
GLP-1 Mediates Regulation of Colonic ACE2 Expression by the Bile Acid Receptor GPBAR1 in Inflammation

 ,
,  ,
,  ,
, 
Abstract
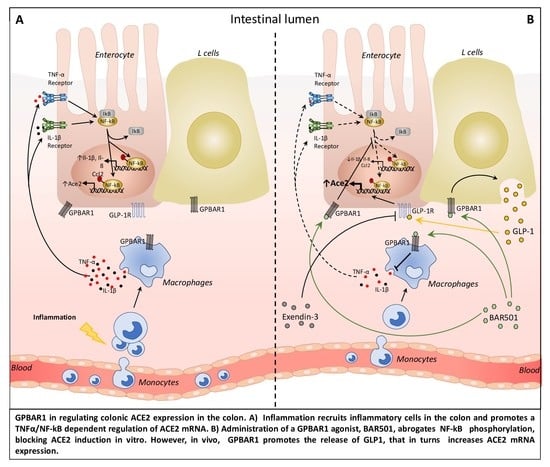
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. Human Tissue Sample
2.2. GEO Data Sets
2.3. Animals
2.4. Mouse Models of Colitis
2.5. Clinical Disease Activity Index (CDAI)
2.6. Mouse Endoscopies
2.7. Intestinal Permeability
2.8. Determination of Blood Glucose Levels
2.9. Histopathology
2.10. Immunohistochemistry
2.11. Cell Culture
2.12. RNA Extraction
2.13. Reverse Transcription of mRNA and Real-Time PCR
2.14. Western Blot
2.15. Statistical Analysis
3. Results
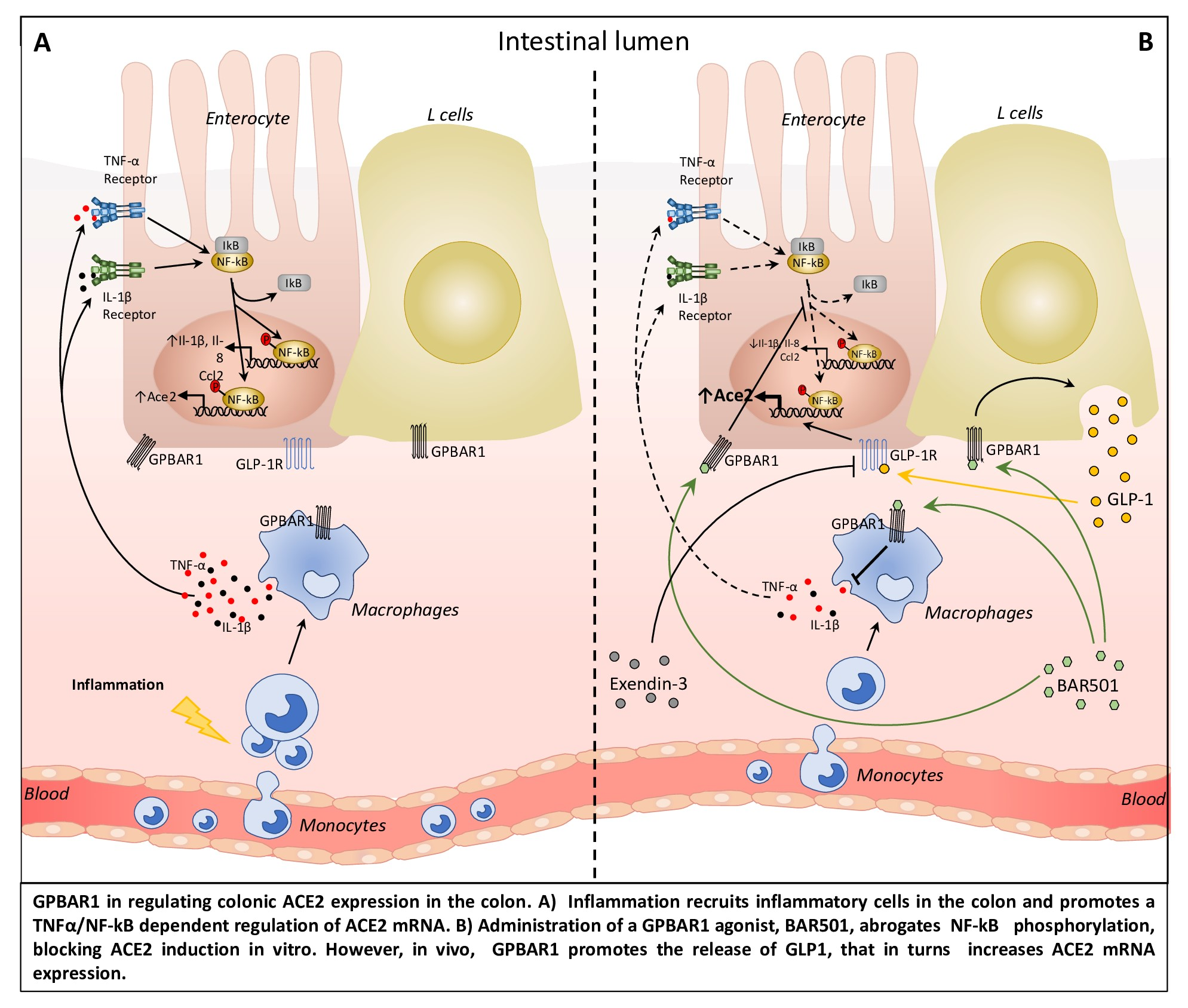
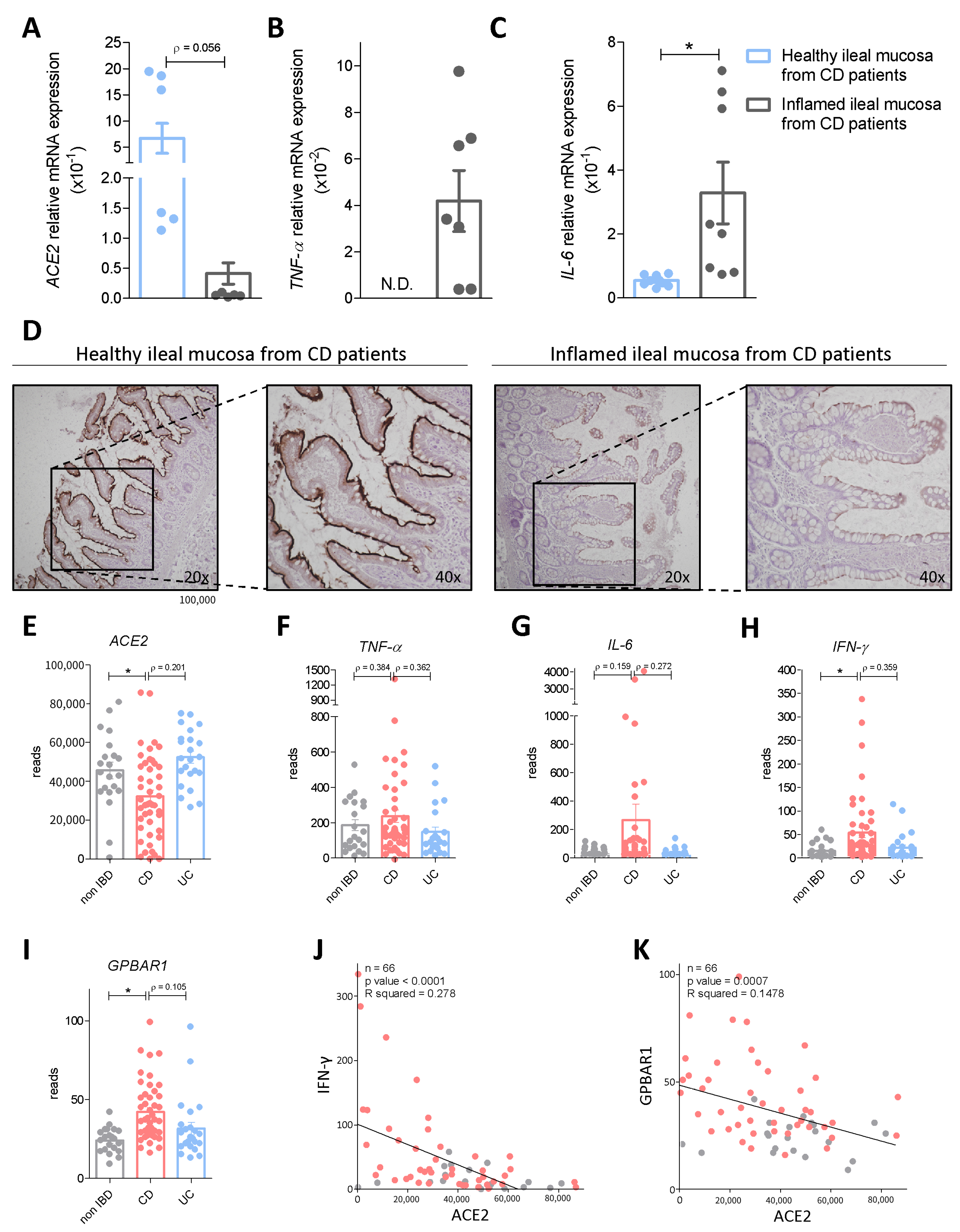
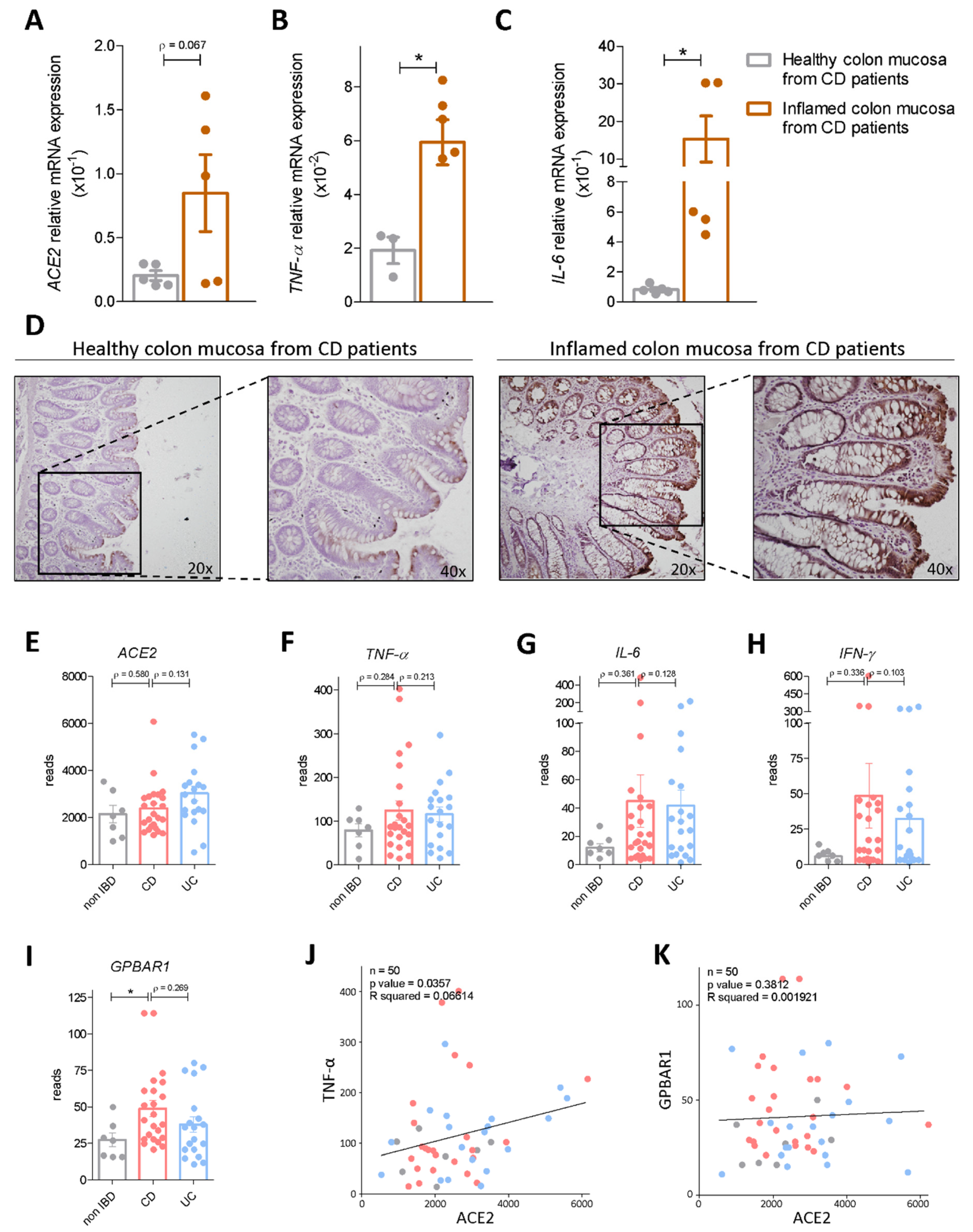
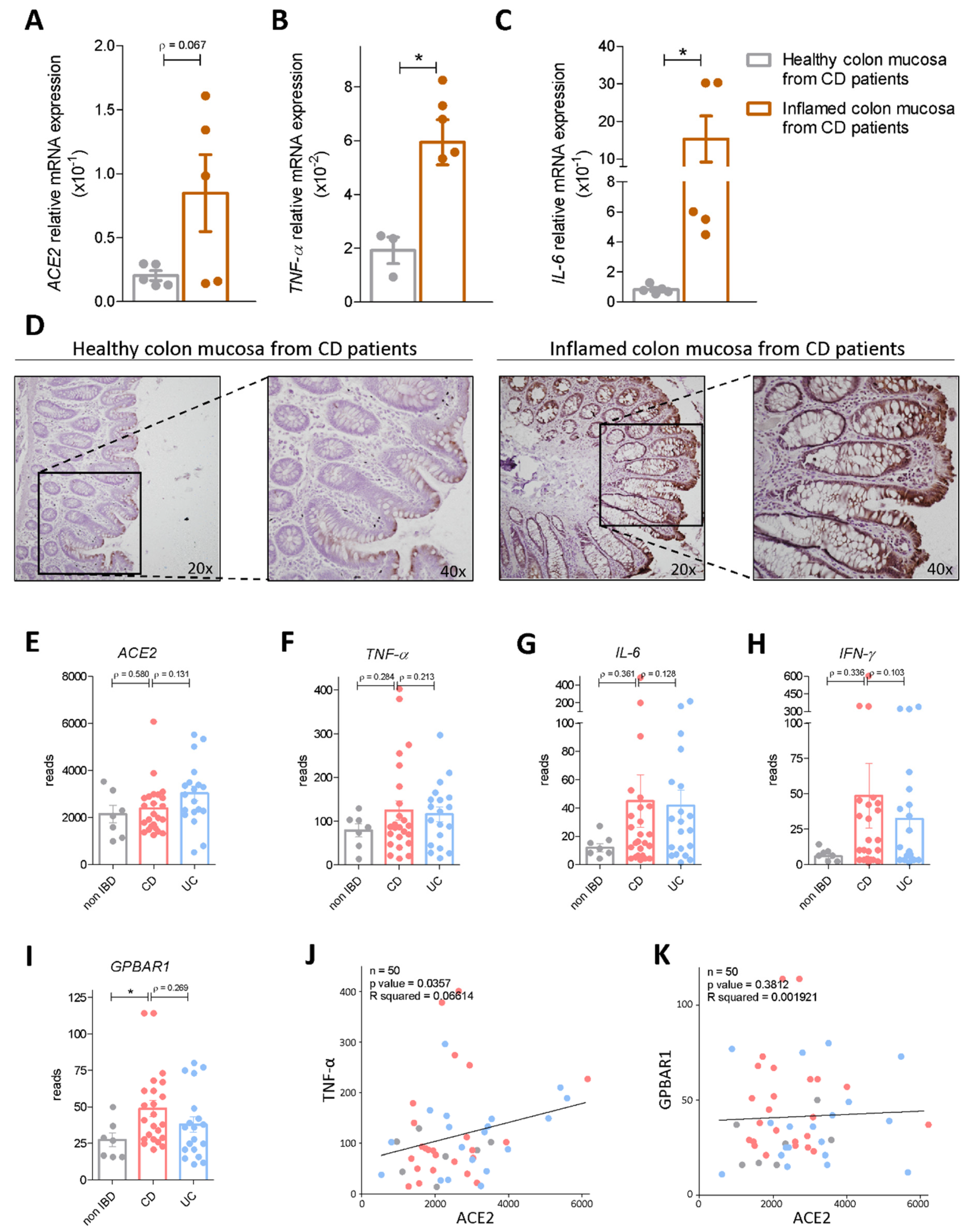
3.1. Regional Variation of ACE2 mRNA Expression in Intact and Inflamed Gut
3.2. Interaction of Bile Acid Signaling and ACE2 in Ileum and Colon
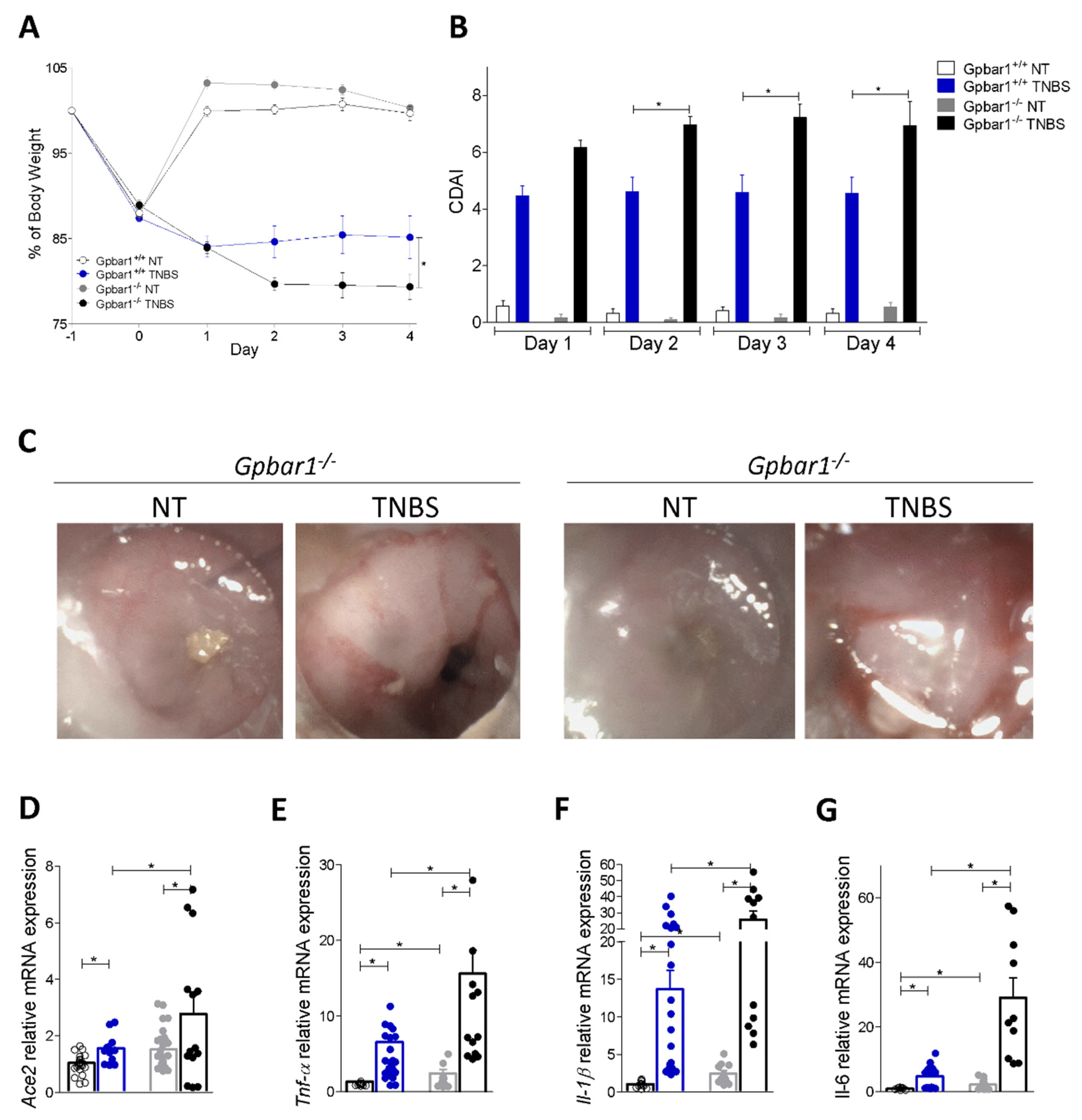
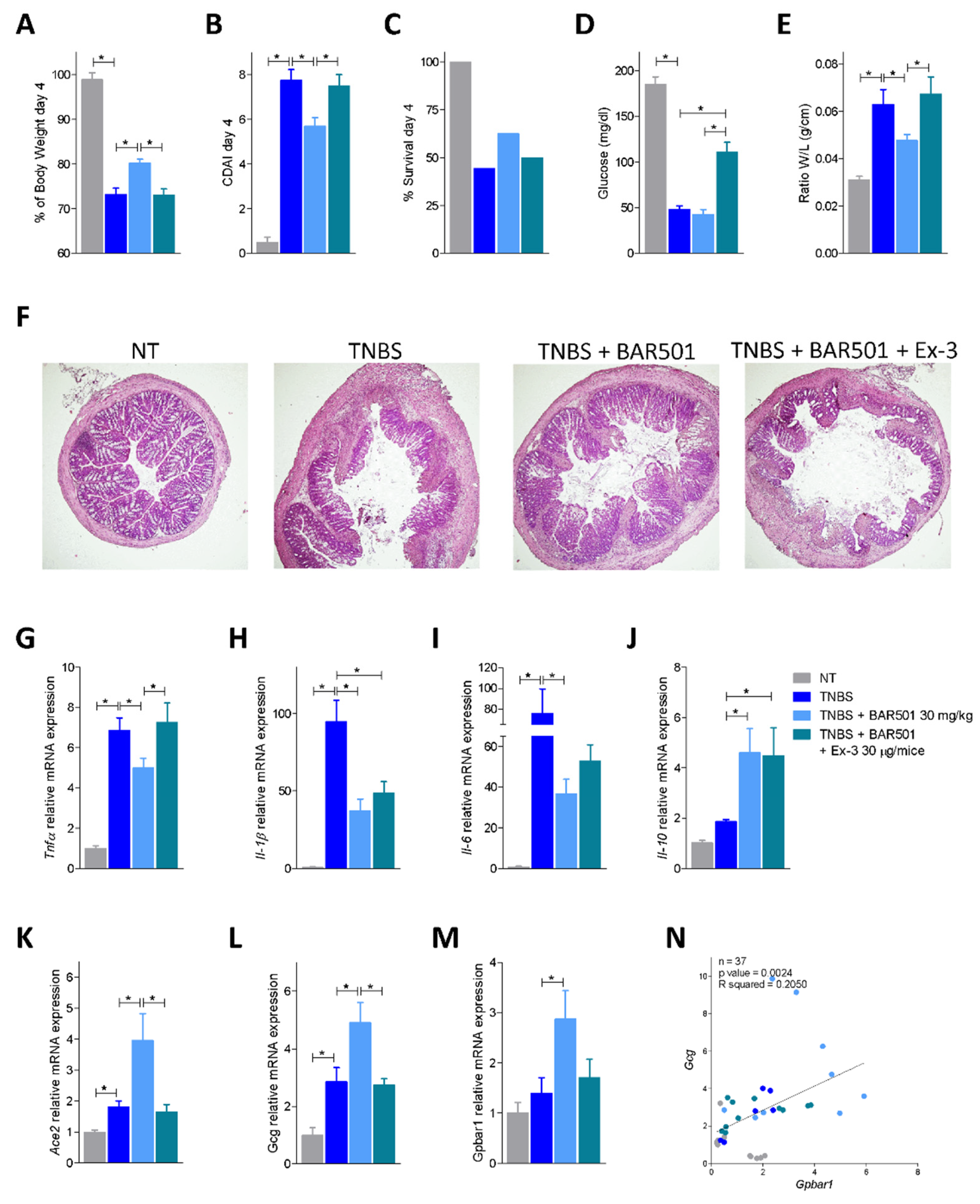
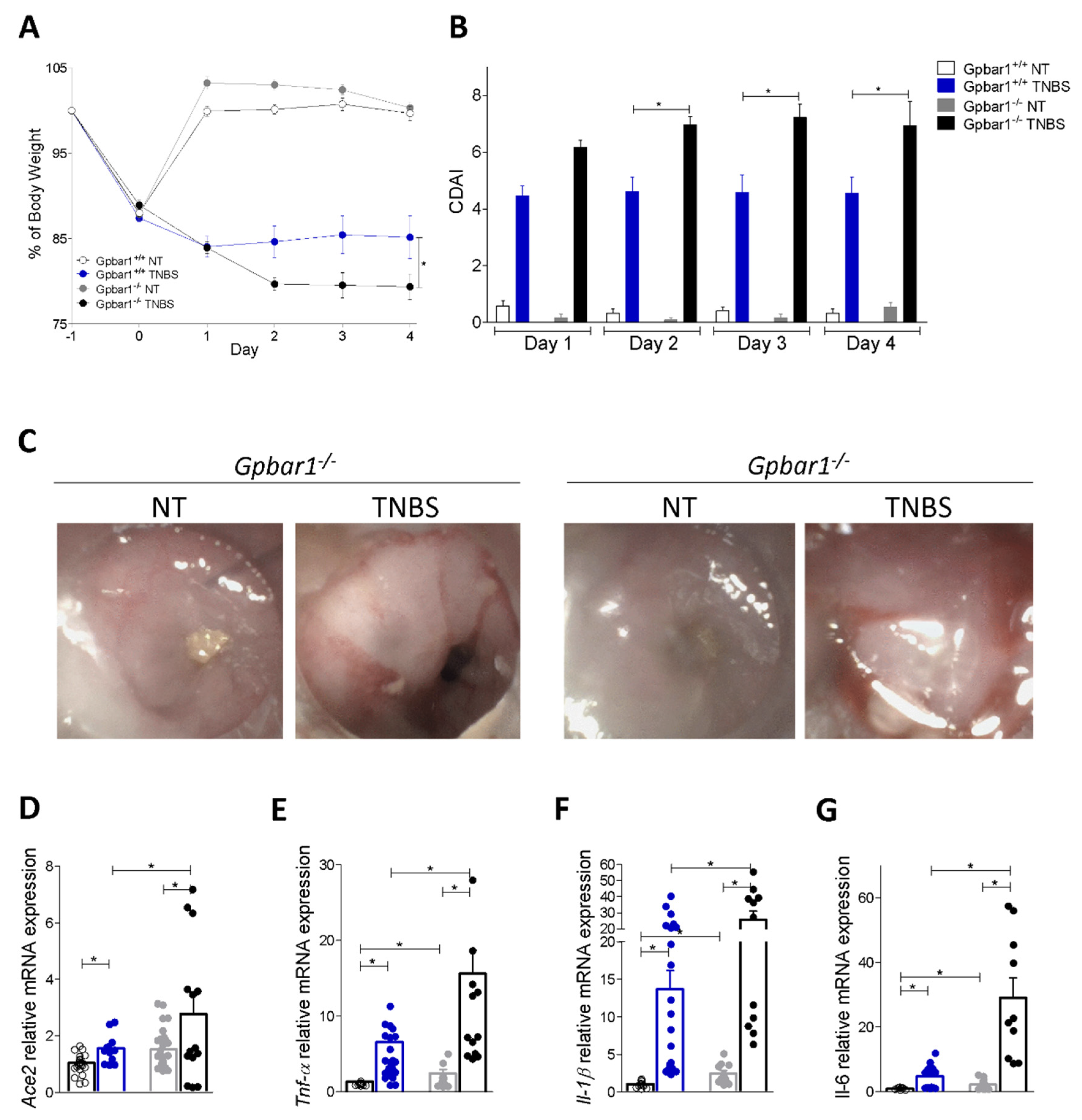
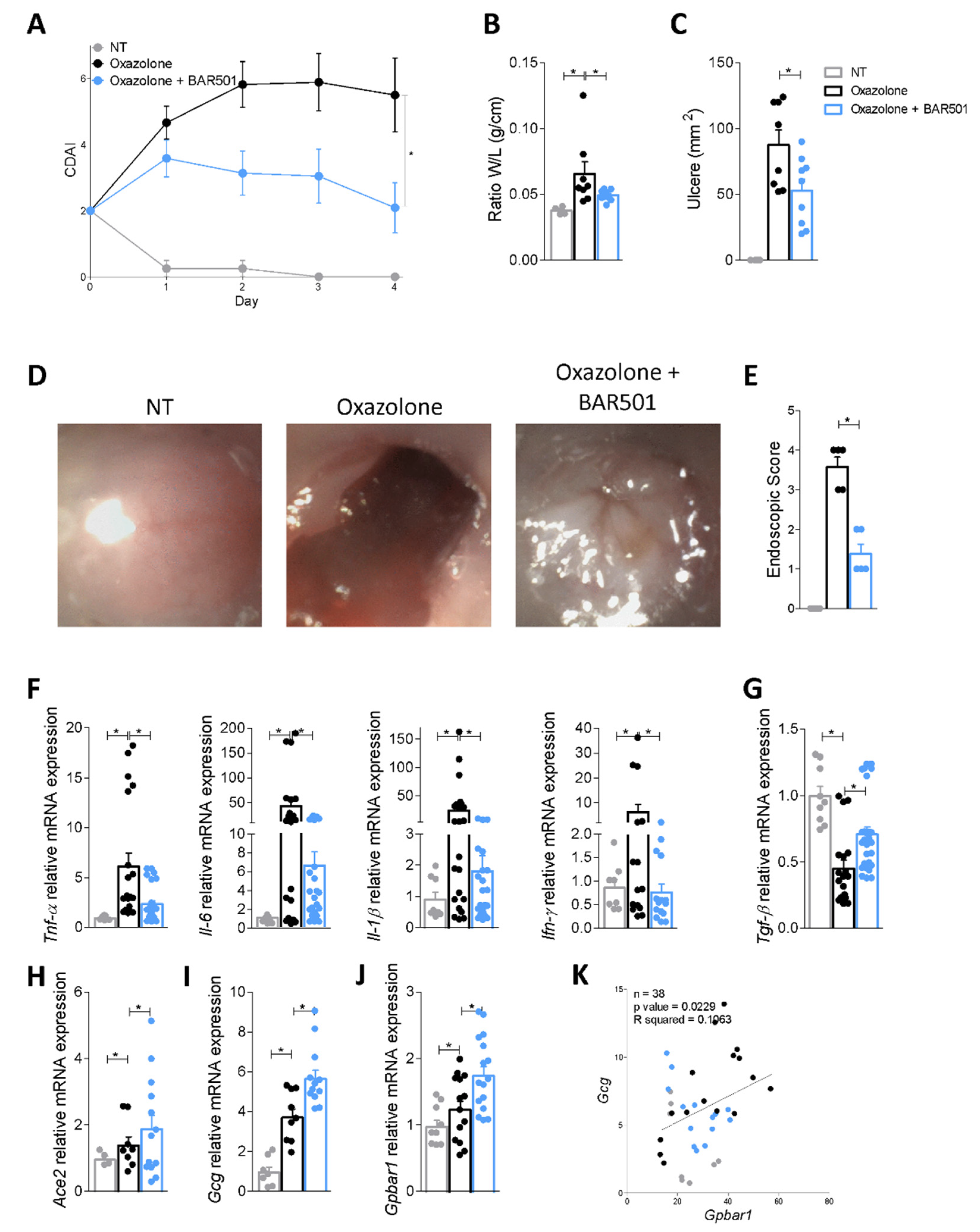
3.3. Regulation of Ace2 Expression by GPBAR1 in Mouse Models of Intestinal Inflammation
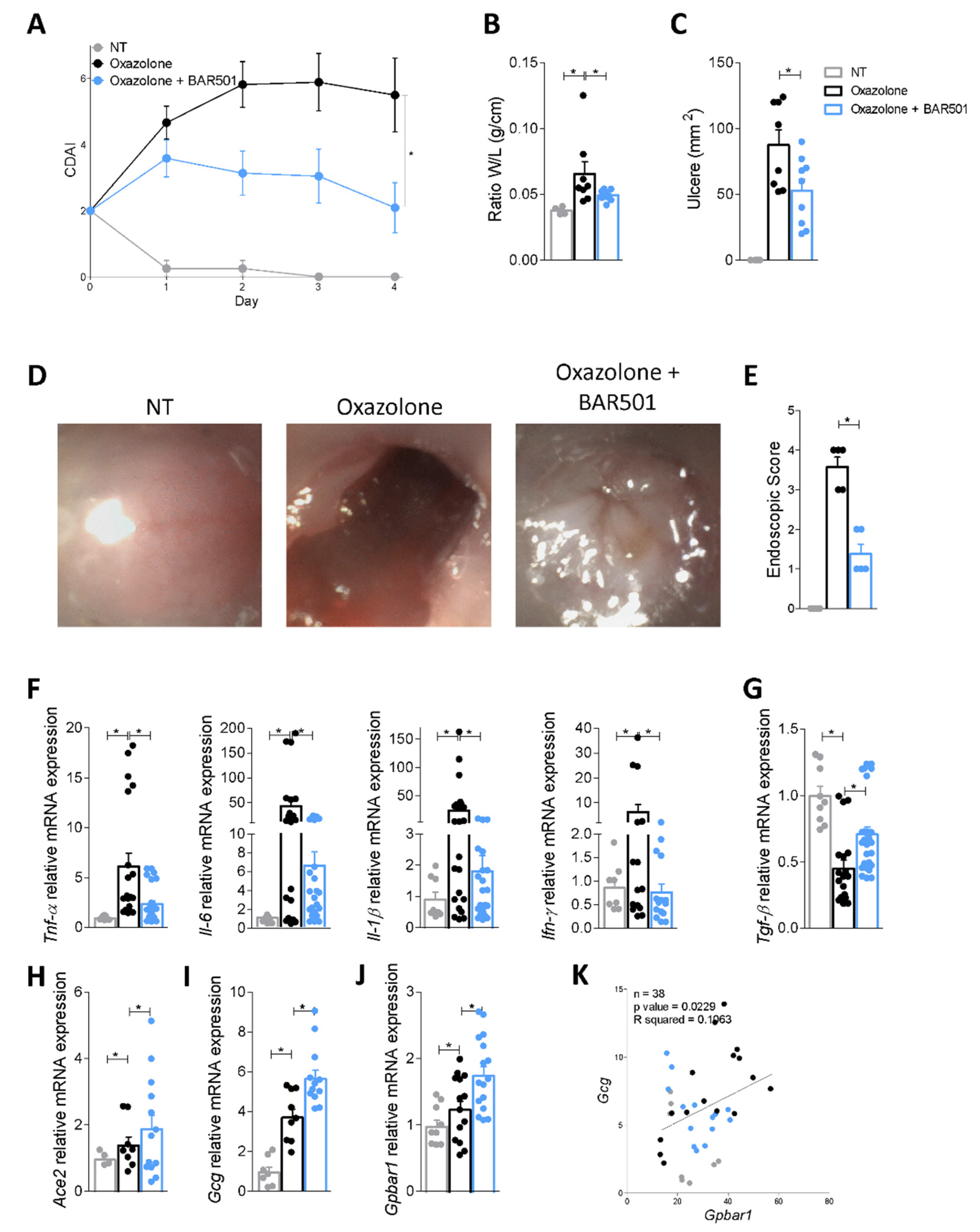
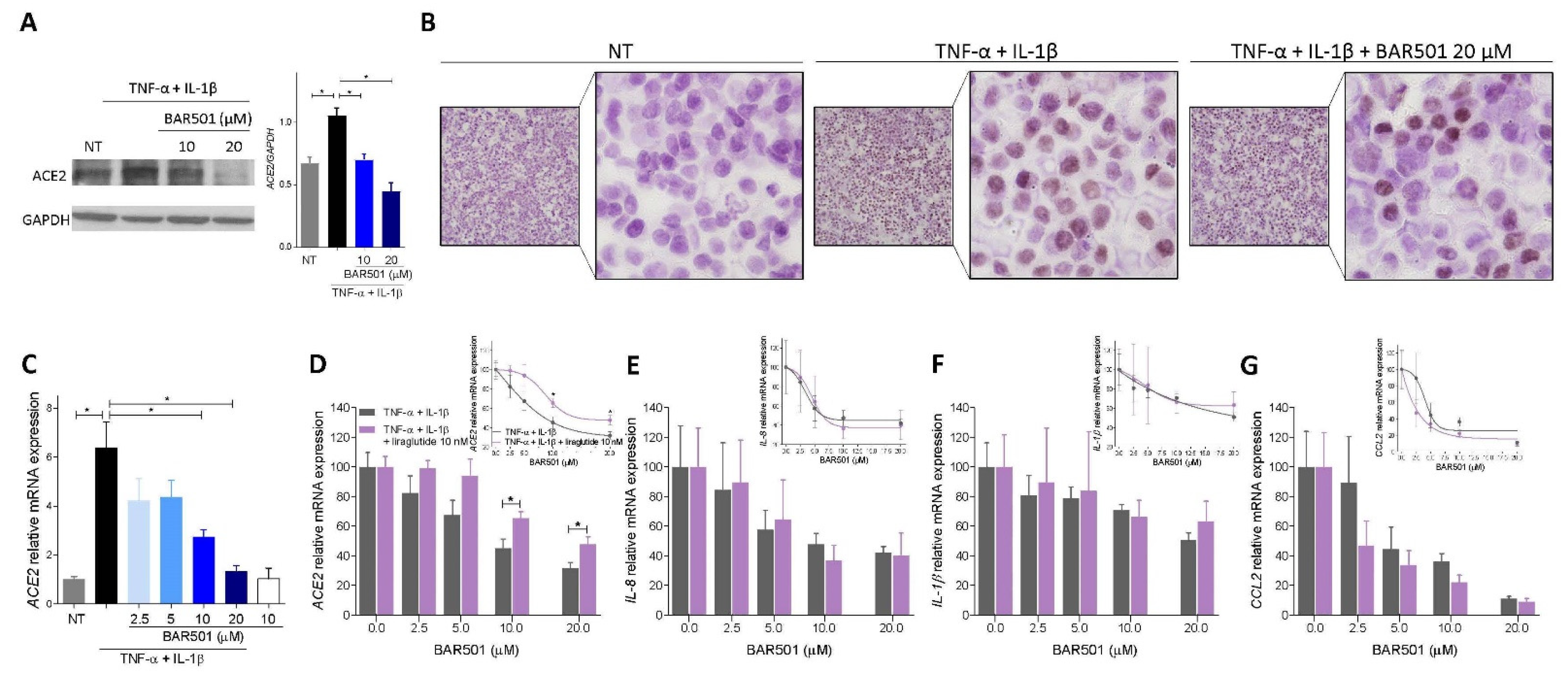
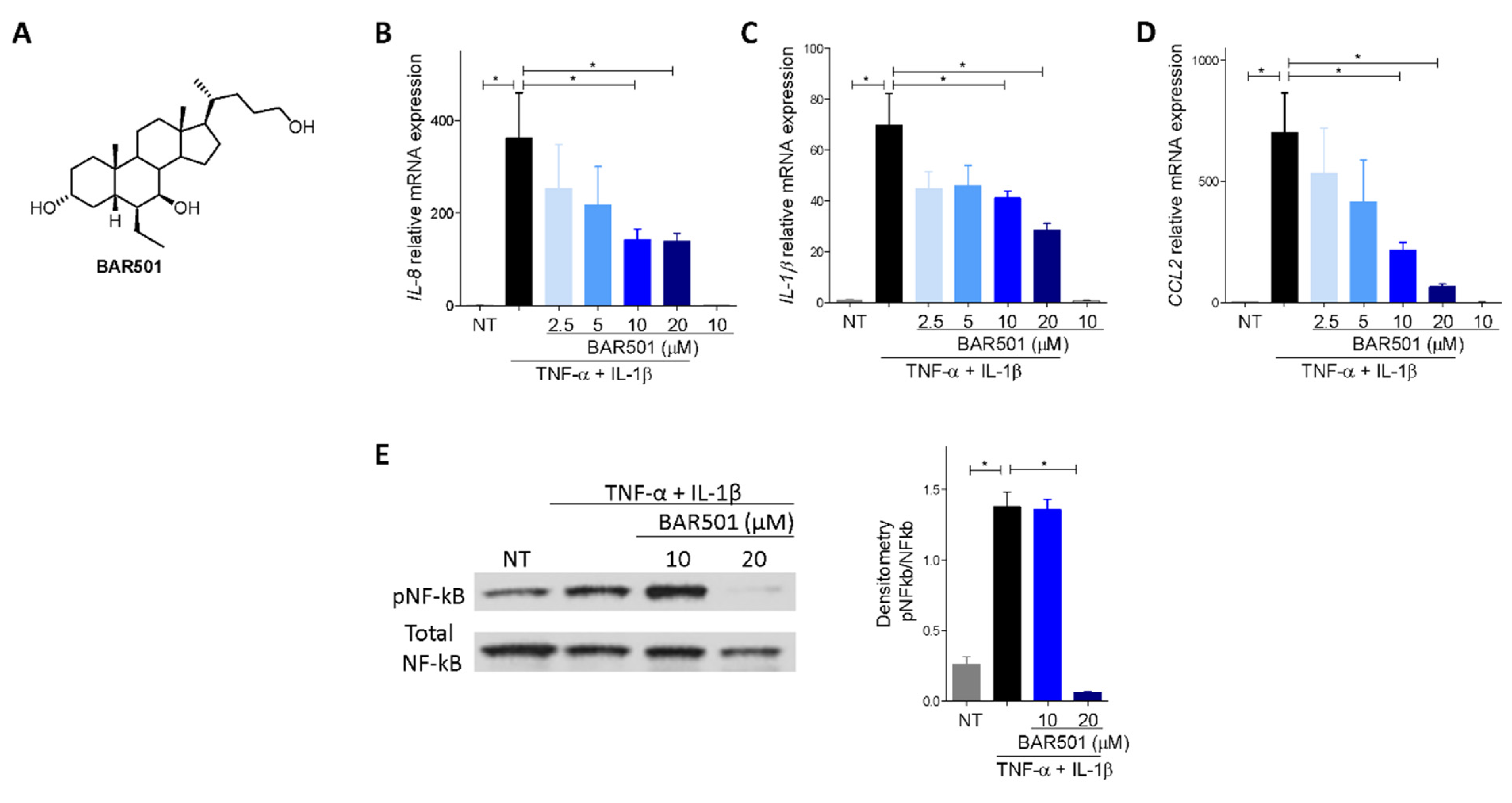
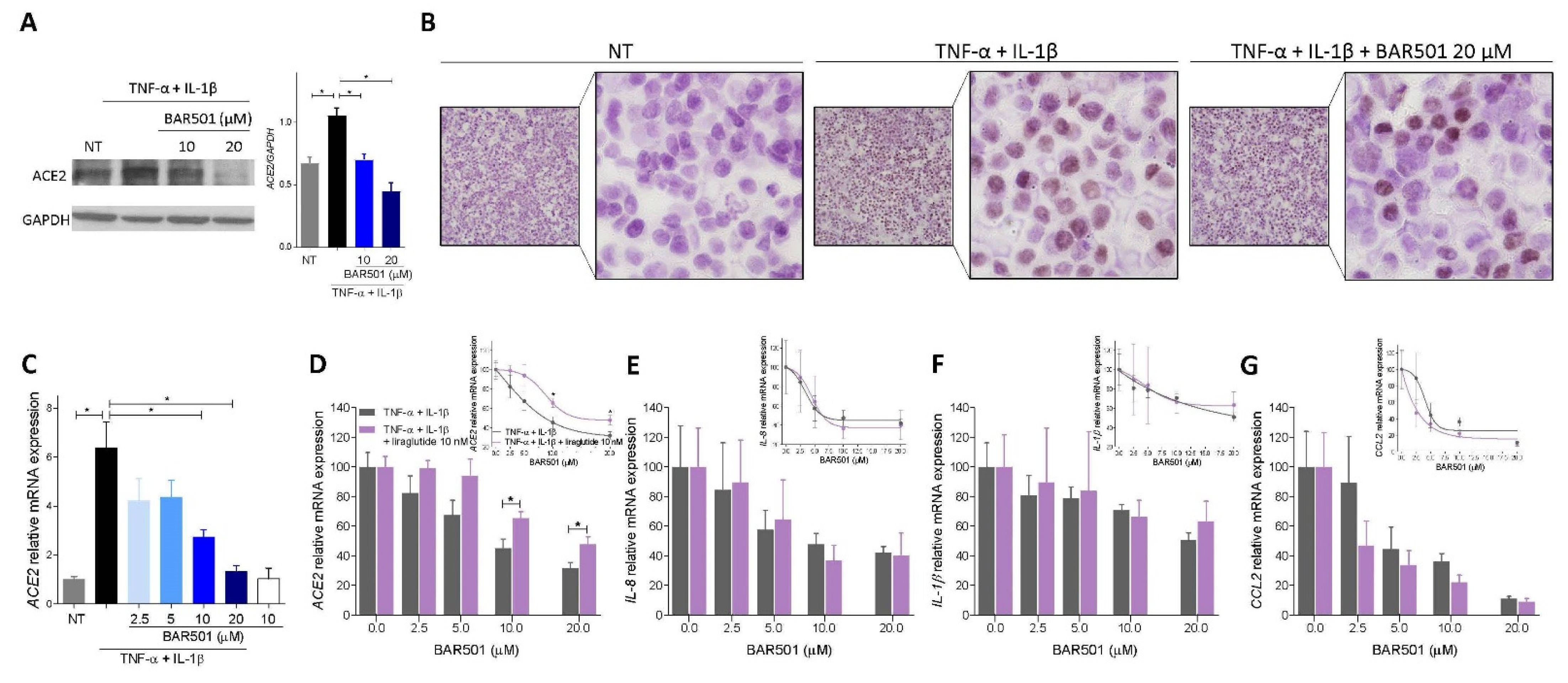
3.4. BAR501, a Selective GPBAR1 Agonist Regulates ACE2 Expression In Vivo
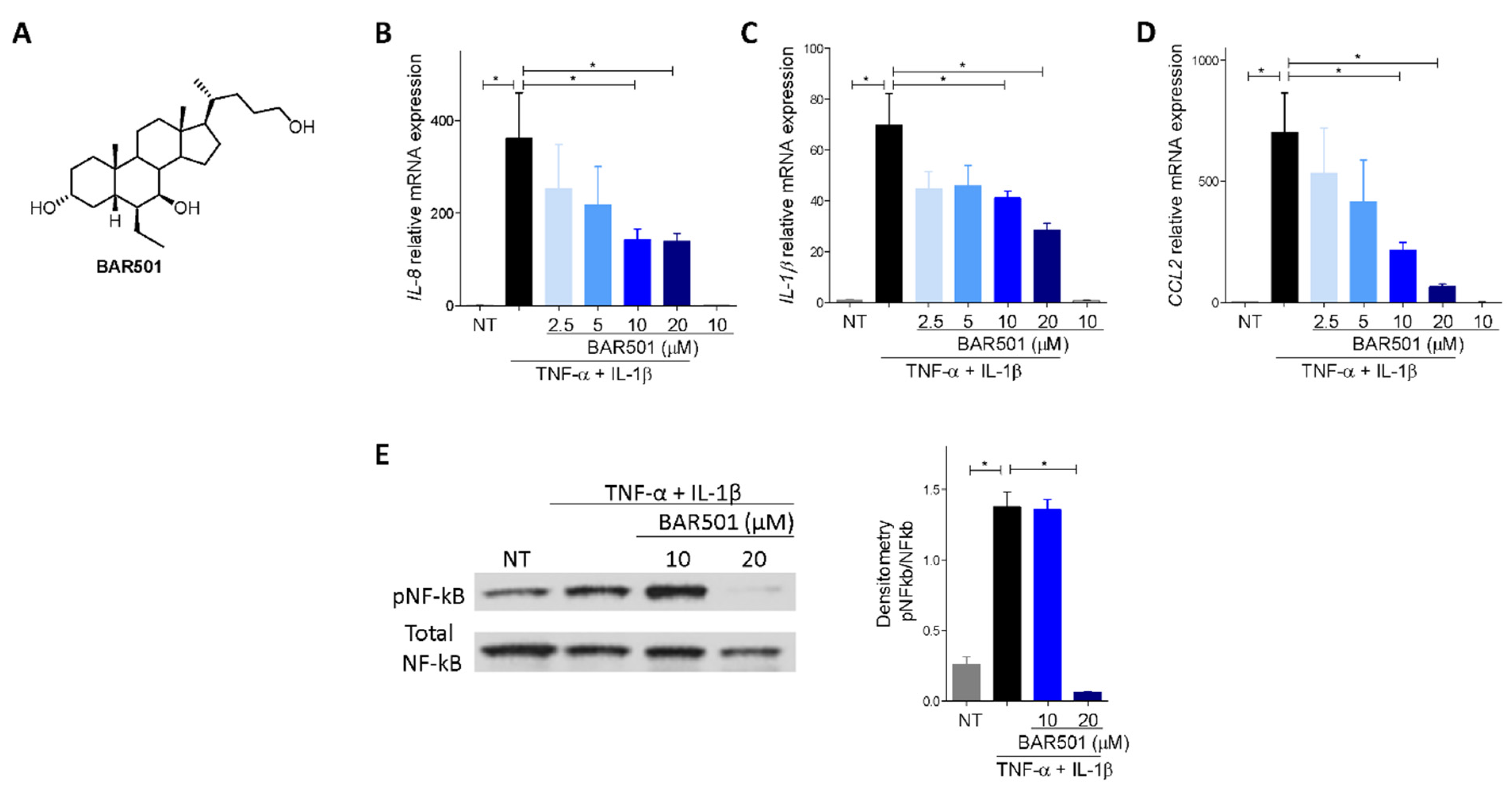
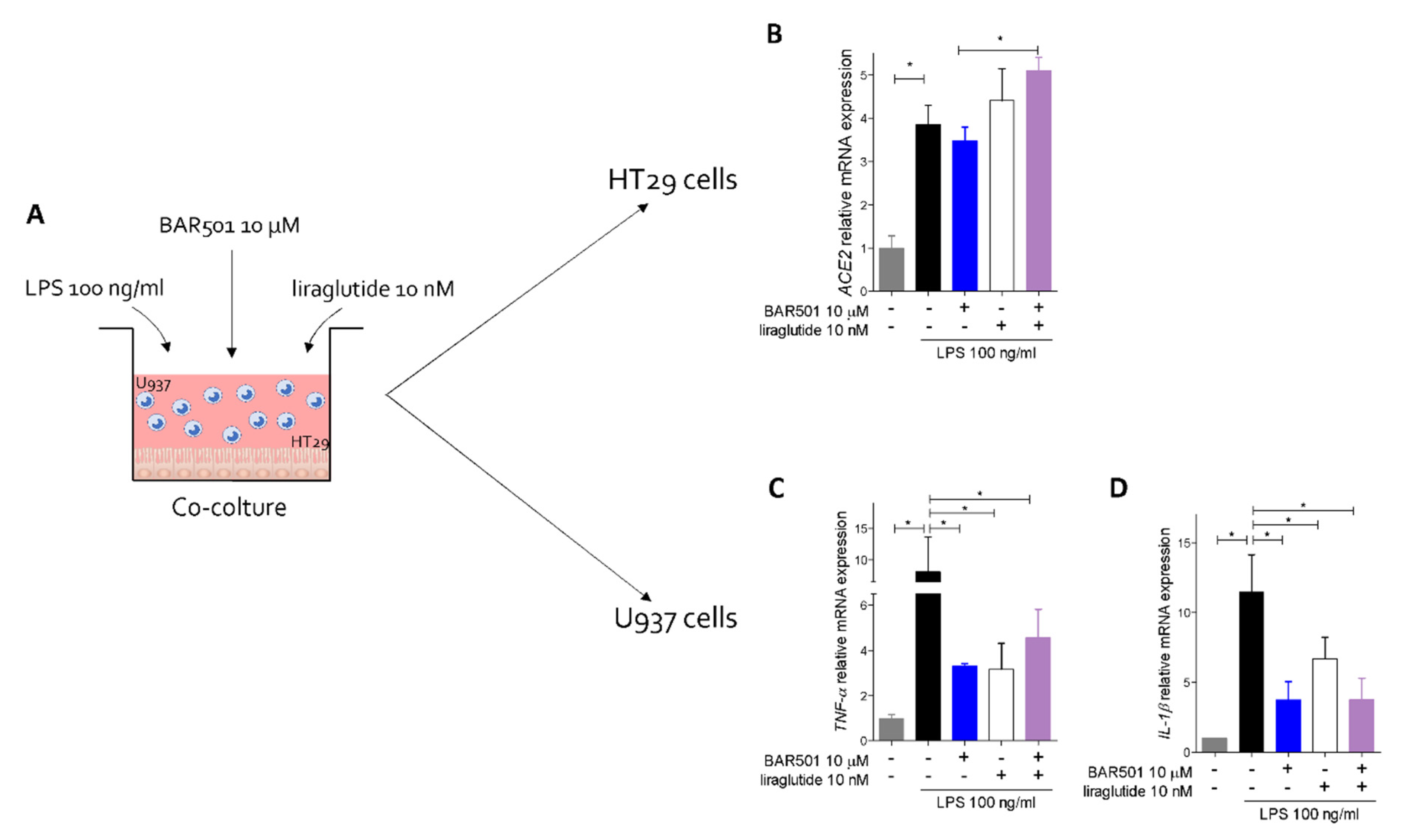
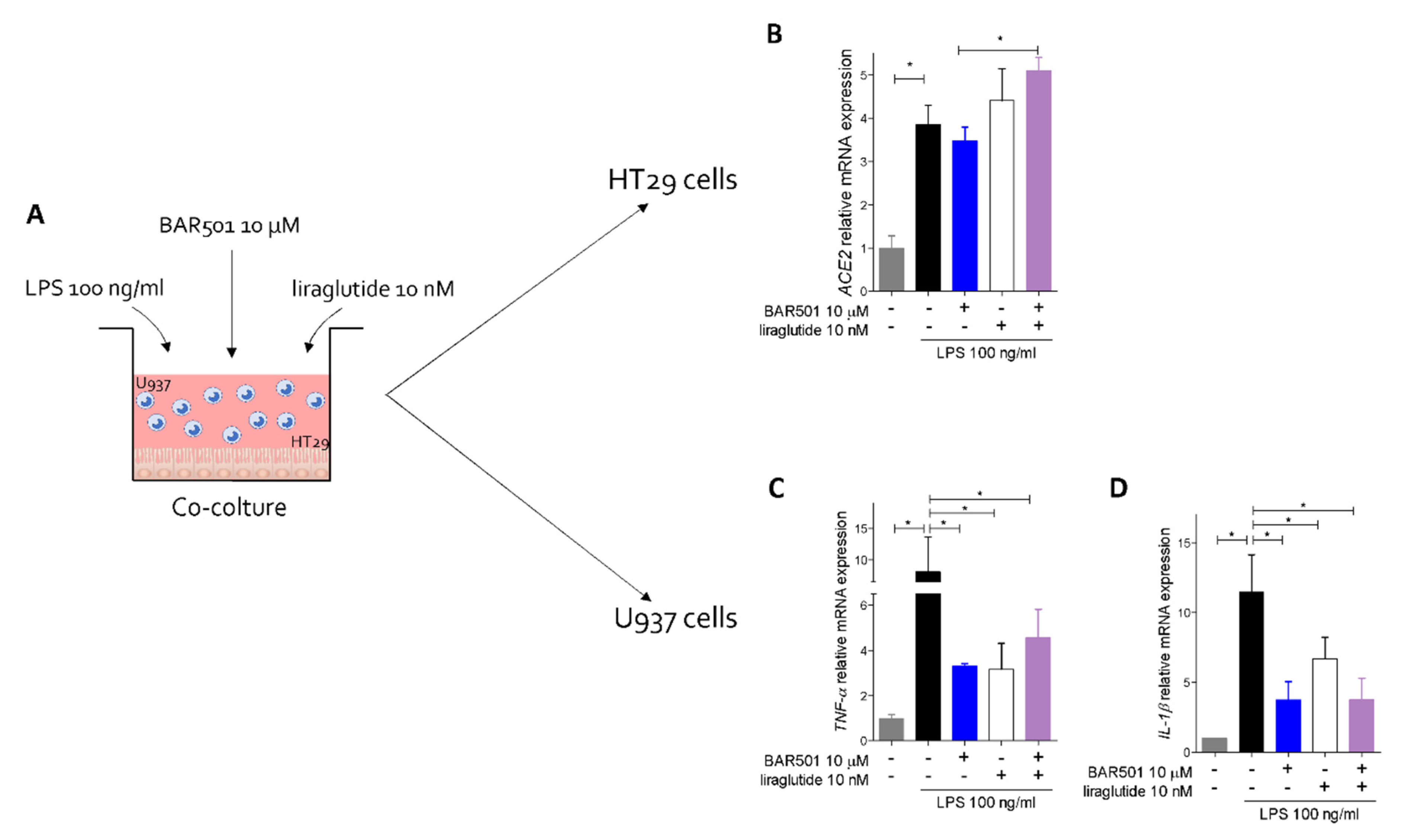
3.5. Effect of Activation of GPBAR1 on the Colorectal Adenocarcinoma HT29 Cell Line
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arendse, L.B.; Danser, A.H.J.; Poglitsch, M.; Touyz, R.M.; Burnett, J.C.J.; Llorens-Cortes, C.; Ehlers, M.R.; Sturrock, E.D. Novel Therapeutic Approaches Targeting the Renin-Angiotensin System and Associated Peptides in Hypertension and Heart Failure. Pharmacol. Rev. 2019, 71, 539–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; He, X.; Zhang, L.; Ran, Q.; Wang, J.; Xiong, A.; Wu, D.; Chen, F.; Sun, J.; Chang, C. Assessing ACE2 Expression Patterns in Lung Tissues in the Pathogenesis of COVID-19. J Autoimmun 2020, 112, 102463. [Google Scholar] [CrossRef] [PubMed]
