
Alterations in Protein Translation and Carboxylic Acid Catabolic Processes in Diabetic Kidney Disease

 , , ,
, , ,
Abstract
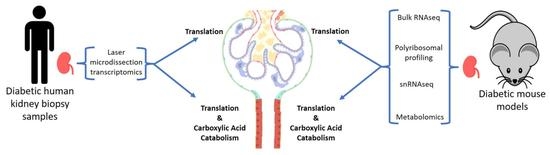
:
1. Introduction
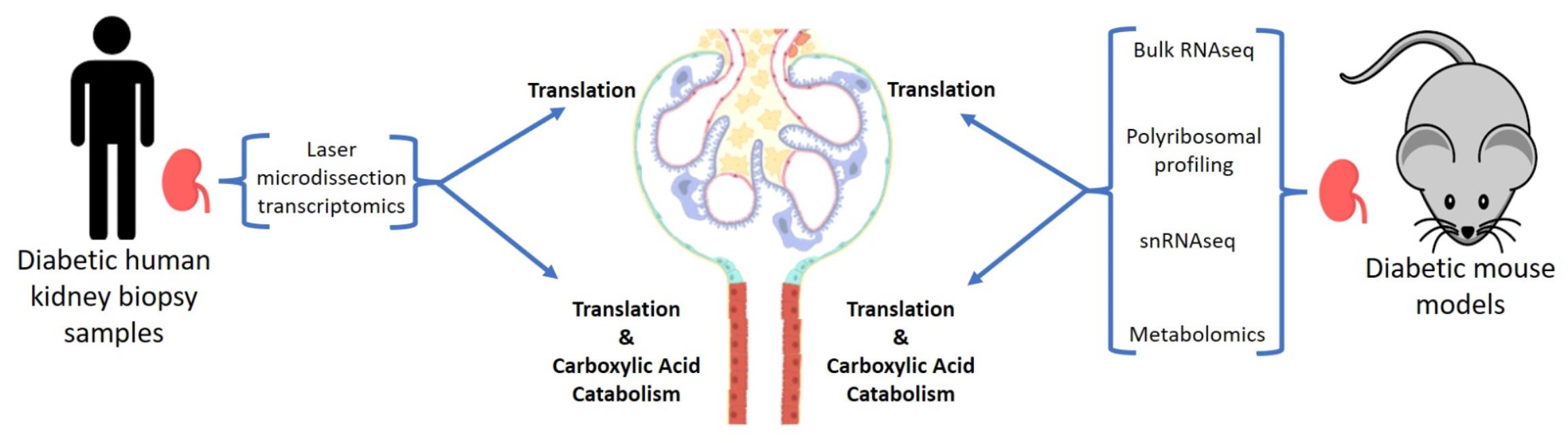
2. Materials and Methods
2.1. Human Subjects
2.2. Laser Microdissection of Human Kidney Tissue and RNA-Sequencing
2.3. Animal Study Approvals
