
Cardiac Cachexia: Unaddressed Aspect in Cancer Patients



Abstract
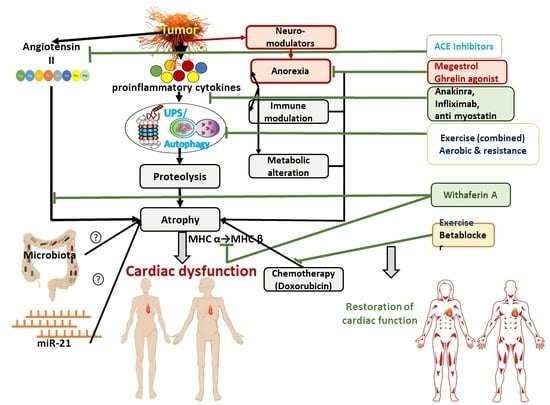
1. Introduction: Definition of Cancer Cachexia
2. Multiple Organ Involvement in Cancer Cachexia
3. Definition and Pathophysiology of Cancer-Induced Cardiac Cachexia
3.1. Inflammation and Immune Modulation
3.2. Hormones and Proteolytic Pathways
3.3. Mitochondrial Dysfunction and Oxidative Stress
4. Angiotensin II-Induced Cardiac Dysfunction Mimics Cardiac Cachexia
5. Diagnosis of Cardiac Cachexia
6. Cancer Induced Cardiac Cachexia: Experimental and Clinical Evidence

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No | Experimental Model | Methods/Tools Used | Major Outcome | References |
|---|---|---|---|---|
| 1. | 10-week-old female CD2F1 (BALB/c × DBA/2 F1) mice were injected with C26 cell line | Western blot analysis, quantitative real time PCR, picrosirius red quantification, hydroxyproline assay | In LV: Significant elevation of mRNA of TIMP 1, MMP-2, MMP-3, and MMP-14; no change in TIMP2 mRNA or in the protein levels of MMP-2, MMP-3, MMP-9, and MMP-14; and TIMP 2 elevated | [70] |
| 2. | Rat AH-130 hepatoma CC model | NMR spectroscopy, infrared monitoring system to monitor movement, echocardiography, invasive hemodynamic assessment, ECG, Luminex-200 system, RIA, ELISA | Decrease in LV mass, heart weight, LVEDD, LVEF, LVFS, LVESP, LVEDP. Maximum response observed 13 days after inoculation | [69] |
| 3. | Colon -26 adenocarcinoma cell line-injected male CD2F1 mice | Transthoracic echocardiography, transmission electron microscopy, RT-PCR, and Western blotting | Decreased heart rate and fractional shortening, disrupted cardiac muscle morphology, increased heart muscle fibrosis, decreased expression of TnI and MHCα, and increased expression of IL-6 and IL-6 R | [71] |
| 4. | Colon -26 adenocarcinoma cell line-injected male CD2F1 8-week-old male and female mice | Echocardiography, electron microscopy, RT-PCR and Western blotting, ubiquitin conjugation assay | Pronounced cardiac atrophy in male mice with reductions in myocyte size and sarcomere protein and autophagy activation | [30] |
| 5. | Ectopic mouse model: 9–10-week-old male Balb/c, C57BL6/N, and Fox Chase SCID inoculated with adenocarcinoma cells; C26 mouse, MC38 mouse, SW480 mouse, and orthotopic mouse model (PDAC): 10 week old male C57BL6/J, genetic mouse model (APC delta 580 mice); diabetic mice: 9–11 week old female ob/ob and db/db C26; MC38 and HEK293A cell culture | Echocardiography and pressure–volume loop measurement, echo magnetic resonance imaging, ELISA, gene expression analysis | Decrease in heart weight, volume, myocyte diameter, and deterioration of cardiac function in C26 transplanted and APC mutant cachectic animals and elevated ataxin-10 levels in C26 and SW480 and no fibrosis in the cachectic animals | [65] |
| 6. | Male and female 7-week-old C57BL/J6 mice inoculated with Lewis lung carcinoma (LLC1) cells | Echocardiography, fractional protein synthesis rate, mRNA sequencing, transmission electron microscopy of mitochondria, immunoblot assay, histology | Decrease in IVS thickness, EF, %FS, fractional protein synthesis, cardiac mitochondrial oxygen consumption rate, and Complex II, III, IV and V proteins and in Apl, Aplr, N-Myc, Egr1 and Sox9 mRNAs; increase in LV ID (S + D), (AR), and (FF); increased Gadd45b; no cardiac fibrosis | [74] |
| 7. | Eight-week-old, male C57BL6/J mice inoculated with Lewis lung carcinoma cells and H9c2 ventricular cardiomyocytes culture | Two-photon excitation fluorescence, immunoblot analysis, gas chromatography–mass spectrometry, bioenergetic flux analysis | Lower heart weight lower (10%); low optical redox ratio (15%); increased COX-IV (50%); decreased VDAC (50%); lower Cytb (mt-DNA) (30%); and lower GPx-3 and GPx-7 (~50%) | [33] |
| 8. | Six-week-old female mice inoculated with A2780 ovarian cancer cells. | Echocardiography, histology with haematoxylin and eosin staining and Masson’s trichrome staining, qPCR for RNA quantification, and ELISA | Decreased heart rate, FS%, CO, LV mass, E/A ratio, E/e’, CSA of cardiomyocytes, cTnI, shiftin MHCα to β and increase in AngII, AT1aR mRNA, IL-6, TNFα, MIP-2, and IFNγ | [18] |
| S. No. | Type of Cancer (Sample Size) | Study Design/Tools Used | Outcome in Cancer Patients | References |
|---|---|---|---|---|
| 1. | Lung cancer (58), pancreatic cancer (60), GI cancer (59) | Retrospective study on deceased cancer patients | Significantly low BMI and HW in cachectic patients, LVWT and RVWT same in both cachectic and non-cachectic patients | [2] |
| 2. | Colorectal cancer (50) | Prospective study by 2D echo, Holter ECG, and treadmill exercise tests and biomarker analysis | Significantly reduced LVEF and HRV (SDNN, SDANN, SDNN index, VLF, and LF) parameters; significantly higher hsTnT | [75] |
| 3. | Ovarian cancer (25) | Case–control study, highly sensitive troponin immunoassay | Significantly high hsTnI and conventional TnI | [76] |
| 4. | Non-small cell lung or colorectal cancer patients | One set: human cadaver heart weights and wall thickness measurement Second set: plasma level of biochemical parameters from cancer patients | Reduction in heart weight (25.6%) and LVWT (12.1%); cardiac fibrosis+, increased plasma Aaldosterone, BNP, and renin levels in cancer patients | [69] |
| 5. | Total: 555 (breast: 146, lung: 61, GI: 67, myelodysplastic: 68, myeloproliferative: 99, brain:23, ENT: 33) | Prospective study with follow up with a median of 25 months; venous blood analysis of cardiovascular functional peptide and morphological markers | (CRP), haptoglobin, fibronectin, (SAA), interleukin 6 (IL-6), hsTnT and NT-proBNP, (MR-proANP), (MR-proADM), CT-proET-1 and stable copeptin: all were elevated | [77] |
| 6. | Chemotherapy naïve breast cancer or lymphoma (381) | Part of prospective study CAPRI (NCT04367220), cardiovascular magnetic resonance imaging | Smaller chamber volume, higher global strain amplitude, increased septal and lateral wall native T1 mapping among patients with cancer | [78] |
| 7. | 122 Solid tumors (gynaecological: 20, breast: 19, GI: 51, sarcoma 13, lungs; 19) | Retrospective study/Echo cardiography with 2D strain analysis of the left ventricle | No difference in LV diameter, wall thickness; siginificant decrease in LVEF and in LV longitudinal, circumferential, and radial strain | [80] |
| 8. | 101 Chemo- and radiotherapy in naïve cancer patients | Retrospective study/echo cardiography with 2D strain analysis of the right ventricle | Significant decrease in global RV longitudinal strain | [81] |
| 9. | 92 Chemo- and radiotherapy in naïve solid cancer patients | Retrospective study/echo cardiography with 2D strain analysis of the left atrium | Significant increase in LAVmin/BSA and LAVpre-a/BSA: decrease in LA Tot EF (%), LA Pass EF (%); decrease in longitudinal as well as systolic and early diastolic strain | [82] |
7. Synergistic Effect of Chemotherapy on Cancer-Induced Cardiac Cachexia (Dysfunctions)
8. Therapeutical Approach for Cardiac Cachexia
8.1. Nonpharmacological Therapy
8.2. Pharmacotherapy
9. Clinical Trials
10. Future Prospects