- Valyaeva, A.A.; Zharikova, A.A.; Kasianov, A.S.; Vassetzky, Y.S.; Sheval, E. V Expression of SARS-CoV-2 Entry Factors in Lung Epithelial Stem Cells and Its Potential Implications for COVID-19. Sci Rep 2020, 10, 17772. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 Entry into Cells. Nat. Rev. Mol. Cell Biol. 2021, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Camargo, S.M.; Singer, D.; Makrides, V.; Huggel, K.; Pos, K.M.; Wagner, C.A.; Kuba, K.; Danilczyk, U.; Skovby, F.; Kleta, R.; et al. Tissue-Specific Amino Acid Transporter Partners ACE2 and Collectrin Differentially Interact with Hartnup Mutations. Gastroenterology 2009, 136, 872–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 Links Amino Acid Malnutrition to Microbial Ecology and Intestinal Inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Chakraborty, S.; Saha, P.; Mell, B.; Cheng, X.; Yeo, J.-Y.; Mei, X.; Zhou, G.; Mandal, J.; Golonka, R.; et al. Gnotobiotic Rats Reveal That Gut Microbiota Regulates Colonic MRNA of Ace2, the Receptor for SARS-CoV-2 Infectivity. Hypertension 2020, 76, e1–e3. [Google Scholar] [CrossRef]
- Round, J.L.; Palm, N.W. Causal Effects of the Microbiota on Immune-Mediated Diseases. Sci. Immunol. 2018, 3, eaao1603. [Google Scholar] [CrossRef] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-Microbe Interactions Have Shaped the Genetic Architecture of Inflammatory Bowel Disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, K.; Antanaviciute, A.; Fawkner-Corbett, D.; Jagielowicz, M.; Aulicino, A.; Lagerholm, C.; Davis, S.; Kinchen, J.; Chen, H.H.; Alham, N.K.; et al. Colonic Epithelial Cell Diversity in Health and Inflammatory Bowel Disease. Nature 2019, 567, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Singer, D.; Camargo, S.M.R.; Ramadan, T.; Schäfer, M.; Mariotta, L.; Herzog, B.; Huggel, K.; Wolfer, D.; Werner, S.; Penninger, J.M.; et al. Defective Intestinal Amino Acid Absorption in Ace2 Null Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G686–G695. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Yang, Z.; Wang, Y.; Zhou, F.; Li, S.; Li, C.; Li, L.; Zhang, W.; Li, X. Recent Advance of ACE2 and Microbiota Dysfunction in COVID-19 Pathogenesis. Heliyon 2021, 7, e07548. [Google Scholar] [CrossRef] [PubMed]
- Potdar, A.A.; Dube, S.; Naito, T.; Li, K.; Botwin, G.; Haritunians, T.; Li, D.; Casero, D.; Yang, S.; Bilsborough, J.; et al. Altered Intestinal ACE2 Levels Are Associated With Inflammation, Severe Disease, and Response to Anti-Cytokine Therapy in Inflammatory Bowel Disease. Gastroenterology 2020, 160, 809–822.e7. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Carino, A.; Baldoni, M.; Santucci, L.; Costanzi, E.; Graziosi, L.; Distrutti, E.; Biagioli, M. Bile Acid Signaling in Inflammatory Bowel Diseases. Dig. Dis. Sci. 2021, 66, 674–693. [Google Scholar] [CrossRef] [PubMed]
- Biagioli, M.; Marchianò, S.; Carino, A.; Di Giorgio, C.; Santucci, L.; Distrutti, E.; Fiorucci, S. Bile Acids Activated Receptors in Inflammatory Bowel Disease. Cells 2021, 10, 1281. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Distrutti, E.; Carino, A.; Zampella, A.; Biagioli, M. Bile Acids and Their Receptors in Metabolic Disorders. Prog. Lipid. Res. 2021, 82, 101094. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Distrutti, E. Bile Acid-Activated Receptors, Intestinal Microbiota, and the Treatment of Metabolic Disorders. Trends Mol. Med. 2015, 21, 702–714. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G Protein-Coupled Receptor Responsive to Bile Acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Itadani, H.; Tanaka, K.; Nakamura, T.T.; Itadani, H.; et al. Identification of Membrane-Type Receptor for Bile Acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Biagioli, M.; Carino, A.; Fiorucci, C.; Marchianò, S.; Di Giorgio, C.; Roselli, R.; Magro, M.; Distrutti, E.; Bereshchenko, O.; Scarpelli, P.; et al. GPBAR1 Functions as Gatekeeper for Liver NKT Cells and Provides Counterregulatory Signals in Mouse Models of Immune-Mediated Hepatitis. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 447–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carino, A.; Cipriani, S.; Marchianò, S.; Biagioli, M.; Scarpelli, P.; Zampella, A.; Monti, M.C.; Fiorucci, S. Gpbar1 Agonism Promotes a Pgc-1α-Dependent Browning of White Adipose Tissue and Energy Expenditure and Reverses Diet-Induced Steatohepatitis in Mice. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Biagioli, M.; Carino, A.; Cipriani, S.; Francisci, D.; Marchianò, S.; Scarpelli, P.; Sorcini, D.; Zampella, A.; Fiorucci, S. The Bile Acid Receptor GPBAR1 Regulates the M1/M2 Phenotype of Intestinal Macrophages and Activation of Gpbar1 Rescues Mice from Murine Colitis. J. Immunol. 2017, 199, 718–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, T.; Tanaka, K.; Suzuki, J.; Miyoshi, H.; Harada, N.; Nakamura, T.; Miyamoto, Y.; Kanatani, A.; Tamai, Y. Targeted Disruption of G Protein-Coupled Bile Acid Receptor 1 (Gpbar1/M-Bar) in Mice. J Endocrinol 2006, 191, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuma, S.; Hirasawa, A.; Tsujimoto, G. Bile Acids Promote Glucagon-like Peptide-1 Secretion through TGR5 in a Murine Enteroendocrine Cell Line STC-1. Biochem Biophys Res Commun 2005, 329, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile Acids Induce Energy Expenditure by Promoting Intracellular Thyroid Hormone Activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, S.; Mencarelli, A.; Chini, M.G.M.G.G.; Distrutti, E.; Renga, B.; Bifulco, G.; Baldelli, F.; Donini, A.; Fiorucci, S. The Bile Acid Receptor GPBAR-1 (TGR5) Modulates Integrity of Intestinal Barrier and Immune Response to Experimental Colitis. PLoS ONE 2011, 6, e25637. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Distrutti, E. The Pharmacology of Bile Acids and Their Receptors. Handb. Exp. Pharmacol. 2019, 256, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Romaní-Pérez, M.; Outeiriño-Iglesias, V.; Moya, C.M.; Santisteban, P.; González-Matías, L.C.; Vigo, E.; Mallo, F. Activation of the GLP-1 Receptor by Liraglutide Increases ACE2 Expression, Reversing Right Ventricle Hypertrophy, and Improving the Production of SP-A and SP-B in the Lungs of Type 1 Diabetes Rats. Endocrinology 2015, 156, 3559–3569. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-Omics of the Gut Microbial Ecosystem in Inflammatory Bowel Diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, S.; Popp, V.; Kindermann, M.; Gerlach, K.; Weigmann, B.; Fichtner-Feigl, S.; Neurath, M.F. Chemically Induced Mouse Models of Acute and Chronic Intestinal Inflammation. Nat. Protoc. 2017, 12, 1295–1309. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Fantini, M.C.; Neurath, M.F. High Resolution Colonoscopy in Live Mice. Nat. Protoc. 2006, 1, 2900–2904. [Google Scholar] [CrossRef] [PubMed]
- Viennois, E.; Tahsin, A.; Merlin, D. Purification of Total RNA from DSS-Treated Murine Tissue via Lithium Chloride Precipitation. Bio. Protoc. 2018, 8, e2829. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Fariñas, M.; Tokuyama, M.; Wei, G.; Huang, R.; Livanos, A.; Jha, D.; Levescot, A.; Irizar, H.; Kosoy, R.; Cording, S.; et al. Intestinal Inflammation Modulates the Expression of ACE2 and TMPRSS2 and Potentially Overlaps With the Pathogenesis of SARS-CoV-2-Related Disease. Gastroenterology 2021, 160, 287–301.e20. [Google Scholar] [CrossRef]
- Biagioli, M.; Marchianò, S.; Roselli, R.; Di Giorgio, C.; Bellini, R.; Bordoni, M.; Gidari, A.; Sabbatini, S.; Francisci, D.; Fiorillo, B.; et al. Discovery of a AHR Pelargonidin Agonist That Counter-Regulates Ace2 Expression and Attenuates ACE2-SARS-CoV-2 Interaction. Biochem. Pharmacol. 2021, 188, 114564. [Google Scholar] [CrossRef]
