Role of Reactive Oxygen Species Originating in Mitochondria. Neurol. Res. Int. 2012, 2012, 542976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Bingol, B.; Sheng, M. Mechanisms of mitophagy: PINK1, Parkin, USP30 and beyond. Free Radic. Biol. Med. 2016, 100, 210–222. [Google Scholar] [CrossRef]
- Puka-Sundvall, M.; Gajkowska, B.; Cholewinski, M.; Blomgren, K.; Lazarewicz, J.W.; Hagberg, H. Subcellular distribution of calcium and ultrastructural changes after cerebral hypoxia-ischemia in immature rats. Dev. Brain Res. 2000, 125, 31–41. [Google Scholar] [CrossRef]
- Northington, F.; Zelaya, M.; O’Riordan, D.; Blomgren, K.; Flock, D.; Hagberg, H.; Ferriero, D.; Martin, L. Failure to complete apoptosis following neonatal hypoxia–ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience 2007, 149, 822–833. [Google Scholar] [CrossRef] [Green Version]
- Hagberg, H.; Mallard, C.; Rousset, C.I.; Thornton, C. Mitochondria: Hub of injury responses in the developing brain. Lancet Neurol. 2014, 13, 217–232. [Google Scholar] [CrossRef]
- Baburamani, A.A.; Hurling, C.; Stolp, H.; Sobotka, K.; Gressens, P.; Hagberg, H.; Thornton, C. Mitochondrial Optic Atrophy (OPA) 1 Processing Is Altered in Response to Neonatal Hypoxic-Ischemic Brain Injury. Int. J. Mol. Sci. 2015, 16, 22509–22526. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, N.; Wilkins, H. Mitophagy and the Brain. Int. J. Mol. Sci. 2020, 21, 9661. [Google Scholar] [CrossRef]
- Doxaki, C.; Palikaras, K. Neuronal Mitophagy: Friend or Foe? Front. Cell Dev. Biol. 2020, 8, 1864. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, L.G.; Angelo, Y.D.S.; Iglesias, A.H.; Peron, J.P.S. Unraveling the Link Between Mitochondrial Dynamics and Neuroinflammation. Front. Immunol. 2021, 12, 624919. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Bouhamida, E.; Danese, A.; Previati, M.; Pinton, P.; Patergnani, S. Relevance of Autophagy and Mitophagy Dynamics and Markers in Neurodegenerative Diseases. Biomedicines 2021, 9, 149. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.; Nieminen, A.-L.; Qian, T.; Trost, L.C.; Elmore, S.P.; Nishimura, Y.; Crowe, R.A.; Cascio, W.E.; Bradham, C.A.; Brenner, D.A.; et al. The mitochondrial permeability transition in cell death: A common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta 1998, 1366, 177–196. [Google Scholar] [CrossRef] [Green Version]
- Leaw, B.; Nair, S.; Lim, R.; Thornton, C.; Mallard, C.; Hagberg, H. Mitochondria, Bioenergetics and Excitotoxicity: New Therapeutic Targets in Perinatal Brain Injury. Front. Cell. Neurosci. 2017, 11, 199. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Spengler, D. The Mitochondrion as Potential Interface in Early-Life Stress Brain Programming. Front. Behav. Neurosci. 2018, 12, 306. [Google Scholar] [CrossRef] [Green Version]
- Ivankovic, D.; Chau, K.; Schapira, A.; Gegg, M.E. Mitochondrial and lysosomal biogenesis are activated following PINK 1/parkin-mediated mitophagy. J. Neurochem. 2016, 136, 388–402. [Google Scholar] [CrossRef]
- Demarest, T.; Waite, E.; Kristian, T.; Puche, A.; Waddell, J.; McKenna, M.; Fiskum, G. Sex-dependent mitophagy and neuronal death following rat neonatal hypoxia–ischemia. Neuroscience 2016, 335, 103–113. [Google Scholar] [CrossRef] [Green Version]
- McWilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zheng, Y.; Liu, M.; Li, Y.; Ma, S.; Tang, W.; Yan, W.; Cao, M.; Zheng, W.; Jiang, L.; et al. BNIP3L/NIX degradation leads to mitophagy deficiency in ischemic brains. Autophagy 2020, 17, 1934–1946. [Google Scholar] [CrossRef]
- Thornton, C.; Jones, A.; Nair, S.; Aabdien, A.; Mallard, C.; Hagberg, H. Mitochondrial dynamics, mitophagy and biogenesis in neonatal hypoxic-ischaemic brain injury. FEBS Lett. 2018, 592, 812–830. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.-Y.; Zhu, S.-H.; Li, V.; Gibson, S.; Xu, X.; Kong, J.-M. BNIP3 Interacting with LC3 Triggers Excessive Mitophagy in Delayed Neuronal Death in Stroke. CNS Neurosci. Ther. 2014, 20, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef] [Green Version]