2.4. Isolation of Mouse Kidney Tissue and Bulk RNA-Sequencing
2.5. Isolation of Mouse Kidney Tissue and Single-Nuclear RNA-Sequencing
2.6. Isolation of Mouse Kidney Tissues and Polyribosomal Profiling
2.7. Metabolomics
2.8. Differential Gene Expression, Pathway Analysis, and Statistics
3. Results
3.1. Human Subjects
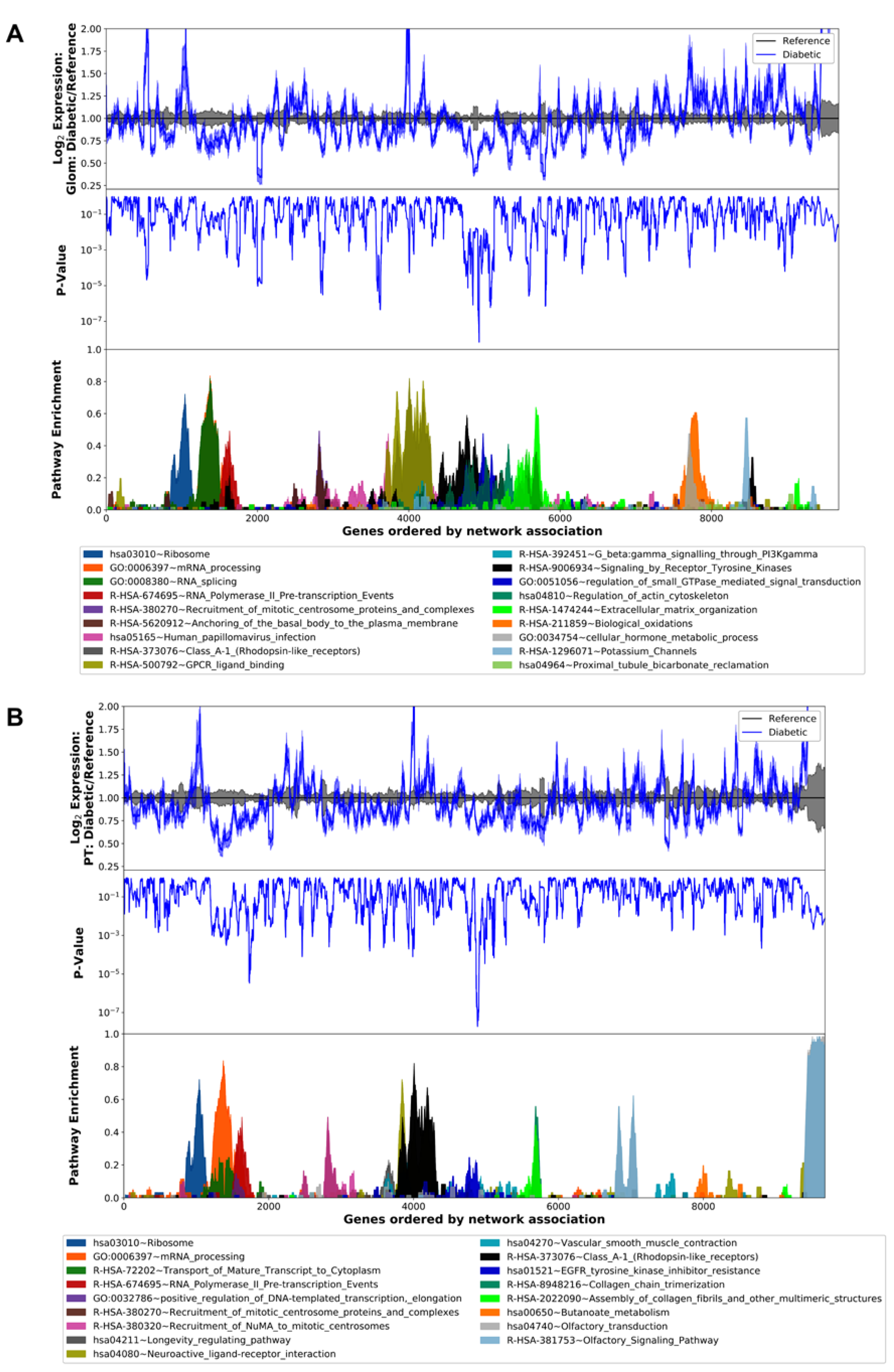