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Barkhudaryan, A.; Scherbakov, N.; Springer, J.; Doehner, W. Cardiac muscle wasting in individuals with cancer cachexia. ESC Heart Fail. 2017, 4, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, T.; Terracina, K.P.; Raza, A.; Matsubara, H.; Takabe, K. Cancer cachexia, mechanism and treatment. World J. Gastrointest. Oncol. 2015, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Schmidt, S.F.; Rohm, M.; Herzig, S.; Diaz, M.B. Cancer cachexia: More than skeletal muscle wasting. Trends Cancer 2018, 4, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Dahlman, I.; Mejhert, N.; Linder, K.; Agustsson, T.; Mutch, D.; Kulyte, A.; Isaksson, B.; Permert, J.; Petrovic, N.; Nedergaard, J. Adipose tissue pathways involved in weight loss of cancer cachexia. Br. J. Cancer 2010, 102, 1541–1548. [Google Scholar] [CrossRef] [PubMed]
- Fouladiun, M.; Körner, U.; Bosaeus, I.; Daneryd, P.; Hyltander, A.; Lundholm, K.G. Body composition and time course changes in regional distribution of fat and lean tissue in unselected cancer patients on palliative care—Correlations with food intake, metabolism, exercise capacity, and hormones. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2005, 103, 2189–2198. [Google Scholar] [CrossRef] [PubMed]
- Peyta, L.; Jarnouen, K.; Pinault, M.; Coulouarn, C.; Guimaraes, C.; Goupille, C.; de Barros, J.-P.P.; Chevalier, S.; Dumas, J.-F.; Maillot, F. Regulation of hepatic cardiolipin metabolism by TNFα: Implication in cancer cachexia. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 1490–1500. [Google Scholar] [CrossRef]
- Goncalves, M.D.; Hwang, S.-K.; Pauli, C.; Murphy, C.J.; Cheng, Z.; Hopkins, B.D.; Wu, D.; Loughran, R.M.; Emerling, B.M.; Zhang, G. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E743–E752. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.C.; Barber, M.D.; Falconer, J.; McMillan, D.C.; Ross, J.A.; Preston, T. Pancreatic cancer as a model: Inflammatory mediators, acute-phase response, and cancer cachexia. World J. Surg. 1999, 23, 584–588. [Google Scholar] [CrossRef]
- Holroyde, C.P.; Skutches, C.L.; Boden, G.; Reichard, G.A. Glucose metabolism in cachectic patients with colorectal cancer. Cancer Res. 1984, 44, 5910–5913. [Google Scholar] [PubMed]
- Kokot, F.; Ficek, R. Effects of neuropeptide Y on appetite. Miner. Electrolyte Metab. 1999, 25, 303–305. [Google Scholar] [CrossRef]
- Argilés, J.M.; Stemmler, B.; López-Soriano, F.J.; Busquets, S. Nonmuscle tissues contribution to cancer cachexia. Mediat. Inflamm. 2015, 2015, 182872. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kumar, R.; Underwood, K.; O’Connor, A.E.; Loveland, K.L.; Seehra, J.S.; Matzuk, M.M. Prevention of cachexia-like syndrome development and reduction of tumor progression in inhibin-deficient mice following administration of a chimeric activin receptor type II-murine Fc protein. Mol. Hum. Reprod. 2007, 13, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Reddel, C.; Allen, J.; Ehteda, A.; Taylor, R.; Chen, V.; Curnow, J.; Kritharides, L.; Robertson, G. Increased thrombin generation in a mouse model of cancer cachexia is partially interleukin-6 dependent. J. Thromb. Haemost. 2017, 15, 477–486. [Google Scholar] [CrossRef]
- Savarese, G.; Lund, L.H. Global public health burden of heart failure. Card. Fail. Rev. 2017, 3, 7. [Google Scholar] [CrossRef]
- Evans, W.J.; Morley, J.E.; Argilés, J.; Bales, C.; Baracos, V.; Guttridge, D.; Jatoi, A.; Kalantar-Zadeh, K.; Lochs, H.; Mantovani, G. Cachexia: A new definition. Clin. Nutr. 2008, 27, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Kelm, N.Q.; Straughn, A.R.; Kakar, S.S. Withaferin A attenuates ovarian cancer-induced cardiac cachexia. PLoS ONE 2020, 15, e0236680. [Google Scholar] [CrossRef] [PubMed]
- Lena, A.; Ebner, N.; Anker, M.S. Cardiac cachexia. Eur. Heart J. Suppl. 2019, 21, L24–L27. [Google Scholar] [CrossRef] [PubMed]
- Martins, T.; Vitorino, R.; Moreira-Gonçalves, D.; Amado, F.; Duarte, J.A.; Ferreira, R. Recent insights on the molecular mechanisms and therapeutic approaches for cardiac cachexia. Clin. Biochem. 2014, 47, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Aukrust, P.; Ueland, T.; Lien, E.; Bendtzen, K.; Müller, F.; Andreassen, A.K.; Nordøy, I.; Aass, H.; Espevik, T.; Simonsen, S. Cytokine network in congestive heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am. J. Cardiol. 1999, 83, 376–382. [Google Scholar] [CrossRef]
- Anker, S.D. Imbalance of catabolic and anabolic pathways in chronic heart failure: Implications for the treatment of cardiac cachexia. Scand. J. Nutr. 2002, 46, 3–10. [Google Scholar] [CrossRef][Green Version]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Von Haehling, S.; Jankowska, E.A.; Anker, S.D. Tumour necrosis factor-α and the failing heart. Basic Res. Cardiol. 2004, 99, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Gulick, T.; Chung, M.K.; Pieper, S.J.; Lange, L.G.; Schreiner, G.F. Interleukin 1 and tumor necrosis factor inhibit cardiac myocyte beta-adrenergic responsiveness. Proc. Natl. Acad. Sci. USA 1989, 86, 6753–6757. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Von Haehling, S. Inflammatory mediators in chronic heart failure: An overview. Heart 2004, 90, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Ausoni, S.; Calamelli, S.; Saccà, S.; Azzarello, G. How progressive cancer endangers the heart: An intriguing and underestimated problem. Cancer Metastasis Rev. 2020, 39, 535–552. [Google Scholar] [CrossRef] [PubMed]
- von Haehling, S.; Doehner, W.; Anker, S.D. Nutrition, metabolism, and the complex pathophysiology of cachexia in chronic heart failure. Cardiovasc. Res. 2007, 73, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Schakman, O.; Gilson, H.; Thissen, J.-P. Mechanisms of glucocorticoid-induced myopathy. J. Endocrinol. 2008, 197, 1–10. [Google Scholar] [CrossRef]
- Cosper, P.F.; Leinwand, L.A. Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res. 2011, 71, 1710–1720. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Rojas, M.; Li, L.; Selzman, C.H.; Tang, R.-H.; Stansfield, W.E.; Rodriguez, J.E.; Glass, D.J.; Patterson, C. Muscle ring finger 1 mediates cardiac atrophy in vivo. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H997–H1006. [Google Scholar] [CrossRef] [PubMed]
- Costelli, P.; De Tullio, R.; Baccino, F.M.; Melloni, E. Activation of Ca 2+-dependent proteolysis in skeletal muscle and heart in cancer cachexia. Br. J. Cancer 2001, 84, 946–950. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.E.; Brown, J.L.; Rosa-Caldwell, M.E.; Perry, R.A.; Brown, L.A.; Haynie, W.S.; Washington, T.A.; Wiggs, M.P.; Rajaram, N.; Greene, N.P. Cancer-induced cardiac atrophy adversely affects myocardial redox state and mitochondrial oxidative characteristics. JCSM Rapid Commun. 2021, 4, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Busquets, S.; Fuster, G.; Ametller, E.; Olivan, M.; Figueras, M.; Costelli, P.; Carbó, N.; Argilés, J.M.; López-Soriano, F.J. Resveratrol does not ameliorate muscle wasting in different types of cancer cachexia models. Clin. Nutr. 2007, 26, 239–244. [Google Scholar] [CrossRef]
- Springer, J.; Tschirner, A.; Hartman, K.; Von Haehling, S.; Anker, S.D.; Doehner, W. The xanthine oxidase inhibitor oxypurinol reduces cancer cachexia-induced cardiomyopathy. Int. J. Cardiol. 2013, 168, 3527–3531. [Google Scholar] [CrossRef] [PubMed]
- Min, K.; Kwon, O.S.; Smuder, A.J.; Wiggs, M.P.; Sollanek, K.J.; Christou, D.D.; Yoo, J.K.; Hwang, M.H.; Szeto, H.H.; Kavazis, A.N. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J. Physiol. 2015, 593, 2017–2036. [Google Scholar] [CrossRef] [PubMed]