- Khajah, M.A.; Fateel, M.M.; Ananthalakshmi, K.V.; Luqmani, Y.A. Anti-Inflammatory Action of Angiotensin 1-7 in Experimental Colitis May Be Mediated through Modulation of Serum Cytokines/Chemokines and Immune Cell Functions. Dev. Comp. Immunol. 2017, 74, 200–208. [Google Scholar] [CrossRef]
- Khajah, M.A.; Fateel, M.M.; Ananthalakshmi, K.V.; Luqmani, Y.A. Anti-Inflammatory Action of Angiotensin 1-7 in Experimental Colitis. PLoS ONE 2016, 11, e0150861. [Google Scholar] [CrossRef] [Green Version]
- Brighton, C.A.; Rievaj, J.; Kuhre, R.E.; Glass, L.L.; Schoonjans, K.; Holst, J.J.; Gribble, F.M.; Reimann, F. Bile Acids Trigger GLP-1 Release Predominantly by Accessing Basolaterally Located G Protein-Coupled Bile Acid Receptors. Endocrinology 2015, 156, 3961–3970. [Google Scholar] [CrossRef] [Green Version]
- Marruecos, L.; Bertran, J.; Álvarez-Villanueva, D.; Mulero, M.C.; Guillén, Y.; Palma, L.G.; Floor, M.; Vert, A.; Arce-Gallego, S.; Pecharroman, I.; et al. Dynamic Chromatin Association of IκBα Is Regulated by Acetylation and Cleavage of Histone H4. EMBO Rep. 2021, 22, e52649. [Google Scholar] [CrossRef]
- Festa, C.; Renga, B.; D’Amore, C.; Sepe, V.; Finamore, C.; De Marino, S.; Carino, A.; Cipriani, S.; Monti, M.C.C.; Zampella, A.; et al. Exploitation of Cholane Scaffold for the Discovery of Potent and Selective Farnesoid X Receptor (FXR) and G-Protein Coupled Bile Acid Receptor 1 (GP-BAR1) Ligands. J. Med. Chem. 2014, 57, 8477–8495. [Google Scholar] [CrossRef]
- Toyonaga, T.; Araba, K.C.; Kennedy, M.M.; Keith, B.P.; Wolber, E.A.; Beasley, C.; Steinbach, E.C.; Schaner, M.R.; Jain, A.; Long, M.D.; et al. Increased Colonic Expression of ACE2 Associates with Poor Prognosis in Crohn’s Disease. Sci. Rep. 2020, 11, 13533. [Google Scholar] [CrossRef] [PubMed]