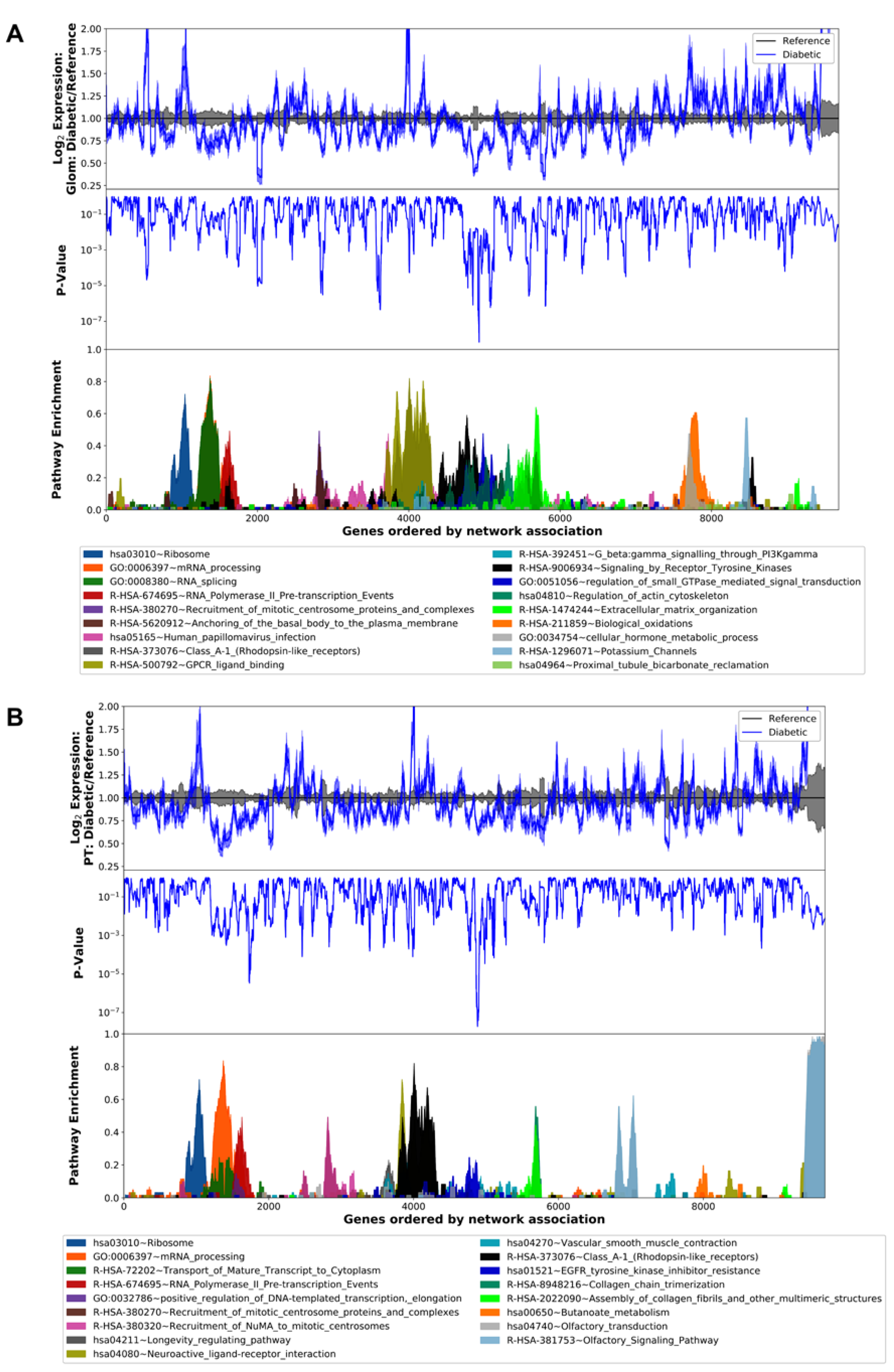
3.2. Differential Gene Expression and Pathway Analysis in the Diabetic Human Glomerulus and Proximal Tubule
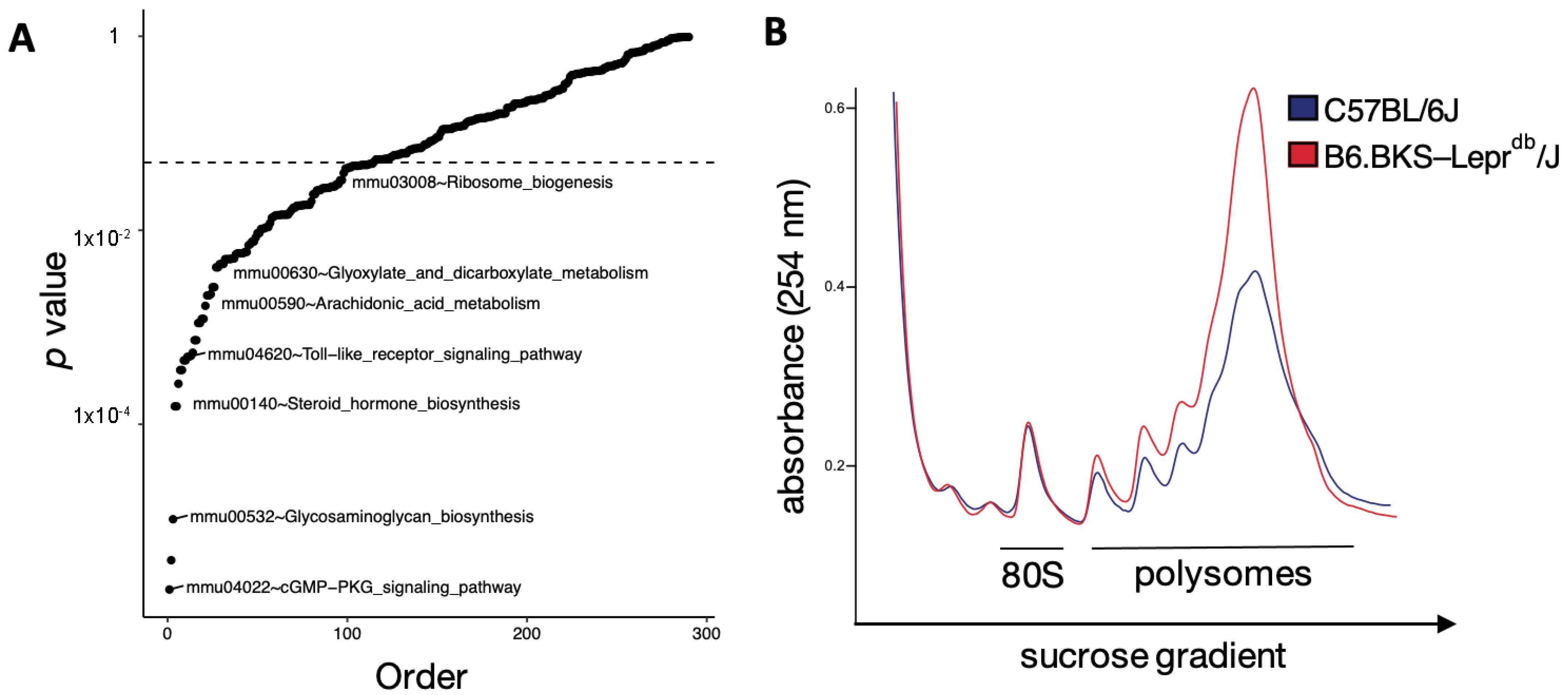
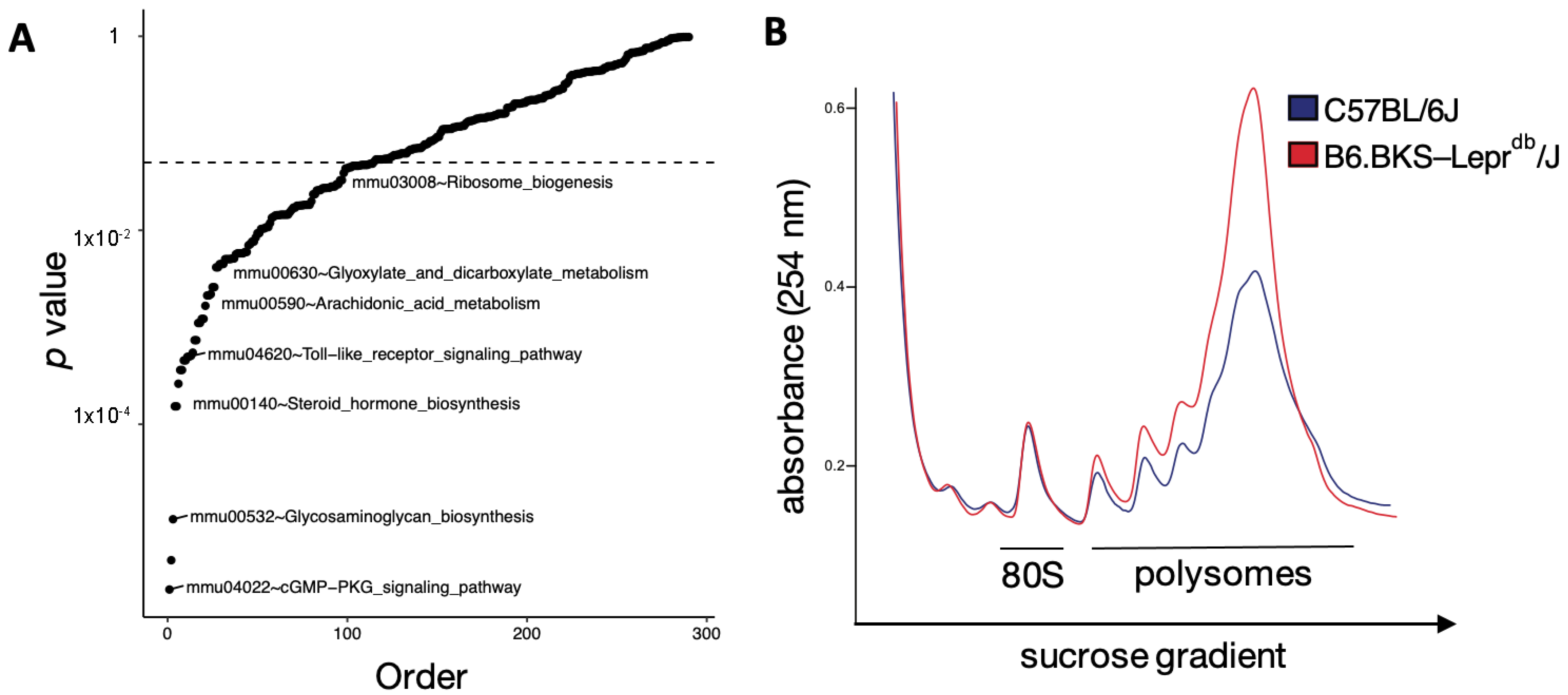
3.3. Translation Is Altered in the Diabetic Mouse Kidney
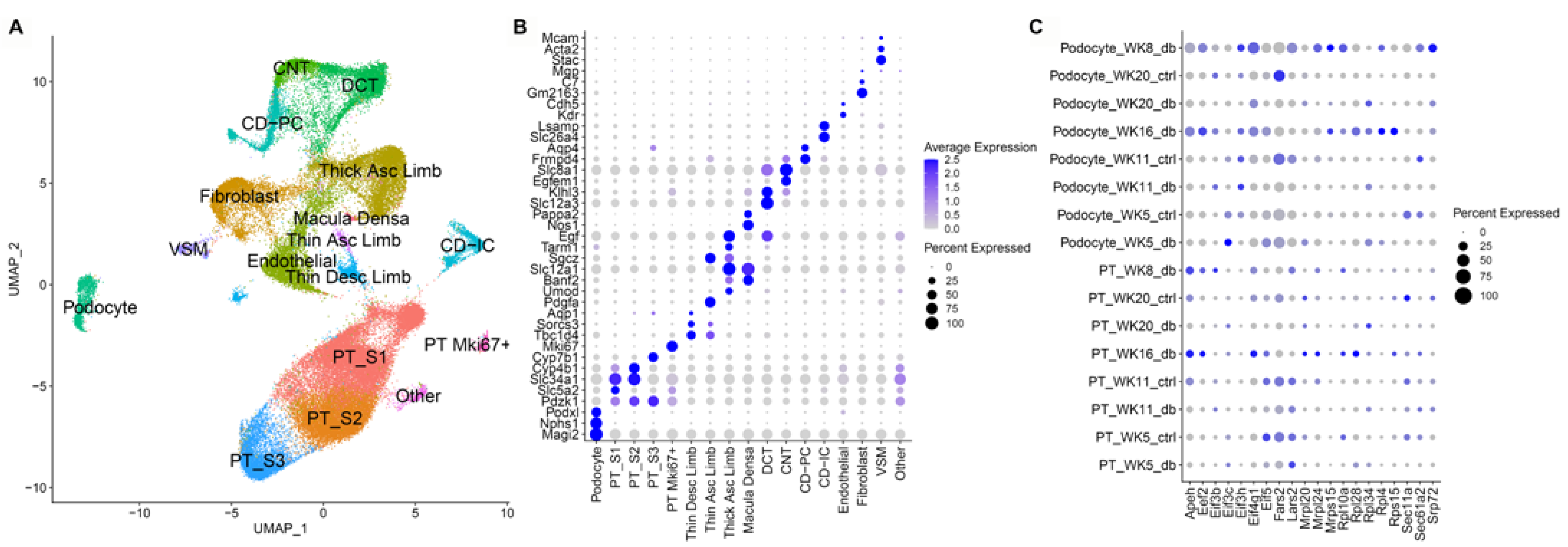