- Belloum, Y.; Rannou-Bekono, F.; Favier, F.B. Cancer-induced cardiac cachexia: Pathogenesis and impact of physical activity. Oncol. Rep. 2017, 37, 2543–2552. [Google Scholar] [CrossRef] [PubMed]
- Ino, K.; Shibata, K.; Kajiyama, H.; Yamamoto, E.; Nagasaka, T.; Nawa, A.; Nomura, S.; Kikkawa, F. Angiotensin II type 1 receptor expression in ovarian cancer and its correlation with tumour angiogenesis and patient survival. Br. J. Cancer 2006, 94, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Lever, A.F.; Hole, D.J.; Gillis, C.R.; McCallum, I.R.; McInnes, G.T.; MacKinnon, P.L.; Meredith, P.A.; Murray, L.S.; Reid, J.L.; Robertson, J.W. Do inhibitors of angiotensin-I-converting enzyme protect against risk of cancer? Lancet 1998, 352, 179–184. [Google Scholar] [CrossRef]
- Anker, S.D.; Negassa, A.; Coats, A.J.; Afzal, R.; Poole-Wilson, P.A.; Cohn, J.N.; Yusuf, S. Prognostic importance of weight loss in chronic heart failure and the effect of treatment with angiotensin-converting-enzyme inhibitors: An observational study. Lancet 2003, 361, 1077–1083. [Google Scholar] [CrossRef]
- Delafontaine, P.; Akao, M. Angiotensin II as candidate of cardiac cachexia. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 220. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shen, C.; Zhou, J.; Wang, X.; Yu, X.-Y.; Liang, C.; Liu, B.; Pan, X.; Zhao, Q.; Song, J.L.; Wang, J. Angiotensin-II-induced muscle wasting is mediated by 25-hydroxycholesterol via GSK3β signaling pathway. EBioMedicine 2017, 16, 238–250. [Google Scholar] [CrossRef]
- Zablocki, D.; Sadoshima, J. Angiotensin II and oxidative stress in the failing heart. Antioxid. Redox Signal. 2013, 19, 1095–1109. [Google Scholar] [CrossRef]
- Zhang, S.; Lu, Y.; Jiang, C. Inhibition of histone demethylase JMJD1C attenuates cardiac hypertrophy and fibrosis induced by angiotensin II. J. Recept. Signal Transduct. 2020, 40, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Han, F. FOXF1 ameliorates angiotensin II-induced cardiac fibrosis in cardiac fibroblasts through inhibiting the TGF-β1/Smad3 signaling pathway. J. Recept. Signal Transduct. 2020, 40, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.H.; Hong, X.; Zhao, N.; Liu, X.H.; Xiao, Y.F.; Chen, T.T.; Deng, L.B.; Wang, X.L.; Wang, J.B.; Ji, G.J. CD38 promotes angiotensin II-induced cardiac hypertrophy. J. Cell. Mol. Med. 2017, 21, 1492–1502. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-W.; Qin, D.-Z.; James, E.; McKallip, R.J.; Wang, N.-P.; Zhang, W.-W.; Zheng, R.-H.; Han, Q.-H.; Zhao, Z.-Q. CD44 deficiency in mice protects the heart against angiotensin II-induced cardiac fibrosis. Shock 2019, 51, 372–380. [Google Scholar] [CrossRef]
- Zheng, C.-B.; Gao, W.-C.; Xie, M.; Li, Z.; Ma, X.; Song, W.; Luo, D.; Huang, Y.; Yang, J.; Zhang, P. Ang II promotes cardiac autophagy and hypertrophy via Orai1/STIM1. Front. Pharmacol. 2021, 12, 622774. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Zhang, M.; Hu, J.; Lin, J.; Feng, X.; Wang, S.; Wang, T.; Gao, E.; Wang, H.; Sun, D. Mst1 knockout enhances cardiomyocyte autophagic flux to alleviate angiotensin II-induced cardiac injury independent of angiotensin II receptors. J. Mol. Cell. Cardiol. 2018, 125, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Delafontaine, P.; Brink, M. The growth hormone and insulin-like growth factor 1 axis in heart failure. In Proceedings of the Annales D’endocrinologie, France, Paris, 1 February 2000; pp. 22–26. [Google Scholar]
- Rausch, V.; Sala, V.; Penna, F.; Porporato, P.E.; Ghigo, A. Understanding the common mechanisms of heart and skeletal muscle wasting in cancer cachexia. Oncogenesis 2021, 10, 1–13. [Google Scholar] [CrossRef]
- Levine, B.D. What do we know, and what do we still need to know? J. Physiol. 2008, 586, 25–34. [Google Scholar] [CrossRef] [PubMed]
- de Boer, R.A.; Hulot, J.S.; Tocchetti, C.G.; Aboumsallem, J.P.; Ameri, P.; Anker, S.D.; Bauersachs, J.; Bertero, E.; Coats, A.J.; Čelutkienė, J. Common mechanistic pathways in cancer and heart failure. A scientific roadmap on behalf of the Translational Research Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur. J. Heart Fail. 2020, 22, 2272–2289. [Google Scholar] [CrossRef] [PubMed]
- Scafoglieri, A.; Clarys, J.P. Dual energy X-ray absorptiometry: Gold standard for muscle mass? J. Cachexia Sarcopenia Muscle 2018, 9, 786. [Google Scholar] [CrossRef] [PubMed]
- Stimpson, S.A.; Leonard, M.S.; Clifton, L.G.; Poole, J.C.; Turner, S.M.; Shearer, T.W.; Remlinger, K.S.; Clark, R.V.; Hellerstein, M.K.; Evans, W.J. Longitudinal changes in total body creatine pool size and skeletal muscle mass using the D3-creatine dilution method. J. Cachexia Sarcopenia Muscle 2013, 4, 217–223. [Google Scholar] [CrossRef]
- Bordignon, C.; Dos Santos, B.S.; Rosa, D.D. Impact of Cancer Cachexia on Cardiac and Skeletal Muscle: Role of Exercise Training. Cancers 2022, 14, 342. [Google Scholar] [CrossRef]
- Skovgaard, D.; Hasbak, P.; Kjaer, A. BNP predicts chemotherapy-related cardiotoxicity and death: Comparison with gated equilibrium radionuclide ventriculography. PLoS ONE 2014, 9, e96736. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Bonetto, A.; Muscaritoli, M.; Costamagna, D.; Minero, V.G.; Bonelli, G.; Fanelli, F.R.; Baccino, F.M.; Costelli, P. Muscle atrophy in experimental cancer cachexia: Is the IGF-1 signaling pathway involved? Int. J. Cancer 2010, 127, 1706–1717. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.B.; Romero, W.G.; de Abreu Carvalho, A.L.R.; Souza, G.A.A.; Claudio, E.R.G.; Abreu, G.R. Effects of treatment with chemotherapy and/or tamoxifen on the biomarkers of cardiac injury and oxidative stress in women with breast cancer. Medicine 2017, 96, e8723. [Google Scholar] [CrossRef] [PubMed]
- Hulmi, J.J.; Nissinen, T.A.; Penna, F.; Bonetto, A. Targeting the activin receptor signaling to counteract the multi-systemic complications of cancer and its treatments. Cells 2021, 10, 516. [Google Scholar] [CrossRef] [PubMed]
- Yndestad, A.; Ueland, T.; Øie, E.; Florholmen, G.; Halvorsen, B.; Attramadal, H.; Simonsen, S.; Frøland, S.S.; Gullestad, L.; Christensen, G. Elevated levels of activin A in heart failure: Potential role in myocardial remodeling. Circulation 2004, 109, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Pérez, A.V.; Doehner, W.; Von Haehling, S.; Schmidt, H.; Zimmermann, A.V.; Volk, H.-D.; Anker, S.D.; Rauchhaus, M. The relationship between tumor necrosis factor-α, brain natriuretic peptide and atrial natriuretic peptide in patients with chronic heart failure. Int. J. Cardiol. 2010, 141, 39–43. [Google Scholar] [CrossRef]
- Anker, S.D.; Chua, T.P.; Ponikowski, P.; Harrington, D.; Swan, J.W.; Kox, W.J.; Poole-Wilson, P.A.; Coats, A.J. Hormonal changes and catabolic/anabolic imbalance in chronic heart failure and their importance for cardiac cachexia. Circulation 1997, 96, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Martins, T.; Vitorino, R.; Amado, F.; Duarte, J.A.; Ferreira, R. Biomarkers for cardiac cachexia: Reality or utopia. Clin. Chim. Acta 2014, 436, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.; Oeing, C.U.; Rohm, M.; Baysal-Temel, E.; Lehmann, L.H.; Bauer, R.; Volz, H.C.; Boutros, M.; Sohn, D.; Sticht, C. Ataxin-10 is part of a cachexokine cocktail triggering cardiac metabolic dysfunction in cancer cachexia. Mol. Metab. 2016, 5, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Kazemi-Bajestani, S.M.; Becher, H.; Fassbender, K.; Chu, Q.; Baracos, V.E. Concurrent evolution of cancer cachexia and heart failure: Bilateral effects exist. J. Cachexia Sarcopenia Muscle 2014, 5, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Roderburg, C.; Loosen, S.H.; Jahn, J.K.; Gänsbacher, J.; Luedde, T.; Kostev, K.; Luedde, M. Heart failure is associated with an increased incidence of cancer diagnoses. ESC Heart Fail. 2021, 8, 3628–3633. [Google Scholar] [CrossRef] [PubMed]