- Patankar, J.V.; Chiriac, M.T.; Lehmann, M.; Kühl, A.A.; Atreya, R.; Becker, C.; Gonzalez-Acera, M.; Schmitt, H.; Gamez-Belmonte, R.; Mahapatro, M.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 Attachment Receptor Angiotensin-Converting Enzyme 2 Is Decreased in Crohn’s Disease and Regulated By Microbial and Inflammatory Signaling. Gastroenterology 2021, 160, 925–928.e4. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular Interaction and Inhibition of SARS-CoV-2 Binding to the ACE2 Receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef] [PubMed]
- Rohilla, S. Designing Therapeutic Strategies to Combat Severe Acute Respiratory Syndrome Coronavirus-2 Disease: COVID-19. Drug Dev. Res. 2021, 82, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Camargo, S.M.R.; Vuille-Dit-Bille, R.N.; Meier, C.F.; Verrey, F. ACE2 and Gut Amino Acid Transport. Clin. Sci. 2020, 134, 2823–2833. [Google Scholar] [CrossRef]
- Garg, M.; Royce, S.G.; Tikellis, C.; Shallue, C.; Batu, D.; Velkoska, E.; Burrell, L.M.; Patel, S.K.; Beswick, L.; Jackson, A.; et al. Imbalance of the Renin-Angiotensin System May Contribute to Inflammation and Fibrosis in IBD: A Novel Therapeutic Target? Gut 2020, 69, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Biagioli, M.; Zampella, A.; Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018, 9, 1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Marino, S.; Finamore, C.; Biagioli, M.; Carino, A.; Marchiano, S.; Roselli, R.; Di Giorgio, C.; Bordoni, M.; Di Leva, F.S.; Novellino, E.; et al. GPBAR1 Activation by C6-Substituted Hyodeoxycholane Analogues Protect against Colitis. ACS Med. Chem. Lett. 2020, 11, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Baldoni, M.; Ricci, P.; Zampella, A.; Distrutti, E.; Biagioli, M. Bile Acid-Activated Receptors and the Regulation of Macrophages Function in Metabolic Disorders. Curr. Opin. Pharmacol. 2020, 53, 45–54. [Google Scholar] [CrossRef]
- Fiorillo, B.; Marchianò, S.; Moraca, F.; Sepe, V.; Carino, A.; Rapacciuolo, P.; Biagioli, M.; Limongelli, V.; Zampella, A.; Catalanotti, B.; et al. Discovery of Bile Acids Derivatives as Potent Ace2 Activators by Virtual Screening and Essential Dynamics. J. Chem. Inf. Model. 2021, 62, 196–209. [Google Scholar] [CrossRef]