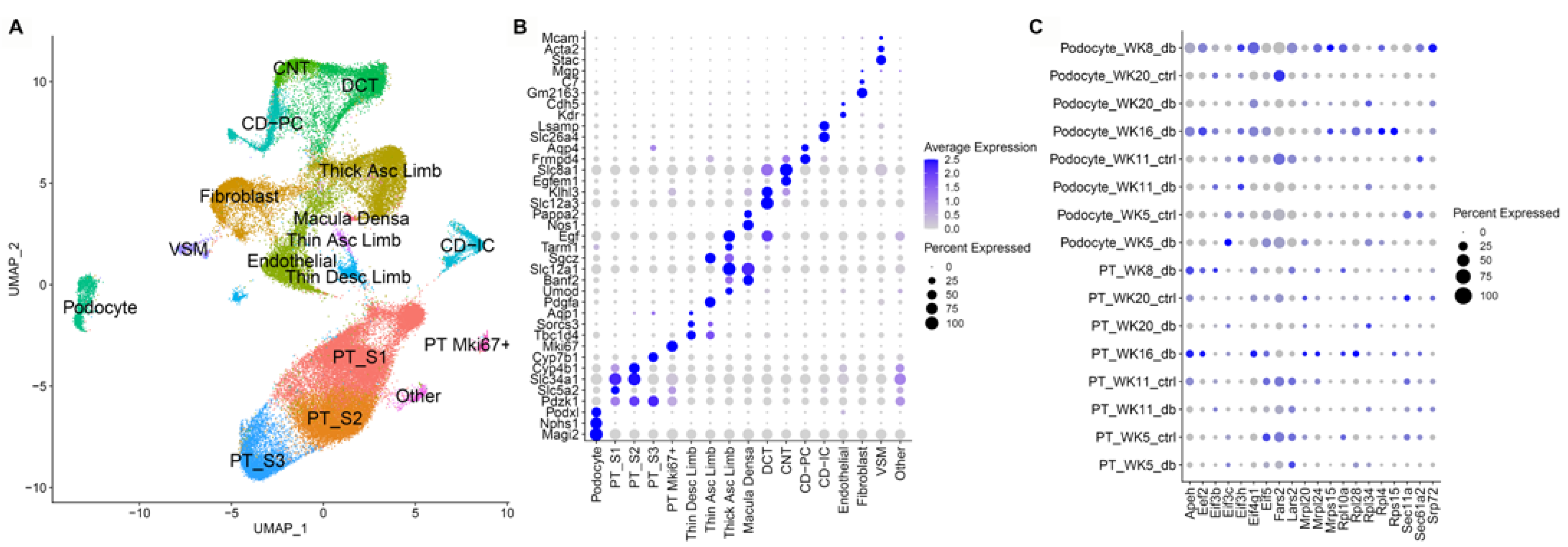
3.4. Single Nuclear RNA Sequencing Reveals Highly Enriched Pathways in the db Mouse Model
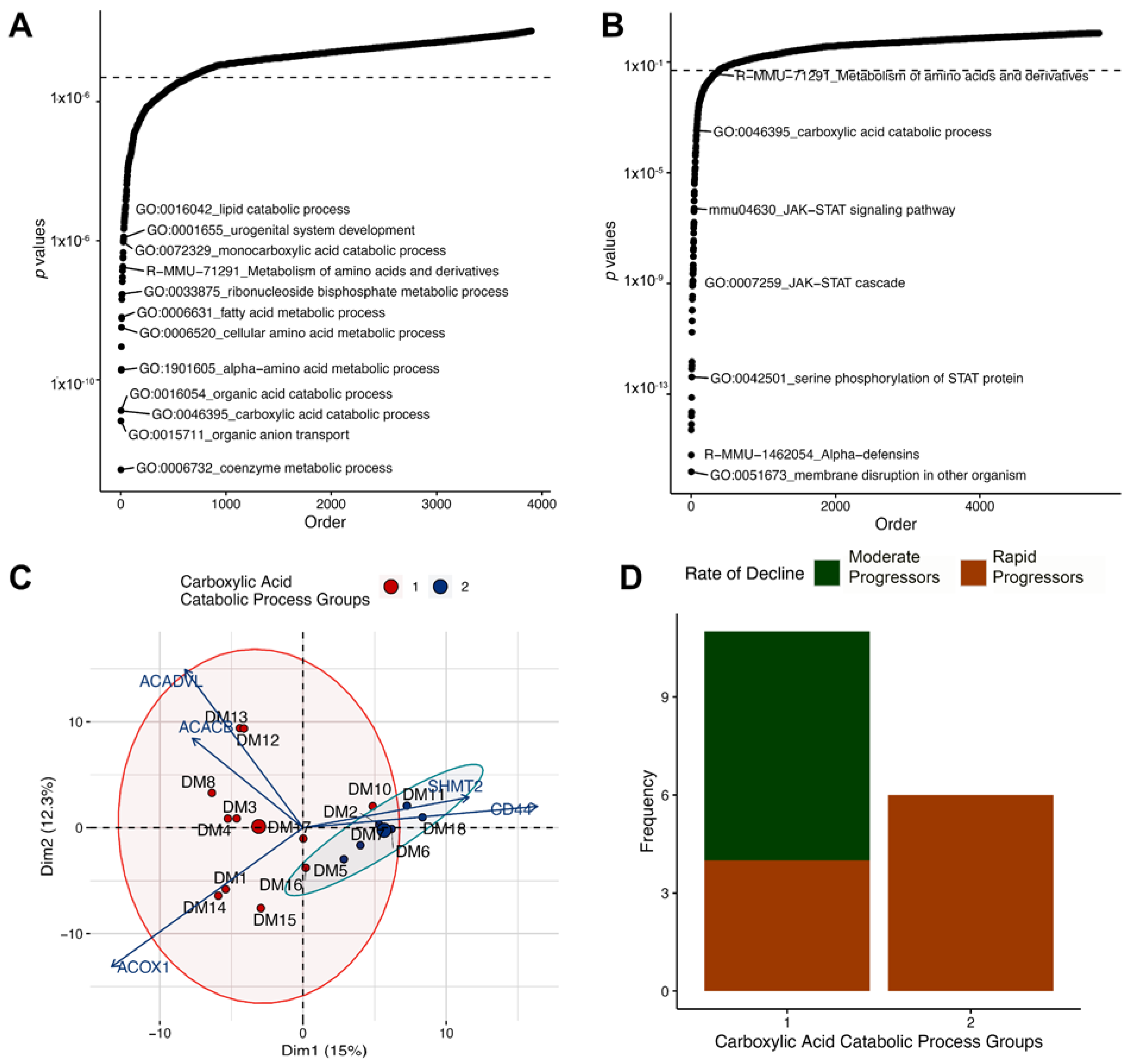
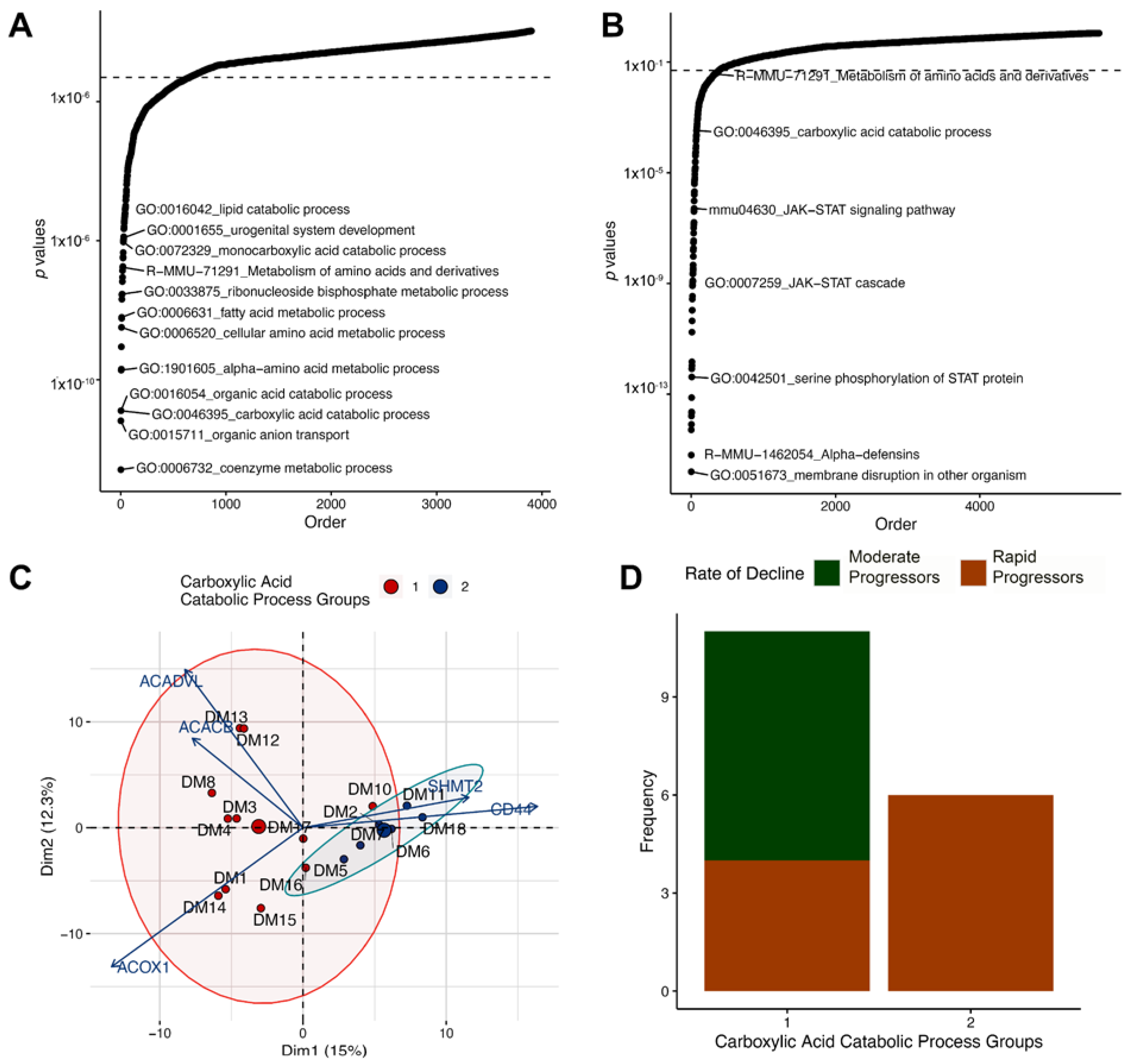
3.5. Altered Regulation of the Carboxylic Acid Catabolic Processes in the Proximal Tubule Is Associated with Progression of Kidney Failure