- Burch, G.; Phillips, J.H.; Ansari, A. The cachectic heart: A clinico-pathologic, electrocardiographic and roentgenographic entity. Dis. Chest 1968, 54, 403–409. [Google Scholar] [CrossRef]
- Springer, J.; Tschirner, A.; Haghikia, A.; Von Haehling, S.; Lal, H.; Grzesiak, A.; Kaschina, E.; Palus, S.; Pötsch, M.; Von Websky, K. Prevention of liver cancer cachexia-induced cardiac wasting and heart failure. Eur. Heart J. 2014, 35, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Devine, R.D.; Bicer, S.; Reiser, P.J.; Velten, M.; Wold, L.E. Metalloproteinase expression is altered in cardiac and skeletal muscle in cancer cachexia. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H685–H691. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Nishijima, Y.; Asp, M.L.; Stout, M.B.; Reiser, P.J.; Belury, M.A. Cardiac alterations in cancer-induced cachexia in mice. Int. J. Oncol. 2010, 37, 347–353. [Google Scholar] [PubMed]
- Trexler, C.L.; Odell, A.T.; Jeong, M.Y.; Dowell, R.D.; Leinwand, L.A. Transcriptome and functional profile of cardiac myocytes is influenced by biological sex. Circ. Cardiovasc. Genet. 2017, 10, e001770. [Google Scholar] [CrossRef] [PubMed]
- Pin, F.; Barreto, R.; Kitase, Y.; Mitra, S.; Erne, C.E.; Novinger, L.J.; Zimmers, T.A.; Couch, M.E.; Bonewald, L.F.; Bonetto, A. Growth of ovarian cancer xenografts causes loss of muscle and bone mass: A new model for the study of cancer cachexia. J. Cachexia Sarcopenia Muscle 2018, 9, 685–700. [Google Scholar] [CrossRef]
- Berent, T.E.; Dorschner, J.M.; Meyer, T.; Craig, T.A.; Wang, X.; Kunz, H.; Jatoi, A.; Lanza, I.R.; Chen, H.; Kumar, R. Impaired cardiac performance, protein synthesis, and mitochondrial function in tumor-bearing mice. PLoS ONE 2019, 14, e0226440. [Google Scholar] [CrossRef] [PubMed]
- Cramer, L.; Hildebrandt, B.; Kung, T.; Wichmann, K.; Springer, J.; Doehner, W.; Sandek, A.; Valentova, M.; Stojakovic, T.; Scharnagl, H. Cardiovascular function and predictors of exercise capacity in patients with colorectal cancer. J. Am. Coll. Cardiol. 2014, 64, 1310–1319. [Google Scholar] [CrossRef]
- Danese, E.; Montagnana, M.; Giudici, S.; Aloe, R.; Franchi, M.; Guidi, G.C.; Lippi, G. Highly-sensitive troponin I is increased in patients with gynecological cancers. Clin. Biochem. 2013, 46, 1135–1138. [Google Scholar] [CrossRef] [PubMed]
- Pavo, N.; Raderer, M.; Hülsmann, M.; Neuhold, S.; Adlbrecht, C.; Strunk, G.; Goliasch, G.; Gisslinger, H.; Steger, G.G.; Hejna, M. Cardiovascular biomarkers in patients with cancer and their association with all-cause mortality. Heart 2015, 101, 1874–1880. [Google Scholar] [CrossRef] [PubMed]
- Labib, D.; Satriano, A.; Dykstra, S.; Hansen, R.; Mikami, Y.; Guzzardi, D.G.; Slavikova, Z.; Feuchter, P.; Flewitt, J.; Rivest, S. Effect of Active Cancer on the Cardiac Phenotype: A Cardiac Magnetic Resonance Imaging-Based Study of Myocardial Tissue Health and Deformation in Patients With Chemotherapy-Naïve Cancer. J. Am. Heart Assoc. 2021, 10, e019811. [Google Scholar] [CrossRef] [PubMed]
- Stoltzfus, K.C.; Zhang, Y.; Sturgeon, K.; Sinoway, L.I.; Trifiletti, D.M.; Chinchilli, V.M.; Zaorsky, N.G. Fatal heart disease among cancer patients. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Tadic, M.; Genger, M.; Baudisch, A.; Kelle, S.; Cuspidi, C.; Belyavskiy, E.; Burkhardt, F.; Venneri, L.; Attanasio, P.; Pieske, B. Left ventricular strain in chemotherapy-naive and radiotherapy-naive patients with cancer. Can. J. Cardiol. 2018, 34, 281–287. [Google Scholar] [CrossRef]
- Tadic, M.; Baudisch, A.; Haßfeld, S.; Heinzel, F.; Cuspidi, C.; Burkhardt, F.; Escher, F.; Attanasio, P.; Pieske, B.; Genger, M. Right ventricular function and mechanics in chemotherapy-and radiotherapy-naïve cancer patients. Int. J. Cardiovasc. Imaging 2018, 34, 1581–1587. [Google Scholar] [CrossRef]
- Tadic, M.; Genger, M.; Cuspidi, C.; Belyavskiy, E.; Frydas, A.; Dordevic, A.; Morris, D.A.; Völkl, J.; Parwani, A.S.; Pieske, B. Phasic left atrial function in cancer patients before initiation of anti-cancer therapy. J. Clin. Med. 2019, 8, 421. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Layard, M.W.; Basa, P.; Davis, H.L., Jr.; Von Hoff, A.L.; Rozencweig, M.; Muggia, F.M. Risk factors for doxorubicin-lnduced congestive heart failure. Ann. Intern. Med. 1979, 91, 710–717. [Google Scholar] [CrossRef] [PubMed]
- de Boer, R.A.; Aboumsallem, J.P.; Bracun, V.; Leedy, D.; Cheng, R.; Patel, S.; Rayan, D.; Zaharova, S.; Rymer, J.; Kwan, J.M. A new classification of cardio-oncology syndromes. Cardio-Oncology 2021, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Huot, J.R.; Essex, A.L.; Gutierrez, M.; Barreto, R.; Wang, M.; Waning, D.L.; Plotkin, L.I.; Bonetto, A. Chronic treatment with multi-kinase inhibitors causes differential toxicities on skeletal and cardiac muscles. Cancers 2019, 11, 571. [Google Scholar] [CrossRef] [PubMed]
- Pouna, P.; Bonoron-Adèle, S.; Gouverneur, G.; Tariosse, L.; Besse, P.; Robert, J. Development of the model of rat isolated perfused heart for the evaluation of anthracycline cardiotoxicity and its circumvention. Br. J. Pharmacol. 1996, 117, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Platel, D.; Pouna, P.; Bonoron-Adèle, S.; Robert, J. Preclinical evaluation of the cardiotoxicity of taxane–anthracycline combinations using the model of isolated perfused rat heart. Toxicol. Appl. Pharmacol. 2000, 163, 135–140. [Google Scholar] [CrossRef]
- Hasinoff, B.B. The cardiotoxicity and myocyte damage caused by small molecule anticancer tyrosine kinase inhibitors is correlated with lack of target specificity. Toxicol. Appl. Pharmacol. 2010, 244, 190–195. [Google Scholar] [CrossRef]
- Albini, A.; Festa, M.M.; Ring, N.; Baci, D.; Rehman, M.; Finzi, G.; Sessa, F.; Zacchigna, S.; Bruno, A.; Noonan, D.M. A polyphenol-rich extract of Olive Mill Wastewater Enhances cancer chemotherapy effects, while mitigating cardiac toxicity. Front. Pharmacol. 2021, 12, 694762. [Google Scholar] [CrossRef]
- Knapik-Czajka, M.; Jurczyk, M.; Bieleń, J.; Aleksandrovych, V.; Gawędzka, A.; Stach, P.; Drąg, J.; Gil, K. Effect of 5-fluorouracil on branched-chain α-keto acid dehydrogenase (BCKDH) complex in rat’s heart. Folia Med. Crac. 2021, 61, 121–129. [Google Scholar]
- Li, Y.; Zhang, Y.; Zhou, X.; Lei, X.; Li, X.; Wei, L. Dynamic observation of 5-fluorouracil-induced myocardial injury and mitochondrial autophagy in aging rats. Exp. Ther. Med. 2021, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Tang, H.; Ai, W.; Zeng, Q.; Yang, H.; Zhu, F.; Wei, Y.; Feng, R.; Wen, L.; Pu, P. Schisandrin B Antagonizes Cardiotoxicity Induced by Pirarubicin by Inhibiting Mitochondrial Permeability Transition Pore (mPTP) Opening and Decreasing Cardiomyocyte Apoptosis. Front. Pharmacol. 2021, 12, 733805. [Google Scholar] [CrossRef]