- Katayama, H.; Kogure, T.; Mizushima, N.; Yoshimori, T.; Miyawaki, A. A Sensitive and Quantitative Technique for Detecting Autophagic Events Based on Lysosomal Delivery. Chem. Biol. 2011, 18, 1042–1052. [Google Scholar] [CrossRef] [Green Version]
- Thornton, C.; Bright, N.J.; Sastre, M.; Muckett, P.J.; Carling, D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid β-peptide exposure. Biochem. J. 2011, 434, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.; Sobotka, K.S.; Joshi, P.; Gressens, P.; Fleiss, B.; Thornton, C.; Mallard, C.; Hagberg, H. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia 2019, 67, 1047–1061. [Google Scholar] [CrossRef] [Green Version]
- Rice, J.E., 3rd; Vannucci, R.C.; Brierley, J.B. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann. Neurol. 1981, 9, 131–141. [Google Scholar] [CrossRef]
- Hedtjärn, M.; Leverin, A.-L.; Eriksson, K.; Blomgren, K.; Mallard, C.; Hagberg, H. Interleukin-18 Involvement in Hypoxic–Ischemic Brain Injury. J. Neurosci. 2002, 22, 5910–5919. [Google Scholar] [CrossRef]
- Svedin, P.; Hagberg, H.; Sävman, K.; Zhu, C.; Mallard, C. Matrix Metalloproteinase-9 Gene Knock-out Protects the Immature Brain after Cerebral Hypoxia–Ischemia. J. Neurosci. 2007, 27, 1511–1518. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Malide, D.; Liu, J.; Rovira, I.I.; Combs, C.A.; Finkel, T. A fluorescence-based imaging method to measure in vitro and in vivo mitophagy using mt-Keima. Nat. Protoc. 2017, 12, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Colella, A.D.; Chegenii, N.; Tea, M.N.; Gibbins, I.L.; Williams, K.A.; Chataway, T.K. Comparison of Stain-Free gels with traditional immunoblot loading control methodology. Anal. Biochem. 2012, 430, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Gürtler, A.; Kunz, N.; Gomolka, M.; Hornhardt, S.; Friedl, A.A.; McDonald, K.; Kohn, J.E.; Posch, A. Stain-Free technology as a normalization tool in Western blot analysis. Anal. Biochem. 2013, 433, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Rona-Voros, K.; Weydt, P. The Role of PGC-1α in the Pathogenesis of Neurodegenerative Disorders. Curr. Drug Targets 2010, 11, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Motori, E.; Atanassov, I.; Kochan, S.M.V.; Folz-Donahue, K.; Sakthivelu, V.; Giavalisco, P.; Toni, N.; Puyal, J.; Larsson, N.-G. Neuronal metabolic rewiring promotes resilience to neurodegeneration caused by mitochondrial dysfunction. Sci. Adv. 2020, 6, eaba8271. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta 2012, 1823, 2297–2310. [Google Scholar] [CrossRef]
- Song, Y.; Du, Y.; Zou, W.; Luo, Y.; Zhang, X.; Fu, J. Involvement of impaired autophagy and mitophagy in Neuro-2a cell damage under hypoxic and/or high-glucose conditions. Sci. Rep. 2018, 8, 3301. [Google Scholar] [CrossRef] [Green Version]
- Mironova, G.D.; Pavlik, L.L.; Kirova, Y.I.; Belosludtseva, N.V.; Mosentsov, A.A.; Khmil, N.V.; Germanova, E.L.; Lukyanova, L.D. Effect of hypoxia on mitochondrial enzymes and ultrastructure in the brain cortex of rats with different tolerance to oxygen shortage. J. Bioenerg. Biomembr. 2019, 51, 329–340. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Talanov, E.Y.; Starinets, V.S.; Tenkov, K.S.; Zakharova, N.M.; Belosludtseva, N.V. Transport of Ca2+ and Ca2+-Dependent Permeability Transition in the Liver and Heart Mitochondria of Rats with Different Tolerance to Acute Hypoxia. Biomolecules 2020, 10, 114. [Google Scholar] [CrossRef] [Green Version]
- Zuo, W.; Zhang, S.; Xia, C.-Y.; Guo, X.-F.; He, W.-B.; Chen, N.-H. Mitochondria autophagy is induced after hypoxic/ischemic stress in a Drp1 dependent manner: The role of inhibition of Drp1 in ischemic brain damage. Neuropharmacology 2014, 86, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Bukowski, M.J.; Wider, J.M.; Reynolds, C.; Calo, L.; Lepore, B.; Tousignant, R.; Jones, M.; Przyklenk, K.; Sanderson, T.H. Mitochondrial dynamics following global cerebral ischemia. Mol. Cell. Neurosci. 2016, 76, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; McEwan, D.G.; Dikic, I. The LC3 interactome at a glance. J. Cell Sci. 2014, 127, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodolfo, C.; Campello, S.; Cecconi, F. Mitophagy in neurodegenerative diseases. Neurochem. Int. 2018, 117, 156–166. [Google Scholar] [CrossRef]