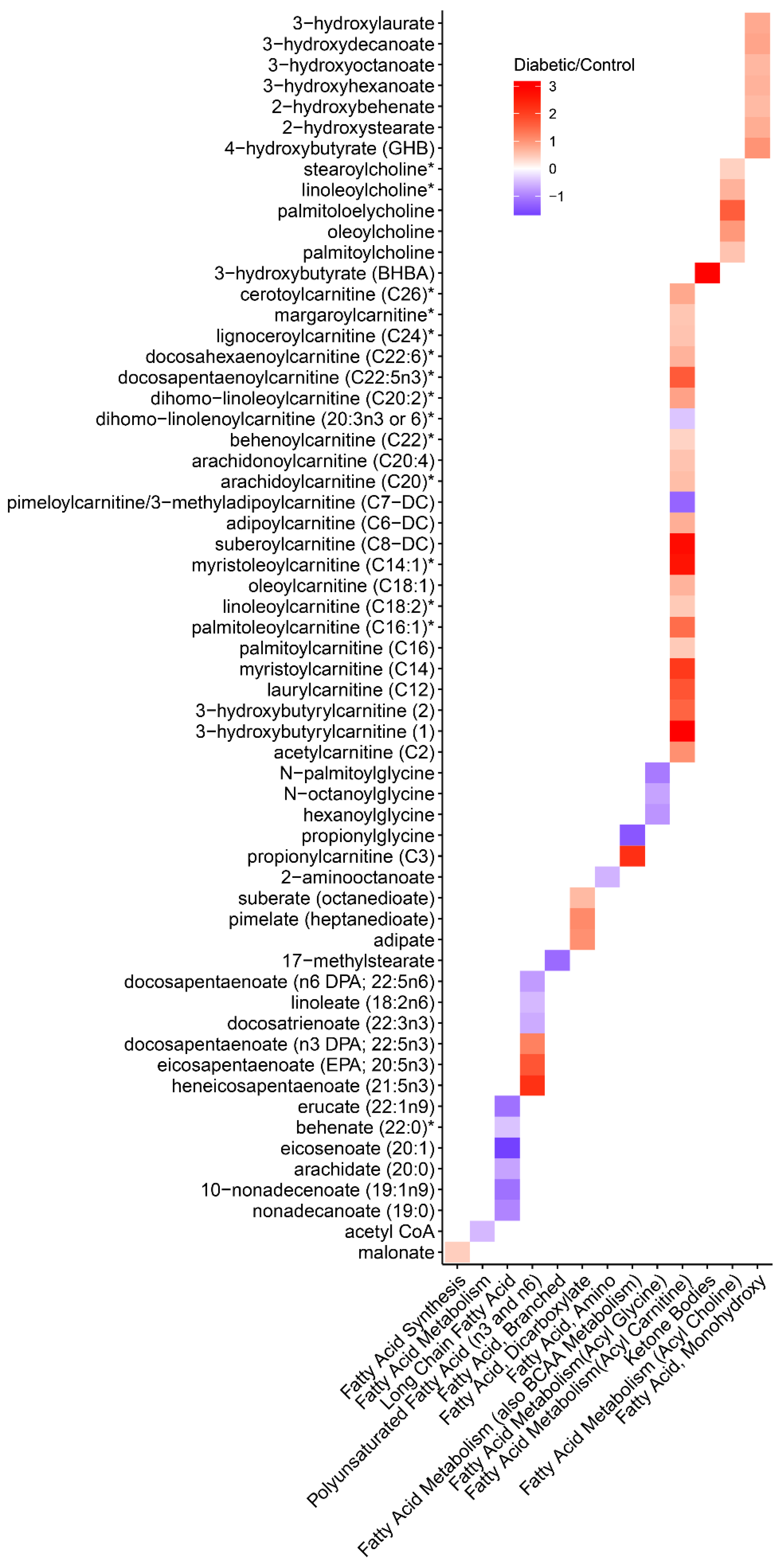
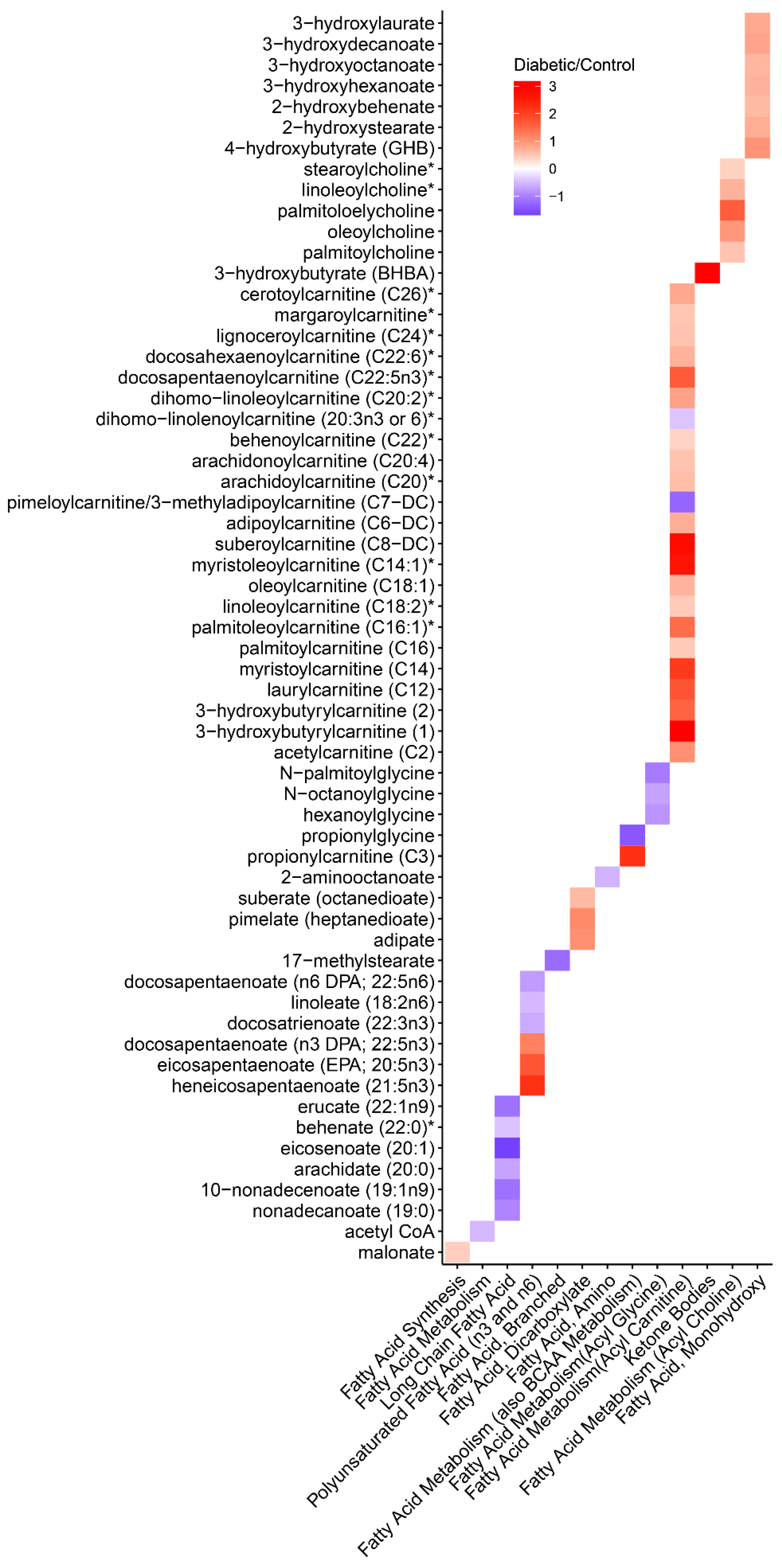
3.6. Metabolomics
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, B.; Lu, Y.; Hajifathalian, K.; Bentham, J.; Di Cesare, M.; Danaei, G.; Bixby, H.; Cowan, M.J.; Ali, M.K.; Taddei, C.; et al. Worldwide trends in diabetes since 1980: A pooled analysis of 751 population-based studies with 4.4 million participants. Lancet 2016, 387, 1513–1530. [Google Scholar] [CrossRef] [Green Version]
- National Diabetes Statistics Report: 2017. Centers for Disease Control and Prevention. National Diabetes Statistics Report Website. Available online: https://www.cdc.gov/diabetes/data/statistics-report/index.html (accessed on 23 January 2022).