- El-Hawwary, A.A.; Omar, N.M. The influence of ginger administration on cisplatin-induced cardiotoxicity in rat: Light and electron microscopic study. Acta Histochem. 2019, 121, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Saleh, D.O.; Mansour, D.F.; Mostafa, R.E. Rosuvastatin and simvastatin attenuate cisplatin-induced cardiotoxicity via disruption of endoplasmic reticulum stress-mediated apoptotic death in rats: Targeting ER-chaperone GRP78 and calpain-1 pathways. Toxicol. Rep. 2020, 7, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Abd-ElRaouf, A.; Nada, A.S.; Mohammed, N.E.-D.A.; Amer, H.A.; Abd-ElRahman, S.S.; Abdelsalam, R.M.; Salem, H.A. Low dose gamma irradiation attenuates cyclophosphamide-induced cardiotoxicity in rats: Role of NF-κB signaling pathway. Int. J. Radiat. Biol. 2021, 97, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Elrashidy, R.A.; Hasan, R.A. Cilostazol preconditioning alleviates cyclophosphamide-induced cardiotoxicity in male rats: Mechanistic insights into SIRT1 signaling pathway. Life Sci. 2021, 266, 118822. [Google Scholar] [CrossRef]
- Kamel, S.S.; Baky, N.A.A.; Karkeet, R.M.; Osman, A.M.M.; Sayed-Ahmed, M.M.; Fouad, M.A. Astaxanthin extenuates the inhibition of aldehyde dehydrogenase and Klotho protein expression in cyclophosphamide-induced acute cardiomyopathic rat model. Clin. Exp. Pharmacol. Physiol. 2022, 49, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Al-Taher, A.Y.; Morsy, M.A.; Rifaai, R.A.; Zenhom, N.M.; Abdel-Gaber, S.A. Paeonol attenuates methotrexate-induced cardiac toxicity in rats by inhibiting oxidative stress and suppressing TLR4-induced NF-κB inflammatory pathway. Mediat. Inflamm. 2020, 2020, 8641026. [Google Scholar] [CrossRef] [PubMed]
- Mohan, N.; Shen, Y.; Endo, Y.; ElZarrad, M.K.; Wu, W.J. Trastuzumab, but not pertuzumab, dysregulates HER2 signaling to mediate inhibition of autophagy and increase in reactive oxygen species production in human cardiomyocytes. Mol. Cancer Ther. 2016, 15, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Novo, G.; Di Lisi, D.; Bronte, E.; Macaione, F.; Accurso, V.; Badalamenti, G.; Rinaldi, G.; Siragusa, S.; Novo, S.; Russo, A. Cardiovascular toxicity in cancer patients treated with tyrosine kinase inhibitors: A real-world single-center experience. Oncology 2020, 98, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Hunt, S.A.; Abraham, W.T.; Chin, M.H.; Feldman, A.M.; Francis, G.S.; Ganiats, T.G.; Jessup, M.; Konstam, M.A.; Mancini, D.M.; Michl, K. 2009 focused update incorporated into the ACC/AHA 2005 guidelines for the diagnosis and management of heart failure in adults: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines developed in collaboration with the International Society for Heart and Lung Transplantation. J. Am. Coll. Cardiol. 2009, 53, e1–e90. [Google Scholar] [PubMed]
- Boyd, A.; Stoodley, P.; Richards, D.; Hui, R.; Harnett, P.; Vo, K.; Marwick, T.; Thomas, L. Anthracyclines induce early changes in left ventricular systolic and diastolic function: A single centre study. PLoS ONE 2017, 12, e0175544. [Google Scholar] [CrossRef]
- Serrano, J.M.; González, I.; Del Castillo, S.; Muñiz, J.; Morales, L.J.; Moreno, F.; Jiménez, R.; Cristóbal, C.; Graupner, C.; Talavera, P. Diastolic dysfunction following anthracycline-based chemotherapy in breast cancer patients: Incidence and predictors. Oncologist 2015, 20, 864. [Google Scholar] [CrossRef]
- Ito, M.; Horimoto, Y.; Sasaki, R.; Miyazaki, S.; Orihata, G.; Saito, M. Cardiotoxicity after Additional Administration of Pertuzumab following Long-Term Trastuzumab: Report of 2 Cases. Case Rep. Oncol. 2021, 14, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Lladó, A.; Bianchi, G.; Cortes, J.; Kellokumpu-Lehtinen, P.-L.; Cameron, D.A.; Miles, D.; Salvagni, S.; Wardley, A.; Goeminne, J.-C. Open-label, phase II, multicenter, randomized study of the efficacy and safety of two dose levels of pertuzumab, a human epidermal growth factor receptor 2 dimerization inhibitor, in patients with human epidermal growth factor receptor 2–negative metastatic breast cancer. J. Clin. Oncol. 2010, 28, 1131. [Google Scholar] [PubMed]
- Mahmood, S.S.; Fradley, M.G.; Cohen, J.V.; Nohria, A.; Reynolds, K.L.; Heinzerling, L.M.; Sullivan, R.J.; Damrongwatanasuk, R.; Chen, C.L.; Gupta, D. Myocarditis in patients treated with immune checkpoint inhibitors. J. Am. Coll. Cardiol. 2018, 71, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Witte, K.K.; Clark, A.L.; Cleland, J.G. Chronic heart failure and micronutrients. J. Am. Coll. Cardiol. 2001, 37, 1765–1774. [Google Scholar] [CrossRef]
- Engelen, M.; Safar, A.; Bartter, T.; Koeman, F.; Deutz, N. High anabolic potential of essential amino acid mixtures in advanced nonsmall cell lung cancer. Ann. Oncol. 2015, 26, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Nystoriak, M.A.; Bhatnagar, A. Cardiovascular effects and benefits of exercise. Front. Cardiovasc. Med. 2018, 5, 135. [Google Scholar] [CrossRef] [PubMed]
- Parry, T.L.; Hayward, R. Exercise Protects against Cancer-induced Cardiac Cachexia. Med. Sci. Sports Exerc. 2018, 50, 1169–1176. [Google Scholar] [CrossRef]
- Fernandes, L.; Tobias, G.; Paixão, A.; Dourado, P.; Voltarelli, V.; Brum, P. Exercise training delays cardiac remodeling in a mouse model of cancer cachexia. Life Sci. 2020, 260, 118392. [Google Scholar] [CrossRef] [PubMed]
- Padrão, A.I.; Nogueira-Ferreira, R.; Vitorino, R.; Carvalho, D.; Correia, C.; Neuparth, M.J.; Pires, M.J.; Faustino-Rocha, A.I.; Santos, L.L.; Oliveira, P.A. Exercise training protects against cancer-induced cardiac remodeling in an animal model of urothelial carcinoma. Arch. Biochem. Biophys. 2018, 645, 12–18. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, M.G.; Winningham, M.L.; Nickel, J.L. Effects of aerobic interval training on cancer patients’ functional capacity. Nurs. Res. 1989, 38, 348–351. [Google Scholar] [CrossRef]
- Courneya, K.S.; Segal, R.J.; Mackey, J.R.; Gelmon, K.; Reid, R.D.; Friedenreich, C.M.; Ladha, A.B.; Proulx, C.; Vallance, J.; Lane, K. Effects of aerobic and resistance exercise in breast cancer patients receiving adjuvant chemotherapy: A multicenter randomized controlled trial. J. Clin. Oncol. 2007, 25, 4396–4404. [Google Scholar] [CrossRef] [PubMed]
- Courneya, K.S.; McKenzie, D.C.; Mackey, J.R.; Gelmon, K.; Friedenreich, C.M.; Yasui, Y.; Reid, R.D.; Cook, D.; Jespersen, D.; Proulx, C. Effects of exercise dose and type during breast cancer chemotherapy: Multicenter randomized trial. J. Natl. Cancer Inst. 2013, 105, 1821–1832. [Google Scholar] [CrossRef] [PubMed]
- Van Waart, H.; Stuiver, M.M.; van Harten, W.H.; Geleijn, E.; Kieffer, J.M.; Buffart, L.M.; de Maaker-Berkhof, M.; Boven, E.; Schrama, J.; Geenen, M.M. Effect of low-intensity physical activity and moderate-to high-intensity physical exercise during adjuvant chemotherapy on physical fitness, fatigue, and chemotherapy completion rates: Results of the PACES randomized clinical trial. J. Clin. Oncol. 2015, 33, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.M.; Iyengar, N.M.; Nilsen, T.S.; Michalski, M.; Thomas, S.M.; Herndon, J.; Sasso, J.; Yu, A.; Chandarlapaty, S.; Dang, C.T. Feasibility, safety, and efficacy of aerobic training in pretreated patients with metastatic breast cancer: A randomized controlled trial. Cancer 2018, 124, 2552–2560. [Google Scholar] [CrossRef] [PubMed]
- Courneya, K.S.; Sellar, C.M.; Stevinson, C.; McNeely, M.L.; Peddle, C.J.; Friedenreich, C.M.; Tankel, K.; Basi, S.; Chua, N.; Mazurek, A. Randomized controlled trial of the effects of aerobic exercise on physical functioning and quality of life in lymphoma patients. J. Clin. Oncol. 2009, 27, 4605–4612. [Google Scholar] [CrossRef] [PubMed]