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta Bioenerg. 2015, 1853, 2784–2790. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.-X.; Ni, H.-M.; Li, M.; Liao, Y.; Chen, X.; Stolz, D.B.; Dorn, G.W., 2nd; Yin, X.-M. Nix Is Critical to Two Distinct Phases of Mitophagy, Reactive Oxygen Species-mediated Autophagy Induction and Parkin-Ubiquitin-p62-mediated Mitochondrial Priming. J. Biol. Chem. 2010, 285, 27879–27890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Klionsky, D.J. Molecular process and physiological significance of mitophagy. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, Aging; Hayat, M.A., Ed.; Academic Press: Amsterdam, The Netherlands, 2014; pp. 49–63. [Google Scholar]
- Koentjoro, B.; Park, J.-S.; Sue, C.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7, srep44373. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, G.; Liang, T.; Pan, P.; Li, X.; Li, H.; Shen, H.; Wang, Z.; Chen, G. Nix Plays a Neuroprotective Role in Early Brain Injury After Experimental Subarachnoid Hemorrhage in Rats. Front. Neurosci. 2020, 14, 245. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Rogov, V.V.; Suzuki, H.; Marinković, M.; Lang, V.; Kato, R.; Kawasaki, M.; Buljubasic, M.; Šprung, M.; Rogova, N.; Wakatsuki, S.; et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 2017, 7, 1131. [Google Scholar] [CrossRef]
- Marinković, M.; Šprung, M.; Novak, I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 2021, 17, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, L.; Cheng, Q.; Li, Y.; Wu, H.; Zhang, W.; Wang, Y.; Sehgal, S.A.; Siraj, S.; Wang, X.; et al. Mitochondrial E3 ligase MARCH 5 regulates FUNDC 1 to fine-tune hypoxic mitophagy. EMBO Rep. 2017, 18, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Vincow, E.S.; Merrihew, G.; Thomas, R.E.; Shulman, N.J.; Beyer, R.P.; MacCoss, M.J.; Pallanck, L.J. The PINK1–Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 6400–6405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Sun, Y.; Guo, S.; Lu, B. The PINK1/Parkin pathway regulates mitochondrial dynamics and function in mammalian hippocampal and dopaminergic neurons. Hum. Mol. Genet. 2011, 20, 3227–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M. Keeping the immune system in check: A role for mitophagy. Immunol. Cell Biol. 2015, 93, 3–10. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Romaní-Aumedes, J.; Canal, M.; Martín-Flores, N.; Sun, X.; Perez-Fernandez, V.; Wewering, S.; Fernández-Santiago, R.; Ezquerra, M.; Pont-Sunyer, C.; Lafuente, A.; et al. Parkin loss of function contributes to RTP801 elevation and neurodegeneration in Parkinson’s disease. Cell Death Dis. 2014, 5, e1364. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Li, L.; Zhan, L.; Zuo, Y.; Li, K.; Qiu, M.; Li, H.; Sun, W.; Xu, E. Hypoxic postconditioning promotes mitophagy against transient global cerebral ischemia via PINK1/Parkin-induced mitochondrial ubiquitination in adult rats. Cell Death Dis. 2021, 12, 630. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, S.; Leverin, A.-L.; Rocha-Ferreira, E.; Sobotka, K.S.; Thornton, C.; Mallard, C.; Hagberg, H. Induction of Mitochondrial Fragmentation and Mitophagy after Neonatal Hypoxia–Ischemia. Cells 2022, 11, 1193. https://doi.org/10.3390/cells11071193
Nair S, Leverin A-L, Rocha-Ferreira E, Sobotka KS, Thornton C, Mallard C, Hagberg H. Induction of Mitochondrial Fragmentation and Mitophagy after Neonatal Hypoxia–Ischemia. Cells. 2022; 11(7):1193. https://doi.org/10.3390/cells11071193
Chicago/Turabian StyleNair, Syam, Anna-Lena Leverin, Eridan Rocha-Ferreira, Kristina S. Sobotka, Claire Thornton, Carina Mallard, and Henrik Hagberg. 2022. "Induction of Mitochondrial Fragmentation and Mitophagy after Neonatal Hypoxia–Ischemia" Cells 11, no. 7: 1193. https://doi.org/10.3390/cells11071193
APA StyleNair, S., Leverin, A.-L., Rocha-Ferreira, E., Sobotka, K. S., Thornton, C., Mallard, C., & Hagberg, H. (2022). Induction of Mitochondrial Fragmentation and Mitophagy after Neonatal Hypoxia–Ischemia. Cells, 11(7), 1193. https://doi.org/10.3390/cells11071193