- Martinez-Castelao, A.; Navarro-Gonzalez, J.F.; Gorriz, J.L.; de Alvaro, F. The Concept and the Epidemiology of Diabetic Nephropathy Have Changed in Recent Years. J. Clin. Med. 2015, 4, 1207–1216. [Google Scholar] [CrossRef] [Green Version]
- Saran, R.; Li, Y.; Robinson, B.; Ayanian, J.; Balkrishnan, R.; Bragg-Gresham, J.; Chen, J.T.; Cope, E.; Gipson, D.; He, K.; et al. US Renal Data System 2014 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am. J. Kidney Dis. 2015, 66 (Suppl. S1), S1–S305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonventre, J.V. Can we target tubular damage to prevent renal function decline in diabetes? Semin. Nephrol. 2012, 32, 452–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, K.A. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am. J. Kidney Dis. 1992, 20, 1–17. [Google Scholar] [CrossRef]
- An, Y.; Xu, F.; Le, W.; Ge, Y.; Zhou, M.; Chen, H.; Zeng, C.; Zhang, H.; Liu, Z. Renal histologic changes and the outcome in patients with diabetic nephropathy. Nephrol. Dial. Transplant. 2015, 30, 257–266. [Google Scholar] [CrossRef] [Green Version]
- Perkovic, V.; Jardine, M.J.; Neal, B.; Bompoint, S.; Heerspink, H.J.L.; Charytan, D.M.; Edwards, R.; Agarwal, R.; Bakris, G.; Bull, S.; et al. Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N. Engl. J. Med. 2019, 380, 2295–2306. [Google Scholar] [CrossRef] [Green Version]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef] [Green Version]
- Spriggs, K.A.; Bushell, M.; Willis, A.E. Translational regulation of gene expression during conditions of cell stress. Mol. Cell 2010, 40, 228–237. [Google Scholar] [CrossRef]
- Jung, M.; Jin, S.G.; Zhang, X.; Xiong, W.; Gogoshin, G.; Rodin, A.S.; Pfeifer, G.P. Longitudinal epigenetic and gene expression profiles analyzed by three-component analysis reveal down-regulation of genes involved in protein translation in human aging. Nucleic Acids Res. 2015, 43, e100. [Google Scholar] [CrossRef] [Green Version]
- Le Bacquer, O.; Petroulakis, E.; Paglialunga, S.; Poulin, F.; Richard, D.; Cianflone, K.; Sonenberg, N. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. J. Clin. Investig. 2007, 117, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Rooijers, K.; Loayza-Puch, F.; Nijtmans, L.G.; Agami, R. Ribosome profiling reveals features of normal and disease-associated mitochondrial translation. Nat. Commun. 2013, 4, 2886. [Google Scholar] [CrossRef] [Green Version]
- Schafer, S.; Adami, E.; Heinig, M.; Rodrigues, K.E.; Kreuchwig, F.; Silhavy, J.; van Heesch, S.; Simaite, D.; Rajewsky, N.; Cuppen, E.; et al. Translational regulation shapes the molecular landscape of complex disease phenotypes. Nat. Commun. 2015, 6, 7200. [Google Scholar] [CrossRef]
- Alpers, C.E.; Hudkins, K.L. Mouse models of diabetic nephropathy. Curr. Opin. Nephrol. Hypertens. 2011, 20, 278–284. [Google Scholar] [CrossRef]
- de Boer, I.H.; Alpers, C.E.; Azeloglu, E.U.; Balis, U.G.J.; Barasch, J.M.; Barisoni, L.; Blank, K.N.; Bomback, A.S.; Brown, K.; Dagher, P.C.; et al. Rationale and design of the Kidney Precision Medicine Project. Kidney Int. 2021, 99, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Eadon, M.T.; Schwantes-An, T.H.; Phillips, C.L.; Roberts, A.R.; Greene, C.V.; Hallab, A.; Hart, K.J.; Lipp, S.N.; Perez-Ledezma, C.; Omar, K.O.; et al. Kidney Histopathology and Prediction of Kidney Failure: A Retrospective Cohort Study. Am. J. Kidney Dis. 2020, 76, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Barwinska, D.; Ferkowicz, M.J.; Cheng, Y.H.; Winfree, S.; Dunn, K.W.; Kelly, K.J.; Sutton, T.A.; Rovin, B.H.; Parikh, S.V.; Phillips, C.L.; et al. Application of Laser Microdissection to Uncover Regional Transcriptomics in Human Kidney Tissue. J. Vis. Exp. 2020, 160, e61371. [Google Scholar] [CrossRef]
- Barwinska, D.; El-Achkar, T.M.; Melo Ferreira, R.; Syed, F.; Cheng, Y.H.; Winfree, S.; Ferkowicz, M.J.; Hato, T.; Collins, K.S.; Dunn, K.W.; et al. Molecular characterization of the human kidney interstitium in health and disease. Sci. Adv. 2021, 7, eabd3359. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Janosevic, D.; Myslinski, J.; McCarthy, T.W.; Zollman, A.; Syed, F.; Xuei, X.; Gao, H.; Liu, Y.L.; Collins, K.S.; Cheng, Y.H.; et al. The orchestrated cellular and molecular responses of the kidney to endotoxin define a precise sepsis timeline. eLife 2021, 10, e62270. [Google Scholar] [CrossRef] [PubMed]
- Ransick, A.; Lindstrom, N.O.; Liu, J.; Zhu, Q.; Guo, J.J.; Alvarado, G.F.; Kim, A.D.; Black, H.G.; Kim, J.; McMahon, A.P. Single-Cell Profiling Reveals Sex, Lineage, and Regional Diversity in the Mouse Kidney. Dev. Cell 2019, 51, 399–413. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, R.M.; Clendenon, S.G.; Richards, W.G.; Boedigheimer, M.; Damore, M.; Rossetti, S.; Harris, P.C.; Herbert, B.S.; Xu, W.M.; Wandinger-Ness, A.; et al. Transcriptome analysis reveals manifold mechanisms of cyst development in ADPKD. Hum. Genomics 2016, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; He, Q.Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Fikri, A.M.; Smyth, R.; Kumar, V.; Al-Abadla, Z.; Abusnana, S.; Munday, M.R. Pre-diagnostic biomarkers of type 2 diabetes identified in the UAE’s obese national population using targeted metabolomics. Sci. Rep. 2020, 10, 17616. [Google Scholar] [CrossRef]
- Dharuri, H.; t Hoen, P.A.; van Klinken, J.B.; Henneman, P.; Laros, J.F.; Lips, M.A.; El Bouazzaoui, F.; van Ommen, G.J.; Janssen, I.; van Ramshorst, B.; et al. Downregulation of the acetyl-CoA metabolic network in adipose tissue of obese diabetic individuals and recovery after weight loss. Diabetologia 2014, 57, 2384–2392. [Google Scholar] [CrossRef]
- Nagai, K.; Matsubara, T.; Mima, A.; Sumi, E.; Kanamori, H.; Iehara, N.; Fukatsu, A.; Yanagita, M.; Nakano, T.; Ishimoto, Y.; et al. Gas6 induces Akt/mTOR-mediated mesangial hypertrophy in diabetic nephropathy. Kidney Int. 2005, 68, 552–561. [Google Scholar] [CrossRef] [Green Version]