- Segal, R.J.; Reid, R.D.; Courneya, K.S.; Sigal, R.J.; Kenny, G.P.; Prud’Homme, D.G.; Malone, S.C.; Wells, G.A.; Scott, C.G.; Slovinec D’Angelo, M.E. Randomized controlled trial of resistance or aerobic exercise in men receiving radiation therapy for prostate cancer. J. Clin. Oncol. 2009, 27, 344–351. [Google Scholar] [CrossRef]
- Pinto, B.; Stein, K.; Dunsiger, S. Peer Mentoring to Promote Exercise Among Cancer Survivors: A Community Partnership: S12-1. Pscyho-Oncology 2013, 22, 115–116. [Google Scholar]
- Rogers, L.Q.; Courneya, K.S.; Anton, P.M.; Hopkins-Price, P.; Verhulst, S.; Vicari, S.K.; Robbs, R.S.; Mocharnuk, R.; McAuley, E. Effects of the BEAT Cancer physical activity behavior change intervention on physical activity, aerobic fitness, and quality of life in breast cancer survivors: A multicenter randomized controlled trial. Breast Cancer Res. Treat. 2015, 149, 109–119. [Google Scholar] [CrossRef]
- Jones, L.W.; Douglas, P.S.; Khouri, M.G.; Mackey, J.R.; Wojdyla, D.; Kraus, W.E.; Whellan, D.J.; O’Connor, C.M. Safety and efficacy of aerobic training in patients with cancer who have heart failure: An analysis of the HF-ACTION randomized trial. J. Clin. Oncol. 2014, 32, 2496. [Google Scholar] [CrossRef]
- Hong, D.S.; Hui, D.; Bruera, E.; Janku, F.; Naing, A.; Falchook, G.S.; Piha-Paul, S.; Wheler, J.J.; Fu, S.; Tsimberidou, A.M. MABp1, a first-in-class true human antibody targeting interleukin-1α in refractory cancers: An open-label, phase 1 dose-escalation and expansion study. Lancet Oncol. 2014, 15, 656–666. [Google Scholar] [CrossRef]
- Lust, J.A.; Lacy, M.Q.; Zeldenrust, S.R.; Dispenzieri, A.; Gertz, M.A.; Witzig, T.E.; Kumar, S.; Hayman, S.R.; Russell, S.J.; Buadi, F.K. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1β-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin. Proc. 2009, 84, 114–122. [Google Scholar] [CrossRef]
- Jatoi, A.; Ritter, H.L.; Dueck, A.; Nguyen, P.L.; Nikcevich, D.A.; Luyun, R.F.; Mattar, B.I.; Loprinzi, C.L. A placebo-controlled, double-blind trial of infliximab for cancer-associated weight loss in elderly and/or poor performance non-small cell lung cancer patients (N01C9). Lung Cancer 2010, 68, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Bayliss, T.J.; Smith, J.T.; Schuster, M.; Dragnev, K.H.; Rigas, J.R. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert Opin. Biol. Ther. 2011, 11, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.J.; Cot, T.R.; Cuffe, M.S.; Kramer, J.M.; Braun, M.M. Case reports of heart failure after therapy with a tumor necrosis factor antagonist. Ann. Intern. Med. 2003, 138, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, G.; Macciò, A.; Madeddu, C.; Serpe, R.; Antoni, G.; Massa, E.; Dessì, M.; Panzone, F. Phase II nonrandomized study of the efficacy and safety of COX-2 inhibitor celecoxib on patients with cancer cachexia. J. Mol. Med. 2010, 88, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Reid, J.; Hughes, C.; Murray, L.; Parsons, C.; Cantwell, M. Non-steroidal anti-inflammatory drugs for the treatment of cancer cachexia: A systematic review. Palliat. Med. 2013, 27, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Solheim, T.S.; Fearon, K.C.; Blum, D.; Kaasa, S. Non-steroidal anti-inflammatory treatment in cancer cachexia: A systematic literature review. Acta Oncol. 2013, 52, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Musolino, V.; Palus, S.; Tschirner, A.; Drescher, C.; Gliozzi, M.; Carresi, C.; Vitale, C.; Muscoli, C.; Doehner, W.; von Haehling, S. Megestrol acetate improves cardiac function in a model of cancer cachexia-induced cardiomyopathy by autophagic modulation. J. Cachexia Sarcopenia Muscle 2016, 7, 555–566. [Google Scholar] [CrossRef]
- Garcia, V.R.; López-Briz, E.; Sanchis, R.C.; Perales, J.L.G.; Bort-Martí, S. Megestrol acetate for treatment of anorexia-cachexia syndrome. Cochrane Database Syst. Rev. 2013, 2013, CD004310. [Google Scholar] [CrossRef]
- Loprinzi, C.L.; Kugler, J.W.; Sloan, J.A.; Mailliard, J.A.; Krook, J.E.; Wilwerding, M.B.; Rowland, K.M., Jr.; Camoriano, J.K.; Novotny, P.J.; Christensen, B.J. Randomized comparison of megestrol acetate versus dexamethasone versus fluoxymesterone for the treatment of cancer anorexia/cachexia. J. Clin. Oncol. 1999, 17, 3299–3306. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.; Schutt, A.; Reitemeier, R.; Hahn, R. Corticosteroid therapy of preterminal gastrointestinal cancer. Cancer 1974, 33, 1607–1609. [Google Scholar] [CrossRef]
- Yavuzsen, T.; Davis, M.P.; Walsh, D.; LeGrand, S.; Lagman, R. Systematic review of the treatment of cancer-associated anorexia and weight loss. Database Abstr. Rev. Eff. Qual. Assess. Rev. 2005, 23, 8500–8511. [Google Scholar] [CrossRef] [PubMed]
- Temel, J.S.; Abernethy, A.P.; Currow, D.C.; Friend, J.; Duus, E.M.; Yan, Y.; Fearon, K.C. Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): Results from two randomised, double-blind, phase 3 trials. Lancet Oncol. 2016, 17, 519–531. [Google Scholar] [CrossRef]
- Roeland, E.J.; Bohlke, K.; Baracos, V.E.; Bruera, E.; Del Fabbro, E.; Dixon, S.; Fallon, M.; Herrstedt, J.; Lau, H.; Platek, M. Management of cancer cachexia: ASCO guideline. J. Clin. Oncol. 2020, 38, 2438–2453. [Google Scholar] [CrossRef] [PubMed]
- Reiche, E.M.V.; Nunes, S.O.V.; Morimoto, H.K. Stress, depression, the immune system, and cancer. Lancet Oncol. 2004, 5, 617–625. [Google Scholar] [CrossRef]
- Elkina, Y.; Palus, S.; Tschirner, A.; Hartmann, K.; Von Haehling, S.; Doehner, W.; Mayer, U.; Coats, A.J.; Beadle, J.; Anker, S.D. Tandospirone reduces wasting and improves cardiac function in experimental cancer cachexia. Int. J. Cardiol. 2013, 170, 160–166. [Google Scholar] [CrossRef]
- Devine, R.D.; Eichenseer, C.M.; Wold, L.E. Minocycline attenuates cardiac dysfunction in tumor-burdened mice. J. Mol. Cell. Cardiol. 2016, 100, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Palus, S.; Von Haehling, S.; Flach, V.C.; Tschirner, A.; Doehner, W.; Anker, S.D.; Springer, J. Simvastatin reduces wasting and improves cardiac function as well as outcome in experimental cancer cachexia. Int. J. Cardiol. 2013, 168, 3412–3418. [Google Scholar] [CrossRef]
- Muscaritoli, M.; Costelli, P.; Bossola, M.; Grieco, G.; Bonelli, G.; Bellantone, R.; Doglietto, G.B.; Rossi-Fanelli, F.; Baccino, F.M. Effects of simvastatin administration in an experimental model of cancer cachexia. Nutrition 2003, 19, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Hatanaka, M.; Konishi, M.; Ishida, J.; Palus, S.; Ebner, N.; Döhner, W.; von Haehling, S.; Anker, S.D.; Springer, J. Erythropoietin improves cardiac wasting and outcomes in a rat model of liver cancer cachexia. Int. J. Cardiol. 2016, 218, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, D.; Strom, J.; Chen, Q.M. Glucocorticoid induced leucine zipper inhibits apoptosis of cardiomyocytes by doxorubicin. Toxicol. Appl. Pharmacol. 2014, 276, 55–62. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Legi, A.; Rodriguez, E.; Eckols, T.K.; Mistry, C.; Robinson, P. Substance P antagonism prevents chemotherapy-induced cardiotoxicity. Cancers 2021, 13, 1732. [Google Scholar] [CrossRef] [PubMed]
- Find a Study. Available online: https://clinicaltrials.gov/ct2/home (accessed on 3 March 2022).