- Beumer, J.; Puschhof, J.; Bauzá-Martinez, J.; Martínez-Silgado, A.; Elmentaite, R.; James, K.R.; Ross, A.; Hendriks, D.; Artegiani, B.; Busslinger, G.A.; et al. High-Resolution MRNA and Secretome Atlas of Human Enteroendocrine Cells. Cell 2020, 181, 1291–1306.e19. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like Peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Calabrone, L.; Carlini, V.; Benedetto, N.; Lombardo, M.; Bruno, A.; Noonan, D.M. Preliminary Evidence for IL-10-Induced ACE2 MRNA Expression in Lung-Derived and Endothelial Cells: Implications for SARS-Cov-2 ARDS Pathogenesis. Front. Immunol. 2021, 12, 718136. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biagioli, M.; Marchianò, S.; Roselli, R.; Di Giorgio, C.; Bellini, R.; Bordoni, M.; Distrutti, E.; Catalanotti, B.; Zampella, A.; Graziosi, L.; et al. GLP-1 Mediates Regulation of Colonic ACE2 Expression by the Bile Acid Receptor GPBAR1 in Inflammation. Cells 2022, 11, 1187. https://doi.org/10.3390/cells11071187
Biagioli M, Marchianò S, Roselli R, Di Giorgio C, Bellini R, Bordoni M, Distrutti E, Catalanotti B, Zampella A, Graziosi L, et al. GLP-1 Mediates Regulation of Colonic ACE2 Expression by the Bile Acid Receptor GPBAR1 in Inflammation. Cells. 2022; 11(7):1187. https://doi.org/10.3390/cells11071187
Chicago/Turabian StyleBiagioli, Michele, Silvia Marchianò, Rosalinda Roselli, Cristina Di Giorgio, Rachele Bellini, Martina Bordoni, Eleonora Distrutti, Bruno Catalanotti, Angela Zampella, Luigina Graziosi, and et al. 2022. "GLP-1 Mediates Regulation of Colonic ACE2 Expression by the Bile Acid Receptor GPBAR1 in Inflammation" Cells 11, no. 7: 1187. https://doi.org/10.3390/cells11071187
APA StyleBiagioli, M., Marchianò, S., Roselli, R., Di Giorgio, C., Bellini, R., Bordoni, M., Distrutti, E., Catalanotti, B., Zampella, A., Graziosi, L., Donini, A., & Fiorucci, S. (2022). GLP-1 Mediates Regulation of Colonic ACE2 Expression by the Bile Acid Receptor GPBAR1 in Inflammation. Cells, 11(7), 1187. https://doi.org/10.3390/cells11071187