- Sataranatarajan, K.; Mariappan, M.M.; Lee, M.J.; Feliers, D.; Choudhury, G.G.; Barnes, J.L.; Kasinath, B.S. Regulation of elongation phase of mRNA translation in diabetic nephropathy: Amelioration by rapamycin. Am. J. Pathol. 2007, 171, 1733–1742. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Feliers, D.; Mariappan, M.M.; Sataranatarajan, K.; Mahimainathan, L.; Musi, N.; Foretz, M.; Viollet, B.; Weinberg, J.M.; Choudhury, G.G.; et al. A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am. J. Physiol. Ren. Physiol. 2007, 292, F617–F627. [Google Scholar] [CrossRef]
- Fu, J.; Akat, K.M.; Sun, Z.; Zhang, W.; Schlondorff, D.; Liu, Z.; Tuschl, T.; Lee, K.; He, J.C. Single-Cell RNA Profiling of Glomerular Cells Shows Dynamic Changes in Experimental Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2019, 30, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Lee, K.; Wang, N.; He, J.C. The Role of Endoplasmic Reticulum Stress in Diabetic Nephropathy. Curr. Diab. Rep. 2017, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.T.; Zhang, Z.J.; Cao, J.Y.; Chen, H.; Zhu, Y.S.; Wu, X.; Nawabi, A.Q.; Liu, X.; Shan, W.; Zhang, Y.; et al. The unique molecular mechanism of diabetic nephropathy: A bioinformatics analysis of over 250 microarray datasets. Clin. Kidney J. 2021, 14, 1626–1638. [Google Scholar] [CrossRef]
- Choi, J.; Ravipati, A.; Nimmagadda, V.; Schubert, M.; Castellani, R.J.; Russell, J.W. Potential roles of PINK1 for increased PGC-1alpha-mediated mitochondrial fatty acid oxidation and their associations with Alzheimer disease and diabetes. Mitochondrion 2014, 18, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Petrie, M.A.; Suneja, M.; Faidley, E.; Shields, R.K. A minimal dose of electrically induced muscle activity regulates distinct gene signaling pathways in humans with spinal cord injury. PLoS ONE 2014, 9, e115791. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, C.; Li, J.; Mei, Y.; Liang, Y. Hypoglycemic and Hypolipidemic Effects of Phellinus Linteus Mycelial Extract from Solid-State Culture in A Rat Model of Type 2 Diabetes. Nutrients 2019, 11, 296. [Google Scholar] [CrossRef] [Green Version]
- Riancho, J.A.; Vazquez, L.; Garcia-Perez, M.A.; Sainz, J.; Olmos, J.M.; Hernandez, J.L.; Perez-Lopez, J.; Amado, J.A.; Zarrabeitia, M.T.; Cano, A.; et al. Association of ACACB polymorphisms with obesity and diabetes. Mol. Genet. Metab. 2011, 104, 670–676. [Google Scholar] [CrossRef] [Green Version]
- Hasib, A.; Hennayake, C.K.; Bracy, D.P.; Bugler-Lamb, A.R.; Lantier, L.; Khan, F.; Ashford, M.L.J.; McCrimmon, R.J.; Wasserman, D.H.; Kang, L. CD44 contributes to hyaluronan-mediated insulin resistance in skeletal muscle of high-fat-fed C57BL/6 mice. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E973–E983. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, S.B.; Liu, J.; Kim, J.; Ren, Y.; Collins, A.R.; Nguyen, L.; Hsueh, W.A. Critical role for osteopontin in diabetic nephropathy. Kidney Int. 2010, 77, 588–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Chen, X.; Zhang, Y.; George, J.; Cobbs, A.; Wang, G.; Li, L.; Emmett, N. Kidney Injury Molecule-1 Is Upregulated in Renal Lipotoxicity and Mediates Palmitate-Induced Tubular Cell Injury and Inflammatory Response. Int. J. Mol. Sci. 2019, 20, 3406. [Google Scholar] [CrossRef] [Green Version]
- Tani, H.; Ohnishi, S.; Shitara, H.; Mito, T.; Yamaguchi, M.; Yonekawa, H.; Hashizume, O.; Ishikawa, K.; Nakada, K.; Hayashi, J.I. Mice deficient in the Shmt2 gene have mitochondrial respiration defects and are embryonic lethal. Sci. Rep. 2018, 8, 425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Garcia Canaveras, J.C.; Chen, Z.; Wang, L.; Liang, L.; Jang, C.; Mayr, J.A.; Zhang, Z.; Ghergurovich, J.M.; Zhan, L.; et al. Serine Catabolism Feeds NADH when Respiration Is Impaired. Cell Metab. 2020, 31, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.S.; Zhang, L.; Mitch, W.E. Molecular mechanisms of insulin resistance in chronic kidney disease. Kidney Int. 2015, 88, 1233–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable Mean ± SD or n (%) | Diabetic Kidney Disease n = 18 | Reference n = 9 |
|---|---|---|
| Age | 55.0 ± 9.3 | 50.0 ± 14.2 |
| Gender, Female | 11 (61.1) | 6 (66.7) |
| Race, Black A | 5 (27.8) | NA |
| Baseline eGFR (mL/min) B | 63.2 ± 26.2 | NA |
| Baseline proteinuria >3 gm | 10 (55.6) | NA |
| Endpoint eGFR (mL/min) | 22.4 ± 19.2 | NA |
| Rate of progression (mL/min/year) | 14.5 ± 19.6 | NA |
| Patients with > 10 mL/min/yr GFR loss | 11 (61.1) | NA |
| Duration of follow-up data (months) | 47.7 ± 23.7 | NA |
| Histopathologic diabetic kidney disease | 18 (100) | 0 (0) |
| Histopathologic arterionephrosclerosis | 18 (100) | 0 (0) |
| Glomerular obsolescence (% of glomeruli affected) | 24.7 ± 18.7 | 23.9 ± 21.2 |
| IFTA (% of cortex affected) | 48.9 ± 12.8 | 20.0 ± 13.8 |
| Arteriolar Hyalinosis severity (scale of 0–3) | 2.4 ± 0.6 | 0.9 ± 0.4 |
| Presence of nodular glomerulosclerosis | 15 (83.3) | 0 (0) |
| Presence of effacement (>80% of foot processes) | 9 (50) | 0 (0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collins, K.S.; Eadon, M.T.; Cheng, Y.-H.; Barwinska, D.; Melo Ferreira, R.; McCarthy, T.W.; Janosevic, D.; Syed, F.; Maier, B.; El-Achkar, T.M.; et al. Alterations in Protein Translation and Carboxylic Acid Catabolic Processes in Diabetic Kidney Disease. Cells 2022, 11, 1166. https://doi.org/10.3390/cells11071166
Collins KS, Eadon MT, Cheng Y-H, Barwinska D, Melo Ferreira R, McCarthy TW, Janosevic D, Syed F, Maier B, El-Achkar TM, et al. Alterations in Protein Translation and Carboxylic Acid Catabolic Processes in Diabetic Kidney Disease. Cells. 2022; 11(7):1166. https://doi.org/10.3390/cells11071166
Chicago/Turabian StyleCollins, Kimberly S., Michael T. Eadon, Ying-Hua Cheng, Daria Barwinska, Ricardo Melo Ferreira, Thomas W. McCarthy, Danielle Janosevic, Farooq Syed, Bernhard Maier, Tarek M. El-Achkar, and et al. 2022. "Alterations in Protein Translation and Carboxylic Acid Catabolic Processes in Diabetic Kidney Disease" Cells 11, no. 7: 1166. https://doi.org/10.3390/cells11071166
APA StyleCollins, K. S., Eadon, M. T., Cheng, Y.-H., Barwinska, D., Melo Ferreira, R., McCarthy, T. W., Janosevic, D., Syed, F., Maier, B., El-Achkar, T. M., Kelly, K. J., Phillips, C. L., Hato, T., Sutton, T. A., & Dagher, P. C. (2022). Alterations in Protein Translation and Carboxylic Acid Catabolic Processes in Diabetic Kidney Disease. Cells, 11(7), 1166. https://doi.org/10.3390/cells11071166