- Cardinale, D.; Sandri, M.T.; Martinoni, A.; Tricca LabTech, A.; Civelli, M.; Lamantia, G.; Cinieri, S.; Martinelli, G.; Cipolla, C.M.; Fiorentini, C. Left ventricular dysfunction predicted by early troponin I release after high-dose chemotherapy. J. Am. Coll. Cardiol. 2000, 36, 517–522. [Google Scholar] [CrossRef]
- Avila, M.S.; Ayub-Ferreira, S.M.; de Barros Wanderley, M.R.; das Dores Cruz, F.; Gonçalves Brandão, S.M.; Rigaud, V.O.C.; Higuchi-dos-Santos, M.H.; Hajjar, L.A.; Kalil Filho, R.; Hoff, P.M. Carvedilol for prevention of chemotherapy-related cardiotoxicity: The CECCY trial. J. Am. Coll. Cardiol. 2018, 71, 2281–2290. [Google Scholar] [CrossRef]
- Straughn, A.R.; Kelm, N.Q.; Kakar, S.S. Withaferin A and Ovarian Cancer Antagonistically Regulate Skeletal Muscle Mass. Front. Cell Dev. Biol. 2021, 9, 354. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- He, W.A.; Calore, F.; Londhe, P.; Canella, A.; Guttridge, D.C.; Croce, C.M. Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proc. Natl. Acad. Sci. USA 2014, 111, 4525–4529. [Google Scholar] [CrossRef] [PubMed]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Bei, Y.; Xiao, J. MicroRNAs in muscle wasting and cachexia induced by heart failure. Nat. Rev. Cardiol. 2017, 14, 566-566. [Google Scholar] [CrossRef] [PubMed]
- Bindels, L.B.; Neyrinck, A.M.; Loumaye, A.; Catry, E.; Walgrave, H.; Cherbuy, C.; Leclercq, S.; Van Hul, M.; Plovier, H.; Pachikian, B. Increased gut permeability in cancer cachexia: Mechanisms and clinical relevance. Oncotarget 2018, 9, 18224. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Guo, C.; Zhang, D.; Zhang, J.; Wang, X.; Geng, C. The altered tight junctions: An important gateway of bacterial translocation in cachexia patients with advanced gastric cancer. J. Interferon Cytokine Res. 2014, 34, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Bindels, L.B.; Beck, R.; Schakman, O.; Martin, J.C.; De Backer, F.; Sohet, F.M.; Dewulf, E.M.; Pachikian, B.D.; Neyrinck, A.M.; Thissen, J.-P. Restoring specific lactobacilli levels decreases inflammation and muscle atrophy markers in an acute leukemia mouse model. PLoS ONE 2012, 7, e37971. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.W.; Li, D.Y.; Hazen, S.L. Dietary metabolism, the gut microbiome, and heart failure. Nat. Rev. Cardiol. 2019, 16, 137–154. [Google Scholar] [CrossRef] [PubMed]





| S.No | Name of the Drug | Experimental Model | Outcome | Reference |
|---|---|---|---|---|
| 1 | Doxorubicin (Doxo)/epirubicin (Epi), pirarubicin, daunorubicin (Dauno) | Perfusion of isolated hearts from treated (for 11 days on alternate day) and untreated 10-week-old male Sprague-Dawley rats that were sacrificed on the 12th day | Doxorubicin and daunorubicin: most toxic agents. Doxo caused a 33% reduction in [LV(dP/dt)max], a 29% decrease in [LV(dP/dt)min, and less changes were observed with Epi and dauno | [86] |
| 2 | Doxorubicin or epirubicin (3 mg/kg/day), paclitaxel or docetaxel (2.5 mg/kg/day) | Perfused isolated hearts from 10–12-week-old male Sprague Dawley rats treated ip on alternate days for 11 days and sacrificed on 12 th day | Doxo: a 20% decrease in [LV(dP/dt)max] and a 33% decrease in [LV(dP/dt)min]; paclitaxel: No significant changes; doxo + paclitaxel: a 39% decrease in [LV(dP/dt)max] and a 46% decrease [LV(dP/dt)min] | [87] |
| 3 | Tyrosine kinase inhibitors: gefitinib, lapatinib ditosylate, dasatinib, sorafenib tosylate, erlotinib, sunitinib, imatinib | Ventricular myocytes were isolated from 2–3-day-old Sprague Dawley rats and plated in a culture dish. On the 5th day, cells were treated with drugs, and after 3 days, cells were lysed to measure LDH activity | Lapatinib, erlotinib, gefitinib: no cardiotoxicity; imatinib, sorafenib: mild; sunitinib, dasatinib: moderate toxicity. Loest LDH activity correlated with lLapatinib, and the highest was correlated with dasatinib. | [88] |
| 4 | Regorafenib (30 mg/kg/day) or sorafenib (60 mg/kg/day) | Eight-week-old CD2F1 male mice were treated with oral drugs for 6 weeks and sacrificed on day 42 | EF and FS: no change in stroke volume, heart weight, or LV mass; LVID: decreased. More severe effect observed with regorafenib. Increased phosphorylation of Akt/mTOR/P70S6K/GSK3β and MEK/ERK1/2 | [85] |
| 5 | Cisplatin (7 mg/kg i. p, twice a week) | 5-week-old male Nu/MRI nude mice with prostate cancer. At day 27, hearts were surgically removed and used for transmission electron microscopy analysis | Increased mitochondrial damage was seen (in terms of morphology, size, organization, and quantity) | [89] |
| 6 | Four doses of 5-FU, 150 mg/kg b.wt. | Wistar male rats weighing 170–200 gms were sacrified 2 weeks after last dose of 5-FU. BCKDH activity was assayed spectrophotometrically. The mRNA levels for E1, PPM1K, and BDK were quantified by real-time PCR | Increase in myocardial BCKDH activity state. mRNA level for BDK decreased, while mRNA levels for PPM1K Increased. This ultimately leads to the deterioration of cardiac functions. | [90] |
| 7 | Twin doses of 5-FU intraperitoneal (i.p.) injection of 25 mg/kg | Sprague Dawley rats aged 2 and 18 months had blood collected from tail vein on days 0, 7, and 14. On the 15th day, the rats were sacrificed | Weight loss and myocardial injury observed in rats. Ventricular enlargement, decrease in myocardial contractile function, and decrease in LVEF in aged rats. Cardiomyocyte apoptosis, myocardial mitochondrial damage, enhanced mitochondrial autophagy | [91] |
| 8 | THP (3 mg/kg) was injected via caudal vein once a week | SD rats (180–200 g) were sacrified at the end of the 8th week. | Decreased body weight and food intake. EF and FS decreased; LVIDd and LVIDs increased; R and T wave decreased, the S wave increased, and the QT interval was prolonged. | [92] |
| 9 | Cisplatin (2 mg/kg/day) daily by intraperitoneal injection for 1 week | Male albino rats (180–220 g) rats sacrificed on 8th day | Serum levels of CK and LDH increased. The muscle fibers showed disarrangement, discontinuities, and apoptotic nuclei (seen by electron microscopy) | [93] |
| 10 | Single dose of cisplatin (CP; 10 mg/kg) | Adult male Wistar rats (weighing 180–200 g) had blood samples collected from retro-orbital venous plexus.Rats sacrificed on 16th day | Elongation of QTc with ↑ ST height and T wave amplitude; ↑ HR. Increase in serum troponin T, LDH, and CK-MB. GSH and SOD activity reduced; caspase 12 gene expresson elevated. | [94] |
| 11 | CYP (200 mg/Kg, i.p.) as a single dose. | Male Wister rats (150 ± 20 g), aged 15–17-weeks old nlood collected from orbital sinus puncture 3 days after CYP | Cardiac Tn-I protein and LDH enzyme and serum levels of IL-1ß, IL-6, and TNF-a increased; wide-spread swelling and granular and vacuolar degeneration as well as myocardial separation with intramuscular edema. | [95] |
| 12 | Single IP injection of CYP (200 mg/kg) | Wistar albino rats (7-week-old) weighing 160–180 g were sacrficed on the 11th day, and blood was collected from abdominal aorta | Body weight loss with increased heart weight to body weight ratio; increased plasma CK, LDH, AST, and cTn I levels; enhanced apoptotic signaling; upregulated PARP1 and p53 in cardiac tissue | [96] |
| 13 | Single IP injection of CYP (200 mg/kg) | Male Wistar albino rats On the 11th day, the were rats sacrificed | Increased serum LDH, CK-MB, and troponin; decreased soluble α Klotho protein and evidence of histopathological lesions in cardiac tissues; decreased gene expression of ALDH2, Klotho protein, mTOR, IGF, AKT, AMPK, and BCL2; and increased expression of BAX and caspase-8. | [97] |
| 14 | Single intraperitoneal dose of 20 mg/kg MTX | Male Wistar rats weighing 180–210 g, and rats were sacrified on the 6th day | Fragmented necrotic muscle Fibers, apoptotic morphology with hyperacidophilic cytoplasm, nuclear pyknosis, and nuclear fragmentation. Hemorrhage and congestion, increase in cardiac NOX-2, MDA, and NO levels, and decrease in the level of GSH and in SOD. Increased TNF -α and IL-6 levels. | [98] |
| Serial No | Name of Drug | Study Design | Significant Outcome | References |
|---|---|---|---|---|
| 1. | Anthracycline (doxorubicin or epirubicin) | Clinical study with 140 prospectively recruited patients assessed by transthoracic echocardiogram within 7 days post-treatment | LVEF, GLS, and global circumferential straindecreased; LVESVincreased; no changes were observed in LVEDV | [102] |
| 2. | Anthracycline or anthracycline plus trastuzumab | Analytical, observational prospective cohort study of 100 breast cancer patients,; echo cardiogram and biochemical markers measured during 4 visits (pre-treatment, immediately after treatment, and 3- and 9-months post treatment) | Significant decrease in LVEF, increase in hsTNT, NTproBNP, and FABP | [103] |
| 3. | Tamoxifen | Prospective observational study of 30 breast cancer patients; cardiac and oxidative stress markers measured before and 6- and 12-months post treatment | Elevated cTnI and AOPP and decreased GPx | [59] |
| 4. | Pertuzumab following trastuzumab | Two Breast cancer patients | Left ventricular dysfunction | [104] |
| 5. | Pertuzumab | Open-label, phase II, multicenter, randomized study, 78HER2 negative metastatic breast cancer patients | 8 Patients had decrease in LVEF | [105] |
| 6. | ICI (pembrolizumab, ipilimumab, atezolizumab, avelumab) | Retrospective and prospective study involving 8 centers and 35 patients with ICI-associated myocarditis and 105 ICI-treated patients without myocarditis followed up with for 102 days | 50% Patients on myocarditis developed MACE cardiovascular death, cardiogenic shock, and cardiac arrest | [106] |
| 7. | Newer generation tyrosine kinase inhibitors (NTKI), such as nilotinib, ponatinib, dasatinib, and older generation TKI (imatinib) | 55 patients with GIST and CML; evaluation by ECG, echo, and arterial scans | More frequent cardiovascular dysfunction with newer generation of TKI | [100] |
| Trial Identifier | Estimated No of Participants | Status/Phase/Results (If) | Primary Outcome | Conditions | Interventions |
|---|---|---|---|---|---|
| NCT00292526 | 114 | Completed/4/Increase in cTnI following HDC predicts the suppression of LVEF [147] | Incidence of chemotherapy-induced cardiotoxicity | Cardiotoxicity | Enalapril |
| NCT03389724 | 200 | Recruiting/3 | The effect of ACE-I in preventing chemotherapy-related cardiotoxicity measuring in troponin I levels and cardiac imaging | Cardiotoxicit, AML in children | Capoten |
| NCT01724450 | 200 | Completed/3/No effect on incidence of LVEF reduction, significant decrease in troponin levels and diastolic dysfunction [148] | Prevention of systolic dysfunction in patients receiving anthracycline | Breast cancer, heart failure | Carvedilol |
| NCT04023110 | 110 | Recruiting/1 | To measure the left ventricular ejection fraction (LVEF) | Cardiotoxicity, breast cancer | Carvedilol |
| NCT03650205 | 160 | Recruiting/not applicable | Reduction in global longitudinal strain of at least 10% (GLS) | Heart failure, chemotherapy | Ivabradine |
| NCT02943590 | 300 | Active/2 | To determine if statins preserve the LVEF at 12 months | Heart failure | Atorvastatin |
| NCT03186404 | 112 | Recruiting/2 | To compare the cardiac MRI measured LVEF between placebo and statin group | Cancer, heart failure, cardiotoxicity | Atorvastatin |
| NCT03949634 | 272 | Unknown/3 | Congestive heart failure with clinical symptoms or no symptoms but an abnormal LVEF | Early breast cancer | Cyclophosphamide, pegylated liposomal doxorubicin |
| NCT03934905 | 70 | Not yet recruiting/1 and 2 | Change in cardiac function (by 2D echo) after DOX therapy with or without sulforaphane | Anthracycline-related cardiotoxicity in breast cancer | Sulforaphane (nutritional supplement) |
| NCT02796365 | 29 | Completed/not available | Left ventricular strain by spectral Doppler | Doxorubicin-induced cardiomyopaty, breast cancer, gastric cancer, leukaemia | Exercise |
| NCT02006979 | 27 | Completed/1/ Exercise reduced LV twist and NTproBNP and increased cTropT and GLS in the intervention group compared to control | Global longitudinal strain by 2D speckle tracking echocardiography | Breast cancer | Exercise < 24 h prior to each cycle of anthracyclines |
| NCT02472353 | 30 | Terminated did not meet target accrual/2 Results available | Whether the addition of metformin will decrease the incidence of change in left ventricle ejection fraction | Breast cancer | Metformin, doxorubicin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, S.; Singh, P.K.; Roy, P.; Kakar, S.S. Cardiac Cachexia: Unaddressed Aspect in Cancer Patients. Cells 2022, 11, 990. https://doi.org/10.3390/cells11060990
Saha S, Singh PK, Roy P, Kakar SS. Cardiac Cachexia: Unaddressed Aspect in Cancer Patients. Cells. 2022; 11(6):990. https://doi.org/10.3390/cells11060990
Chicago/Turabian StyleSaha, Sarama, Praveen Kumar Singh, Partha Roy, and Sham S. Kakar. 2022. "Cardiac Cachexia: Unaddressed Aspect in Cancer Patients" Cells 11, no. 6: 990. https://doi.org/10.3390/cells11060990
APA StyleSaha, S., Singh, P. K., Roy, P., & Kakar, S. S. (2022). Cardiac Cachexia: Unaddressed Aspect in Cancer Patients. Cells, 11(6), 990. https://doi.org/10.3390/cells11060990